

Abstract
Background
Microalgae have been demonstrated to be among the most promising phototrophic species for producing renewable biofuels and chemicals. Ethanol and butanol are clean energy sources with good chemical and physical properties as alternatives to gasoline. However, biosynthesis of these two biofuels has not been achieved due to low tolerance of algal cells to ethanol or butanol.
Results
With an eye to circumventing these problems in the future and engineering the robust alcohol-producing microalgal hosts, we investigated the metabolic responses of the model green alga Chlamydomonas reinhardtii to ethanol and butanol. Using a quantitative proteomics approach with iTRAQ-LC–MS/MS technologies, we detected the levels of 3077 proteins; 827 and 730 of which were differentially regulated by ethanol and butanol, respectively, at three time points. In particular, 41 and 59 proteins were consistently regulated during at least two sampling times. Multiple metabolic processes were affected by ethanol or butanol, and various stress-related proteins, transporters, cytoskeletal proteins, and regulators were induced as the major protection mechanisms against toxicity of the organic solvents. The most highly upregulated butanol response protein was Cre.770 peroxidase.
Conclusions
The study is the first comprehensive view of the metabolic mechanisms employed by C. reinhardtii to defend against ethanol or butanol toxicity. Moreover, the proteomic analysis provides a resource for investigating potential gene targets for engineering microalgae to achieve efficient biofuel production.
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-017-0931-9) contains supplementary material, which is available to authorized users.
Keywords: Ethanol, Butanol, Proteomics, Chlamydomonas, Microalgae
Background
Renewable biofuels are the obvious alternatives to fossil fuels in order to promote sustainable development of the economy and society as well as to reduce carbon dioxide emission [1]. The first generation of biofuels was produced from terrestrial crops such as corn, sugarcane, and rapeseed [2, 3]. However, excessive consumption of these agricultural products may pose a significant threat to the food supply around the world. Therefore, various forms of lignocellulosic biomass were used in the production of the second generation of biofuels [2]. Nonetheless, there is still possible consequence of forest destruction or competing land use. Microalgae are photosynthetic microorganisms that can grow using carbon dioxide as sole carbon source and with minimal nutrient requirements. Diverse microalgal species have been found to be of high photoefficiency and productivity, greatly exceeding that of agricultural crops. Thus, microalgae are attractive cell factories that can produce large amounts of carbohydrates and lipids during short periods of cultivating time in a relatively small water area without competing for arable land [1, 4, 5]. The algal biomass can be processed in place of terrestrial plants for renewable energy production, namely the third generation biofuels [2].
Based on the advances in genome annotation and genetic engineering of microalgae, the fourth generation of biofuels has been suggested to be produced through metabolic engineering of microalgae [6, 7]. Another economic strategy is to allow fuels or precursors to be directly secreted into the growth medium by manipulating the biology of algal cells as reported for Saccharomyces cerevisiae [8]. Secretion is more feasible for low molecular weight and hydrophobic compounds, such as hydrogen, alkanes, ethanol, and butanol. The production of hydrogen and alkanes have been reported in microalgae [9, 10], but the biosynthesis of ethanol or butanol has not been explored in this kind of organism.
As alternatives to gasoline, ethanol and butanol have been on the list of desirable biofuels of all generations [11–13]. Ethanol is currently the most common renewable biofuel produced from fermentation of food starches or lignocellulose by S. cerevisiae or Zymomonas mobilis. Considering the drawbacks of traditional biofuel production processes, it was attractive to directly couple ethanol production to carbon fixation in photosynthetic microorganisms, including cyanobacteria and eukaryotic microalgae [4]. In a previous study, ethanol synthesis was obtained by introducing pyruvate decarboxylase and alcohol dehydrogenase genes from Z. mobilis into a unicellular cyanobacterium, Synechococcus [14], indicating the feasibility of constructing an integrated system for converting solar energy and inorganic carbon source into biofuel directly. Butanol is also an industrialized biofuel with high energy density and good storage property compared with ethanol. Besides, butanol has been produced from the fermentation of sugar or starch using solventogenic clostridial strains [15]. Because bacteria of the genus Clostridium usually has a low growth rate and is difficult to be genetically manipulated, the butanol biosynthesis pathway has been introduced into S. cerevisiae [16], Escherichia coli [17] and Synechococcus [18] for further research. It is also promising to create similar metabolic pathways for ethanol or butanol production in microalgae. The obvious problem for alcohol bioproduction is that, as organic solvents, ethanol and butanol are toxic to microbes including the producers [19]. Therefore, it is important to select highly tolerant microbial strains for industrial production of these biofuels.
Both omics methods and genetic manipulation have been used to examine the organic solvent tolerance of microbes [20–25]. By quantitative transcriptomic analysis and mutant examination, two genes (i.e., slr0724 and sll1392) were validated to be involved in ethanol resistance of Synechocystis [26]. Based on the results of transcriptomic and proteomic analysis, a novel regulator Slr1037 and three butanol response genes: sll0690, slr0947, and slr1295 have been explored by gene knockout experiments and were found to be involved in butanol resistance in Synechocystis [27, 28]. Ethanol tolerance of S. cerevisiae was enhanced by mutagenesis of the TATA-binding protein gene [29]. Improvement in isobutanol tolerance was observed in E. coli by simultaneous disruption of five unrelated genes [30]. Together these studies hint at the complexity of microbial resistance mechanisms in response to the stress of a single biofuel. Therefore, the tolerance mechanisms of microalgae should be fully investigated before optimal engineering this organism for ethanol or butanol production can take place.
Chlamydomonas reinhardtii is currently used as a model alga for revealing molecular mechanisms of biofuel production in eukaryotic microalgae [31] and various molecular techniques have been developed to manipulate the nuclear and organellar genomes of this organism [32]. In this study, a quantitative proteomics method with isobaric tag for relative and absolute quantitation (iTRAQ) technique and liquid chromatography-tandem mass spectrometry (LC–MS/MS) was applied to explore the global metabolic responses of C. reinhardtii under ethanol or butanol exposure. Protein annotation, gene ontology (GO), and KEGG-enrichment analysis were performed to find response proteins. The expression differences of proteins were further verified by real-time quantitative reverse transcription PCR (qRT-PCR).
Results and discussion
Effects of ethanol and butanol on growth of C. reinhardtii CC-849
The growth of C. reinhardtii CC-849 supplemented with different concentrations of ethanol or butanol was assessed to determine appropriate ethanol or butanol concentrations for proteomic analysis (Fig. 1a, b). All the tested concentrations of ethanol or butanol were found to be inhibitory. The results also showed that 1.8% ethanol and 0.3% butanol, respectively, caused 50% growth decrease at 48 h (corresponding to middle-exponential phase), and therefore these two concentrations were selected for further analysis in this study. The tolerance level of C. reinhardtii CC-849 to ethanol was slightly higher than the 1.5% ethanol tolerance level of the cyanobacterium Synechocystis sp. PCC 6803 [22] and was much lower than the famously high 25% ethanol tolerance level of S. cerevisiae [33]. The tolerance level of C. reinhardtii CC-849 to butanol was 50% higher than the 0.2% butanol tolerance level of Synechocystis sp. PCC 6803 [23]. However, the butanol tolerance of C. reinhardtii CC-849 was weaker than that of the traditional butanol producer Clostridium acetobutylicum [21] and other microbial hosts including E. coli, Z. mobilis, Pseudomonas putida, and S. cerevisiae [34–36].
Fig. 1.
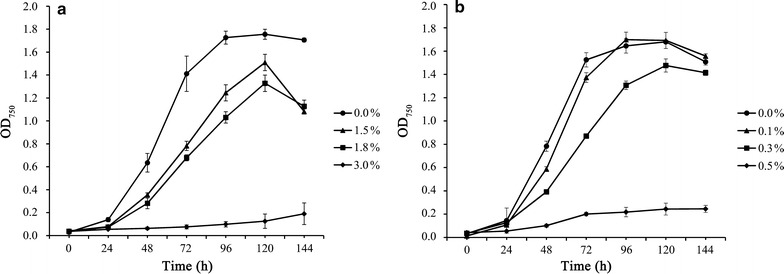
Effects of ethanol (a) and butanol (b) on the growth of C. reinhardtii CC-849
Cell morphology in ethanol, butanol, and control treatments were checked under the light microscope, and the results showed that the density of individual cells under ethanol or butanol stress was found to be lower than those of control treatments (Additional file 1: Figure S1). On the contrary, visible aggregation of a large number of cells was found for Synechocystis sp. PCC 6803 under ethanol or butanol treated conditions [22, 23]. This morphology likely coincides with the lower tolerance of Synechocystis to biofuels than C. reinhardtii.
For proteomic analysis, cultivations at 1.8% ethanol, 0.3% butanol, and control treatments were conducted in triplicate. Algal cells were collected at early exponential (24 h), middle exponential (48 h) and exponential-stationary transition (72 h) phases of the cell growth, respectively (Fig. 1a, b). Parallel cell samples were mixed resulting in proteomic samples at three time points for each treatment.
Overview of quantitative proteomics analysis
After data filtering to eliminate peptides without labeling and reverse-matching peptides, a total of 3077 unique proteins were detected (Additional file 2: Table S1), representing approximately 20% of the 15,143 predicted proteins in the draft genome of C. reinhardtii CC-503 [37]. This percentage was close to the overlapping proteome coverage of 25% using several genome annotation databases in a previous study [38], suggesting the methodology we used in the study is reliable. Analysis on eukaryotic cluster of orthologous groups of proteins (KOG) classified 2301 proteins into 25 functional categories, covering almost every aspect of C. reinhardtii metabolism (Fig. 2). According to the number of unique proteins identified in each functional category, the three most frequently detected functional categories were “posttranslational modification, protein turnover, chaperones”; “translation, ribosomal structure, and biogenesis”; and “general function prediction only,” each representing 12.33, 10.00, and 9.86% of all the proteins identified. Other well-detected functional categories included “signal transduction mechanisms”; “intracellular trafficking, secretion, and vesicular transport”; “energy production and conversion”; “amino acid transport and metabolism”; and “RNA processing and modification,” each representing over 5% of all the proteins identified.
Fig. 2.
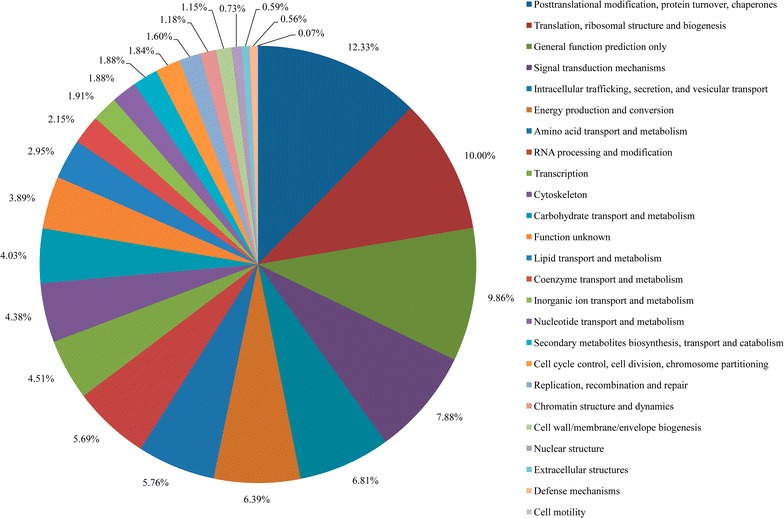
KOG coverage of the proteins detected
Using a cutoff of 2.0-fold change and a P value less than 0.05, we determined that 827 unique proteins were differentially regulated between ethanol and control treatments. The numbers of differential proteins were 89, 193, and 713 at 24, 48, and 72 h, respectively, and far greater differences were observed at 72 h (Fig. 3a, b, Additional file 3: Table S2). During at least two sampling times, 10 and 31 proteins were consistently upregulated and downregulated, respectively (Fig. 3a, b, Table 1).
Fig. 3.
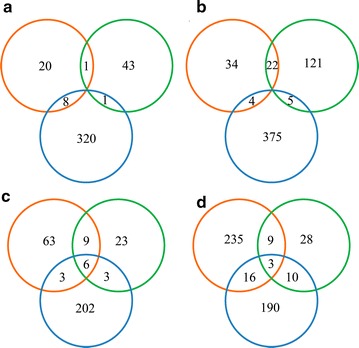
Distribution of upregulated (a, c) and downregulated (b, d) proteins at three time points in ethanol (a, b) and butanol (c, d) treatments. The three sampling times, 24, 48, and 72 h, are represented by orange, green, and blue circles, respectively
Table 1.
Differentially regulated proteins in ethanol treatment
| UniProtKB identifiera | Ethanol vs control | Description | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Upregulated proteins | ||||
| A8JID6 | 2.68 | 0.41 | 9.20 | Plastidic ADP/ATP translocase, AAA1 |
| A8JG58 | 2.38 | 2.33 | Hypothetical protein | |
| A8JC09 | 2.36 | 2.15 | Flagellar-associated protein, FAP151 | |
| A8IKQ0 | 2.36 | 9.46 | Fructose-1, 6-bisphosphatase, FBP1 | |
| A8IL29 | 2.21 | 3.53 | Small ARF-related GTPase, ARFA1A | |
| P22675 | 2.19 | 4.13 | Argininosuccinate lyase, ARG7 | |
| Q5NKW4 | 2.09 | 2.47 | 0.25 | Photosystem I reaction center subunit II, PsaD |
| Q7X7A7 | 2.07 | 9.04 | 14-3-3-like protein, Erb14 | |
| P06007 | 2.01 | 8.87 | Photosystem II protein D2, PsbD | |
| A8IRL8 | 6.61 | 3.16 | Hypothetical protein | |
| Downregulated proteins | ||||
| A8JDR3 | 0.46 | 0.41 | 3.13 | Ribonucleoside-diphosphate reductase, RIR1 |
| A8J567 | 0.45 | 0.48 | 4.13 | Cytosolic ribosomal protein L7a, RPL7a |
| A8IZV9 | 0.45 | 0.11 | Flagellar-associated protein, FAP102 | |
| A8JDV9 | 0.45 | 0.48 | 11.59 | F1F0 ATP synthase gamma subunit, ATP3 |
| A8IVE2 | 0.45 | 0.46 | Cytosolic ribosomal protein L7, RPL7 | |
| A8IVP7 | 0.44 | 0.42 | Predicted protein | |
| A8J9E9 | 0.43 | 0.48 | Carotenoid isomerase | |
| Q1WLZ0 | 0.43 | 0.48 | 14.59 | 60S ribosomal protein L11 |
| A8JAG1 | 0.41 | 0.28 | Hypothetical protein | |
| A8J387 | 0.41 | 0.43 | 16.14 | Hypothetical protein |
| A8HVQ1 | 0.39 | 0.47 | 16.00 | Cytosolic ribosomal protein S8, RPS8 |
| A8I7P5 | 0.38 | 0.41 | 6.49 | Magnesium chelatase subunit H, CHLH1 |
| A8HVM3 | 0.36 | 0.46 | 8.87 | Hypothetical protein |
| A8HS48 | 0.34 | 0.15 | 20.32 | 40S ribosomal protein S3a, RPS3a |
| E3SC57 | 0.34 | 0.19 | 18.88 | 60S ribosomal protein L3, RPL3 |
| A8JHB4 | 0.32 | 0.33 | 7.94 | Ferredoxin-dependent glutamate synthase, GSF1 |
| A8J9F3 | 0.32 | 0.31 | 16.00 | Hypothetical protein |
| A8IWJ5 | 0.29 | 0.46 | 11.70 | Predicted protein |
| A8IGY1 | 0.29 | 0.22 | 13.68 | Cytosolic ribosomal protein S13, RPS13 |
| A8J914 | 0.27 | 0.47 | UDP-glucose dehydrogenase, UGD2 | |
| A8HMG7 | 0.26 | 0.34 | 12.47 | Cytosolic ribosomal protein L26, RPL26 |
| A8JGX5 | 0.25 | 0.36 | 16.00 | Protein arginine N-methyltransferase, PRMT2 |
| A8I647 | 0.24 | 0.47 | 7.94 | Zeta-carotene desaturase, ZDS1 |
| A8HYU2 | 0.23 | 0.49 | Vacuolar H+ ATPase V1 sector, subunit C, ATPvC | |
| A8JHC3 | 0.21 | 0.29 | 9.38 | Cytosolic ribosomal protein S11, RPS11 |
| A8J8M5 | 0.21 | 0.25 | 10.47 | Plastid ribosomal protein S5, PRPS5 |
| A8HQP0 | 0.46 | 0.38 | Transaldolase, TAL1 | |
| A8ILP2 | 0.48 | 0.19 | Predicted protein | |
| A8J9H8 | 0.13 | 0.15 | Nucleoside diphosphate kinase, FAP103 | |
| Q2HZ22 | 0.32 | 0.07 | Putative ferredoxin, FDX5 | |
| Q42690 | 0.45 | 0.20 | Chloroplastic fructose-bisphosphate aldolase 1, FBA3 | |
aProteins upregulated or downregulated during at least two sampling times are displayed
Comparison between butanol and control treatments revealed that 730 unique proteins were differentially regulated. The numbers of differential proteins were 344, 91, and 433 at 24, 48, and 72 h, respectively (Fig. 3c, d, Additional file 4: Table S3). During at least two sampling times, 21 and 38 proteins were consistently upregulated and downregulated, respectively (Fig. 3c, d, Table 2). Only one downregulated hypothetical protein (A8IVP7) was shared between ethanol and butanol treatments (Tables 1 and 2), indicating different metabolic responses to these two organic solvents in C. reinhardtii.
Table 2.
Differentially regulated proteins in butanol treatment
| UniProtKB identifiera | Butanol vs control | Description | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Upregulated proteins | ||||
| A8J7T7 | 6.03 | 2.44 | Cysteine endopeptidase, CEP1 | |
| L8B958 | 4.49 | 3.11 | Pyruvate ferredoxin oxidoreductase, PFO | |
| Q66YD0 | 4.41 | 3.08 | Chloroplast vesicle-inducing protein in plastids 1, VIPP1 | |
| O64925 | 4.06 | 3.91 | 2.63 | Granule-bound starch synthase, STA2 |
| A8JHZ9 | 3.66 | 5.30 | 2.27 | Hypothetical protein |
| A8I980 | 3.47 | 2.51 | 2.36 | Delta-aminolevulinic acid dehydratase, ALAD |
| O49822 | 3.44 | 2.61 | Ascorbate peroxidase, Apx1 | |
| A8IX35 | 2.91 | 2.36 | 6.61 | Hypothetical protein |
| A8J0W9 | 2.88 | 2.29 | NAD-dependent malate dehydrogenase, MDH3 | |
| A8JHP0 | 2.78 | 2.17 | Oxidoreductase-like protein, CPLD35 | |
| A8J6Y3 | 2.70 | 2.91 | 2.03 | Hypothetical protein |
| A8J000 | 2.58 | 2.23 | Acetate kinase, ACK1 | |
| A8IYS5 | 2.54 | 2.94 | 2.42 | Septin-like protein, SEP1 |
| A8I0K9 | 2.33 | 3.28 | Dehydroascorbate reductase, DHAR | |
| A8IZU0 | 2.29 | 2.05 | Heat shock protein 70C, Hsp70C | |
| A8HVU5 | 2.27 | 2.36 | Phosphoglycerate mutase, PGM1a|PGM1b | |
| Q9ZSJ4 | 2.19 | 4.02 | Light-harvesting complex II protein, Lhcb3 | |
| O22448 | 2.01 | 3.66 | Glutathione peroxidase homolog, Gpxh | |
| A8IUI1 | 3.91 | 2.75 | Hypothetical protein | |
| A8JBW0 | 3.44 | 2.91 | Hypothetical protein | |
| A8JGL7 | 3.98 | 38.37 | Heme peroxidase-related protein, Cre.770 | |
| Downregulated proteins | ||||
| A8J597 | 0.49 | 0.29 | Cytosolic ribosomal protein L12, RPL12 | |
| A8IZ36 | 0.47 | 0.16 | Cytosolic ribosomal protein S25, RPS25 | |
| A8JGI9 | 0.46 | 0.33 | Cytosolic ribosomal protein S7, RPS7 | |
| A8JE91 | 0.46 | 0.40 | Chaperonin 60B1, CPN60B1 | |
| A8HTY0 | 0.44 | 0.09 | Plastid ribosomal protein L7/L12, PRPL7/L12 | |
| A8JIB7 | 0.43 | 0.49 | Chaperonin 60A, CPN60A | |
| A8IAN1 | 0.40 | 0.47 | 0.15 | Transketolase, TRK1 |
| A8J768 | 0.39 | 0.28 | Ribosomal protein S14, RPS14 | |
| A8JDM1 | 0.39 | 0.34 | Hypothetical protein | |
| A8IV98 | 0.39 | 0.36 | DEAD box RNA helicase | |
| A8J503 | 0.38 | 0.24 | Plastid ribosomal protein L6, PRPL6 | |
| A8IVZ9 | 0.36 | 0.32 | Glutamine synthetase, GLN2 | |
| A8JAP7 | 0.34 | 0.20 | Hypothetical protein | |
| A8IAT4 | 0.34 | 0.47 | 0.24 | Acetohydroxy acid isomeroreductase, AAI1 |
| A8JCQ8 | 0.33 | 0.34 | Acetyl CoA synthetase, ACS2 | |
| A8J9D9 | 0.33 | 0.38 | Plastid ribosomal protein L24, PRPL24 | |
| A8I403 | 0.33 | 0.19 | Cytosolic ribosomal protein S19, RPS19 | |
| A8JFR9 | 0.30 | 0.23 | 0.28 | Acetyl CoA synthetase, ACS3 |
| A8J785 | 0.30 | 0.14 | Chloroplastic ATP synthase subunit b’, ATPG | |
| A8J841 | 0.29 | 0.48 | Hydroxymethylpyrimidine phosphate synthase, THICb|THICa | |
| A8IVS6 | 0.28 | 0.40 | Hypothetical protein | |
| A8ITX0 | 0.28 | 0.46 | Copper response target 1 protein, CRD1 | |
| A8IUV7 | 0.27 | 0.36 | Cytosolic ribosomal protein L13, RPL13 | |
| A8JGT1 | 0.24 | 0.15 | RNA helicase, RHE | |
| A8IW20 | 0.22 | 0.49 | Eukaryotic initiation factor, EIF4G | |
| A8I0R6 | 0.22 | 0.47 | Hypothetical protein | |
| A8HP90 | 0.19 | 0.39 | Cytosolic ribosomal protein L6, RPL6 | |
| A8I9M5 | 0.17 | 0.31 | 10.86 | Hypothetical protein |
| A8HUK0 | 0.45 | 0.28 | Peptidyl-prolyl cis–trans isomerase, FKB12 | |
| A8I495 | 0.39 | 0.38 | Obg-like ATPase 1, OLA1 | |
| A8IK91 | 0.45 | 0.34 | Translocon component Tic40-related protein, TIC40 | |
| A8IPS8 | 0.35 | 0.16 | Hypothetical protein | |
| A8IVP7 | 0.26 | 0.43 | Hypothetical protein | |
| A8J2S0 | 0.29 | 0.43 | Citrate synthase, CIS2 | |
| A8J6J6 | 0.34 | 0.18 | Acetyl-CoA acyltransferase, ATO1 | |
| A8JCW5 | 0.40 | 0.38 | Hypothetical protein | |
| A8JHB7 | 0.33 | 0.18 | Hypothetical protein | |
| Q9ZTA7 | 0.49 | 0.33 | Protoporphyrinogen oxidase, Ppx1 | |
aProteins upregulated or downregulated during at least two sampling times are displayed
qRT-PCR validation of the proteomic analysis
To examine the reliability of iTRAQ analysis, a subset of 20 genes were selected for qRT-PCR analysis, ten genes for ethanol and butanol treatments, respectively. These genes were chosen based on the expression levels of their corresponding proteins. Among them, ten proteins were upregulated (i.e., AAA1, FAP151, FBP1, ARFA1A, PsaD, PsbD, ALAD, MDH3, Hsp70C, and Cre.770), and ten proteins were downregulated (i.e., RIR1, FAP102, CHLH1, PRMT2, RPL12, CPN60A, GLN2, AAI1, RPL6, and CIS2) according to the proteomic analyses. As displayed in Fig. 4, the values higher than zero represents upregulation and those lower than zero represents downregulation. The RNA level of differential proteins varied with the same pattern to the regulation of protein level during at least two sampling times. Although the correlation between RNA expression and protein abundance is usually very weak, visible positive correlation was observed between proteomic analysis and qRT-PCR analysis, suggesting an overall good quality of the iTRAQ results.
Fig. 4.
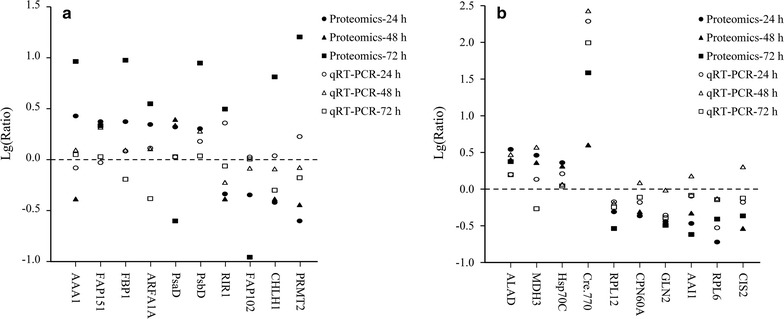
Comparison of ratios calculated from proteomic and qRT-PCR analyses. a Ethanol treatment; b butanol treatment. The values were calculated by lg(Ratio)
Enrichment of GO terms
GO analysis was performed by matching the identified proteins to the proteins annotated with GO terms, and 2310 proteins were annotated (Additional file 5: Table S4). GO enrichment analysis was then carried out to determine the affected cellular metabolisms. As a result, 129 and 126 unique GO terms were enriched for the ethanol and butanol treatments, respectively (Additional file 6: Table S5 and Additional file 7: Table S6). For ethanol treatment, 21 GO terms were shared by at least two time points, including six terms for molecular function and 15 terms for biological process (Table 3). The results showed that three main categories of molecular functions, including “structural constituent of ribosome”; “structural molecule activity”; and “ligase activity, forming carbon–oxygen bonds, aminoacyl-tRNA, and related compounds” were affected by ethanol. Several key biological processes, including “translation and its regulation,” “amino acid activation and tRNA aminoacylation,” “amino acid metabolic process,” “protein metabolic process and its regulation,” and “macromolecule and organic substance biosynthetic processes” were also confirmed to be affected by ethanol. Therefore, protein biosynthesis was most seriously affected by ethanol. In a previous RNA-seq study, genes related with protein biosynthesis were also found to be affected by ethanol in Synechocystis sp. PCC6803 [26].
Table 3.
GO enrichment analysis of differentially regulated proteins in ethanol treatment
| GO terma | GO ID | P valueb | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Molecular function | ||||
| Structural constituent of ribosome | GO:0003735 | 0.002 | 0.003 | 0.000 |
| Aminoacyl-tRNA ligase activity | GO:0004812 | 0.000 | 0.011 | |
| Structural molecule activity | GO:0005198 | 0.001 | 0.000 | 0.000 |
| Ligase activity | GO:0016874 | 0.002 | 0.002 | |
| Ligase activity, forming carbon–oxygen bonds | GO:0016875 | 0.000 | 0.011 | |
| Ligase activity, forming aminoacyl-tRNA and related compounds | GO:0016876 | 0.000 | 0.011 | |
| Biological process | ||||
| Translation | GO:0006412 | 0.000 | 0.000 | 0.000 |
| Regulation of translation | GO:0006417 | 0.002 | 0.015 | |
| Cellular amino acid metabolic process | GO:0006520 | 0.008 | 0.032 | |
| Metabolic process | GO:0008152 | 0.042 | 0.005 | |
| Biosynthetic process | GO:0009058 | 0.046 | 0.001 | |
| Macromolecule biosynthetic process | GO:0009059 | 0.007 | 0.002 | 0.003 |
| Protein metabolic process | GO:0019538 | 0.001 | 0.002 | 0.000 |
| Regulation of cellular protein metabolic process | GO:0032268 | 0.008 | 0.005 | |
| Cellular macromolecule biosynthetic process | GO:0034645 | 0.007 | 0.002 | 0.003 |
| Amino acid activation | GO:0043038 | 0.000 | 0.010 | |
| tRNA aminoacylation | GO:0043039 | 0.000 | 0.010 | |
| Cellular metabolic process | GO:0044237 | 0.043 | 0.000 | |
| Cellular protein metabolic process | GO:0044267 | 0.001 | 0.001 | 0.000 |
| Regulation of protein metabolic process | GO:0051246 | 0.018 | 0.008 | |
| Organic substance biosynthetic process | GO:1901576 | 0.032 | 0.000 | |
aGO terms present during at least two sampling times are displayed
b P values were calculated by Chi-square test
For butanol treatment, 22 GO terms were shared by at least two time points, including seven terms for molecular function and 15 terms for biological process (Table 4). The results revealed that six main categories of molecular functions, including “ribonucleoside binding,” “adenyl ribonucleotide binding,” “structural constituent of ribosome,” “structural molecule activity,” “rRNA binding,” and “tetrapyrrole binding” were affected by butanol. Several key biological processes, including “gene expression”; “translation”; “protein, monosaccharide, hexose, and organic substance metabolic process”; and “macromolecule and organic substance biosynthetic process” were also confirmed to be affected by butanol. Effects of butanol on the “structural molecule activity,” “RNA binding,” and “gene expression processes” were also reported for Synechocystis sp. PCC6803 [23].
Table 4.
GO enrichment analysis of differentially regulated proteins in butanol treatment
| GO terma | GO ID | P valueb | ||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Molecular function | ||||
| Nucleoside binding | GO:0001882 | 0.043 | 0.020 | |
| Structural constituent of ribosome | GO:0003735 | 0.000 | 0.000 | |
| Structural molecule activity | GO:0005198 | 0.000 | 0.000 | |
| rRNA binding | GO:0019843 | 0.035 | 0.028 | |
| Ribonucleoside binding | GO:0032549 | 0.043 | 0.020 | |
| Adenyl ribonucleotide binding | GO:0032559 | 0.045 | 0.003 | |
| Tetrapyrrole binding | GO:0046906 | 0.042 | 0.007 | |
| Biological process | ||||
| Monosaccharide metabolic process | GO:0005996 | 0.033 | 0.019 | |
| Translation | GO:0006412 | 0.000 | 0.000 | |
| Metabolic process | GO:0008152 | 0.042 | 0.024 | |
| Biosynthetic process | GO:0009058 | 0.000 | 0.004 | |
| Macromolecule biosynthetic process | GO:0009059 | 0.000 | 0.002 | |
| Gene expression | GO:0010467 | 0.000 | 0.049 | |
| Hexose metabolic process | GO:0019318 | 0.029 | 0.015 | |
| Protein metabolic process | GO:0019538 | 0.000 | 0.000 | |
| Cellular macromolecule biosynthetic process | GO:0034645 | 0.000 | 0.002 | |
| Cellular metabolic process | GO:0044237 | 0.002 | 0.009 | |
| Primary metabolic process | GO:0044238 | 0.009 | 0.006 | |
| Cellular biosynthetic process | GO:0044249 | 0.000 | 0.004 | |
| Cellular protein metabolic process | GO:0044267 | 0.000 | 0.000 | |
| Organic substance metabolic process | GO:0071704 | 0.003 | 0.021 | |
| Organic substance biosynthetic process | GO:1901576 | 0.000 | 0.001 | |
aGO terms present during at least two sampling times are displayed
b P values were calculated by Chi-square test
Enrichment of KEGG pathways
KEGG analysis was performed to reveal the affected metabolic pathways. For ethanol treatment, the “ribosome” pathway was significantly affected in three growth phases, which coincided with the enriched GO terms and suggesting that this pathway was very active upon ethanol exposure (Table 5). In addition, the “aminoacyl-tRNA biosynthesis” pathway was active in early-exponential phase, while the “oxidative phosphorylation” and “porphyrin and chlorophyll metabolism” pathways were active in the exponential-stationary transition phase. For butanol treatment, the “ribosome,” “microbial metabolism in diverse environments,” “carbon metabolism,” and “carbon fixation in photosynthetic organisms” pathways were significantly affected in at least two growth phases, suggesting that these pathways were very active under butanol stress (Table 5). In addition, the “biosynthesis of secondary metabolites,” “porphyrin and chlorophyll metabolism,” and “RNA degradation” pathways were active in middle-exponential phase, while the “biosynthesis of amino acids,” “photosynthesis,” and “methane metabolism” pathways were active in exponential-stationary transition phase. Association of the ethanol- or butanol-responsive proteins with multiple GO terms and KEGG pathways suggested that algal cells employ multiple and synergistic mechanisms in resistance to single biofuel stress. Adaptations in a diversity of cellular processes were also observed in evolved isobutanol-tolerant E. coli strains [39] and a mutant C. acetobutylicum strain with enhanced butanol tolerance and yield [21].
Table 5.
KEGG pathway enrichment analysis of differentially regulated proteins in ethanol and butanol treatments
| Pathway term | Pathway ID | Protein number | P valuea | ||||
|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 72 h | 24 h | 48 h | 72 h | ||
| Ethanol treatment | |||||||
| Ribosome | ko03010 | 11 | 17 | 58 | 0.007 | 0.012 | 0.000 |
| Aminoacyl-tRNA biosynthesis | ko00970 | 8 | 0.000 | ||||
| Oxidative phosphorylation | ko00190 | 32 | 0.011 | ||||
| Porphyrin and chlorophyll metabolism | ko00860 | 20 | 0.011 | ||||
| Butanol treatment | |||||||
| Ribosome | ko03010 | 53 | 42 | 0.000 | 0.000 | ||
| Biosynthesis of secondary metabolites | ko01110 | 16 | 0.015 | ||||
| Microbial metabolism in diverse environments | ko01120 | 10 | 43 | 0.026 | 0.034 | ||
| Carbon metabolism | ko01200 | 9 | 38 | 0.014 | 0.010 | ||
| Porphyrin and chlorophyll metabolism | ko00860 | 4 | 0.019 | ||||
| Carbon fixation in photosynthetic organisms | ko00710 | 4 | 13 | 0.027 | 0.046 | ||
| RNA degradation | ko03018 | 4 | 0.011 | ||||
| Biosynthesis of amino acids | ko01230 | 33 | 0.039 | ||||
| Photosynthesis | ko00195 | 18 | 0.005 | ||||
| Methane metabolism | ko00680 | 11 | 0.041 | ||||
a P values were calculated by Chi-square test
Butanol affects stress-related proteins
Exposure to organic solvent is an environmental stress to microbes such as E. coli, C. acetobutylicum, Synechocystis, Z. mobilis and S. cerevisiae [19, 22, 23, 40–42]. Prokaryotes and eukaryotes may respond to environmental stress by upregulation of heat shock proteins (Hsps). Previous studies have revealed that Hsps were relevant to ethanol or butanol tolerance in Synechocystis [22, 23]. The Hsps are chaperons that mediate the correct folding of proteins to prevent the aggregation of misfolded proteins and repair intracellular injury during a variety of stress conditions [43]. In this study, Hsp70C was upregulated in C. reinhardtii under butanol exposure. Hsp70C is localized to mitochondria and is inducible by heat shock and light [44]. In contrast, Chaperonin 60A and Chaperonin 60B1 were downregulated by butanol treatment. Cpn60A and Cpn60B1 could be induced by heat shock conditions in C. reinhardtii [45] and they are required for the formation of a normal plastid division apparatus in Arabidopsis thaliana [46].
It was reported that ethanol and butanol are toxic to microbial cells (i.e. Synechocystis, E. coli, and S. cerevisiae) as they induce production of reactive oxygen species and oxidative stress [22, 23, 47, 48]. Our proteomic analysis showed that oxidative stress response was also induced in C. reinhardtii by butanol. The upregulated antioxidant enzymes were ascorbate peroxidase Apx1, dehydroascorbate reductase DHAR, glutathione peroxidase homolog Gpxh and heme peroxidase-related protein Cre.770. Apx1 is a hydrogen peroxide-scavenging enzyme that is specific to plants and algae, and is found to be indispensable to protect chloroplasts and other cell constituents from oxidative damage [49]. For instance, Apx1 plays an important role in protecting C. reinhardtii against oxidative damage imposed by salt stress [50]. In addition, DHAR catalyzes the reduction of dehydroascorbate to ascorbate and would provide sufficient substrate for Apx1. It has been reported that the tolerance of tobacco to ozone and drought stresses was largely enhanced by overexpression of DHAR in its cytosol [51]. Gpxh is a potential isoenzyme of glutathione peroxidase that plays a key role in antioxidant system of various microbes [22, 23, 48, 52]. Similarly, Cre.770 is probable a heme-containing peroxidase, which was greatly induced at both the protein and RNA levels (Table 2 and Fig. 4b), indicating an important role in resistance to oxidative stress in C. reinhardtii.
Ethanol and butanol affect transporters and cytoskeleton-related proteins
Membrane transporters have been suggested to be one important mechanism against toxic chemicals by microbes [19, 43, 53]. Several transporters are involved in tolerance to ethanol, butanol, and hexane in Synechocystis [22, 23, 25, 40]. In the presence of ethanol, we found the plastidic ADP/ATP translocase AAA1 and a potential transporter FAP151 were upregulated. AAA1 exchanges plastid ADP for ATP present in the eukaryotic cytoplasm [54] and the induction of this protein implies a compensate mechanism for ATP supply in chloroplasts when photophosphorylation was inhibited by alcohol. FAP151 contains two conserved domains of ABC transporters that are ubiquitous membrane proteins coupling the transport of diverse substrates across cellular membranes to the hydrolysis of ATP [55]. In addition, the vacuolar H+ ATPase ATPvC was downregulated by ethanol treatment. ATPvC is a universal proton pump of eukaryotes and is required for endocytic and secretory trafficking in Arabidopsis [56, 57]. A flagellar-associated protein FAP102 was downregulated by ethanol treatment but the function of FAP102 is still unknown. A septin-like protein SEP1 was upregulated by butanol treatment, and this protein functions in cells by localizing other proteins to special cell sites, indicating improvement of intracellular trafficking under butanol stress. Another investigation also showed a clear induction of proteins involved in intracellular movements in C. reinhardtii growing on metal-rich natural acidic water [58].
Ethanol and butanol affect the regulatory systems
Responses of microbes to environmental stress were often mediated by regulatory systems such as two-component systems, kinases, GTPases, methylases, and transcriptional regulators [22, 23, 25, 59]. After ethanol treatment of C. reinhardtii, the small ADP-ribosylation factor-related GTPase ARFA1A and 14-3-3-like protein Erb14 were upregulated, while the protein arginine N-methyltransferase PRMT2 was downregulated. The homologous protein of ARFA1A in mouse regulates protein trafficking between intracellular organelles and is essential for lipid droplet growth in adipose cells [60]. Members of the Erb14 family have been identified as regulatory elements in intracellular signaling pathways and cell cycle control [61]. Protein methylation at arginine or lysine is an important posttranslational modification and methylation of eyespot proteins of C. reinhardtii influences the size and position of the eyespot [62]. PRMTs are a family of enzymes that function by transferring a methyl group to the guanidine group of arginine residues in target proteins, and the most notable is the PRMT-mediated methylation of histone proteins, which causes chromatin remodeling and influencing gene transcription. The physiological role of PRMT2 is still unknown while PRMT1 was reported to modulate light-harvesting antenna translation in C. reinhardtii [63]. Butanol treatment of C. reinhardtii caused downregulation of peptidyl-prolyl cis–trans isomerase FKB12, translocon component Tic40-related protein TIC40 and Obg-like ATPase 1 OLA1. Chlamydomonas FKB12 exhibits high affinity to rapamycin in vivo and is a signal protein mediating rapamycin action to inhibit cell growth [64, 65]. Thus, downregulation of FKB12 may induce the cell resistance to a toxic metabolite. TIC40 may function as a co-chaperone that facilitates protein translocation across the inner membrane [66]. OLA1 is a cytosolic ATPase, and multiple roles of this protein have been reported, for instance, protecting mammalian cells by stabilizing Hsp70 during heat shock [67].
Ethanol and butanol affect photosynthesis
The photosystem of photosynthetic microbes are vulnerable under stress conditions such as salt, heavy metals, and organic solvents [58, 68, 69]. However, in previous investigations, a set of photosynthesis-related proteins were upregulated by ethanol or butanol treatment in Synechocystis [22, 23, 25]. Interestingly, we also found the photosystem I reaction center subunit PsaD and the photosystem II protein PsbD were upregulated by ethanol treatment in C. reinhardtii. In contrast, four photosynthesis-related proteins were downregulated by ethanol treatment, including magnesium chelatase subunit CHLH1 involved in chlorophyll biosynthesis [70], zeta-carotene desaturase ZDS1 involved in carotenoid biosynthesis, carotenoid isomerase, and putative ferredoxin FDX5. Carotenoid isomerase was previously found to be involved in photoprotection in rice [71] and may has a similar function in C. reinhardtii. FDX5 plays a critical role in maintaining thylakoid membrane structure and dark metabolism of C. reinhardtii [72].
In the case of butanol treatment, three photosynthesis-related proteins were upregulated, including chloroplast vesicle-inducing protein VIPP1, light-harvesting complex II protein Lhcb3, and delta-aminolevulinic acid dehydratase ALAD. VIPP1 is essential for the biogenesis of thylakoid membranes [73] in Chlamydomonas and photosystem I in Synechococcus [74]. Its induction is beneficial for maintaining the integrity of photosynthetic system. ALAD is a chlorophyll biosynthetic enzyme, and its activity could be induced by light [75]. Two photosynthetic proteins were downregulated by butanol treatment, including copper response target 1 protein CRD1 involved in chlorophyll biosynthesis [76] and protoporphyrinogen oxidase Ppx1, which is involved in porphyrin and chlorophyll metabolism [77].
Ethanol and butanol affect protein synthesis
A set of ribosomal proteins were significantly downregulated by ethanol or butanol treatment, which explains the slow growth of C. reinhardtii under these stress conditions. Reduction of ribosomal proteins was also observed for Synechocystis when exposed to ethanol, butanol, and hexane [22, 23, 40], suggesting common resistance mechanism used by different microbes against environmental stress, such as adopting slowdown of protein biosynthesis and slower growth. Similarly, the protein synthesis rate was found to be decreased in C. reinhardii under salt stress [78]. We found several proteins involved in amino acid biosynthesis were also downregulated, such as ferredoxin-dependent glutamate synthase GSF1 with ethanol, and glutamine synthetase GLN2 and acetohydroxy acid isomeroreductase AAI1 with butanol. The eukaryotic initiation factor for translation EIF4G was also downregulated by butanol treatment. However, the l-arginine synthetase argininosuccinate lyase ARG7 was upregulated by ethanol treatment.
Ethanol and butanol affect carbohydrate and ATP metabolism
In a previous investigation, proteins involved in carbohydrate metabolism were mostly unchanged in Synechocystis under ethanol stress conditions [22]. However, carbohydrate metabolic processes were significantly affected in C. reinhardtii under metal-rich stress conditions [58]. In this study, the fructose-1, 6-bisphosphatase FBP1 was upregulated, while chloroplastic fructose-bisphosphate aldolase 1 FBA3, UDP-glucose dehydrogenase UGD2, and transaldolase TAL1 were downregulated by ethanol treatment. FBP1 is critical in gluconeogenesis and Calvin cycle, and TAL1 is important for the balance of metabolites in the pentose-phosphate pathway.
Under butanol treatment, five proteins involved in carbohydrate metabolism were upregulated, including phosphoglycerate mutase PGM1, NAD-dependent malate dehydrogenase MDH3, acetate kinase ACK1, pyruvate ferredoxin oxidoreductase PFO and granule-bound starch synthase STA2. It was reported that PFO of C. reinhardtii was located in chloroplasts and was central to anaerobic energy metabolism through pyruvate decarboxylation and formation of acetyl-coenzyme A with concomitant reduction of low-potential ferredoxins or flavodoxins [79]. Since biosynthesis of butanol is also an anaerobic process, the upregulation of PFO may enhance the energy supply for butanol synthesis. On the other side, five proteins were downregulated by butanol treatment, including citrate synthase, acetyl CoA synthetases, ACS2 and ACS3, acetyl-CoA acyltransferase ATO1, and transketolase TRK1. Two proteins involved in ATP synthesis were downregulated, including F1F0 ATP synthase gamma subunit ATP3 in ethanol treatment and chloroplastic ATP synthase subunit b’ in butanol treatment.
Ethanol and butanol affects other central metabolic processes
Several proteins involved in other primary metabolism of C. reinhardtii were also significantly affected by ethanol or butanol stress. Ribonucleoside diphosphate reductase RIR1 and nucleoside diphosphate kinase FAP103 were downregulated by ethanol. RIR1 catalyzes the biosynthesis of deoxyribonucleotides in the DNA replication pathway and FAP103 is crucial for the homeostasis of cellular nucleoside di- and triphosphate composition and may control many cellular functions. Butanol treatment caused cysteine endopeptidase CEP1 involved in proteolysis and an oxidoreductase-like protein CPLD35 to be upregulated, while hydroxymethylpyrimidine phosphate synthase THICb involved in thiamine biosynthesis and two RNA helicases were downregulated.
Effects of ethanol and butanol on uninvestigated proteins
Two hypothetical proteins (A8JG58 and A8IRL8) were found upregulated, and seven (A8IVP7, A8JAG1, A8J387, A8HVM3, A8J9F3, A8IWJ5, and A8ILP2) were downregulated by ethanol. Five hypothetical proteins (A8JHZ9, A8IX35, A8J6Y3, A8IUI1, and A8JBW0) were upregulated, while nine (A8JDM1, A8JAP7, A8IVS6, A8I0R6, A8I9M5, A8IPS8, A8IVP7, A8JCW5, and A8JHB7) were downregulated by butanol. These proteins were predicted in genome sequencing project of C. reinhardtii [37] and their strong regulation upon the exposure of ethanol or butanol may help to elucidate their functions. The high percentage of functionally unknown proteins detected is not unreasonable, considering more than 71% of proteins in the C. reinhardtii genome are still annotated as hypothetical proteins.
Conclusions
Quantitative proteomics analysis revealed that ethanol or butanol exposure caused significant slowdown of primary metabolisms in C. reinhardtii, such as photosynthesis, protein synthesis, carbohydrate, and ATP metabolism, and other central metabolic processes. The expression of stress-related proteins, transporters, cytoskeleton-related proteins, and regulators were upregulated in the algal cells as major protection mechanisms against ethanol or butanol toxicity. In addition, antioxidant enzymes were significantly upregulated by butanol treatment compared with ethanol treatment, implying that butanol may cause strong oxidative stress in C. reinhardtii. These metabolic responses are much similar to what has been observed in other microbes, such as E. coli, C. acetobutylicum, Z. mobilis, Synechocystis, and S. cerevisiae [19, 22, 23, 41, 42], indicating the presence of possible common resistance strategies to ethanol or butanol among various species.
In summary, these data provide the first comprehensive view of metabolic responses employed by a model green alga C. reinhardtii to defend against ethanol or butanol stress. This proteomic analysis will provide a resource for investigating potential target genes/proteins for manipulating the algal cells to improve their ethanol or butanol tolerance and thus aid in their biofuel production. Further work will be required to determine the exact relationship between these genes and organic solvent tolerance. The probable heme-containing peroxidase Cre.770 and the pyruvate ferredoxin oxidoreductase PFO are the most promising targets for metabolic engineering of C. reinhardtii to achieve efficient biofuel production.
Methods
Algal culture conditions and biofuel treatment
The cell wall-deficient strain C. reinhardtii CC-849 was obtained from the Chlamydomonas Genetic Center (Duke University). The strain was cultured in liquid TAP medium [80] at 22 °C by providing continuous white light at a density of 30 μmol m−2 s−1. For growth and biofuel treatment, 5 mL fresh algal cells at exponential phase (1 × 106 cells mL−1) were incubated into 50 mL TAP medium in 250-mL conical flasks. Biofuel was added into the medium at the beginning of cultivation with varying final concentrations, 0.0, 1.5, 1.8, and 3.0% (v/v) for ethanol and 0.0, 0.1, 0.3, and 0.5% (v/v) for butanol. The treatment of each concentration was performed in triplicate, and the 0.0% concentration was used as control treatment. One milliliter culture sample was collected every 24 h over a culture period of 6 days, and the optical density at 750 nm (OD750) was measured by NanoDrop 2000 spectrophotometer (Thermo Fisher) for growth monitoring. Analytic purity ethanol and n-butanol were purchased from Aladdin (China). Algal cells for proteomic and qRT-PCR analysis were collected at 24, 48, and 72 h. The cells from triplicate samples were mixed and centrifuged at 8000×g for 5 min at 4 °C. The cell pellets were rinsed in PBS and stored in liquid nitrogen before protein preparation.
Protein extraction and digestion
For each sample, cell pellets were resuspended in lysis buffer (8 M urea, 4% CHAPS, 40 mM Tris–HCl, 2 mM EDTA) with 1 mM protease inhibitor PMSF. The algal cells were lysed by vigorously vortexing for 5 min. Then, dithiothreitol was added to the lysis solution at a final concentration of 10 mM and mixed thoroughly. The sample was centrifuged at 20,000×g for 30 min, and the supernatant was mixed with ice-cold acetone (1:4, v/v) containing 30 mM dithiothreitol. After repeating the lysis step twice, the supernatants were combined, and proteins were harvested by centrifugation at 10,000×g for 30 min after precipitation at − 20 °C overnight. The protein pellets were resuspended in 100 mM triethylammonium bicarbonate solution, and total protein concentration was measured by Bradford assay (Beyotime, China). Before digestion, 200 μg of protein from each lysate was reduced, and a cysteine-blocking reaction was performed using reagents provided in the iTRAQ array kit (AB Sciex, USA). The solution was then filtered through an ultracentrifugal filter (Sartorious, 10 kDa) to remove reagent residues. Proteins remaining on the filter were digested with trypsin (1:50 w/w, AB Sciex) in a 50 μL reaction volume overnight at 37 °C. Peptides were harvested by centrifugation at 12,000×g for 20 min. The centrifugation step was repeated after 50 μL dissolution buffer (AB Sciex) was added to the filter and the filtrates were combined.
iTRAQ labling
The iTRAQ labeling of 100 μg peptides from digestion of each sample was performed using iTRAQ Reagent 8-plex and 4-plex Kit (AB Sciex) according to the manufacturer’s protocol. The peptides were labeled with respective isobaric tags by incubation at room temperature for 2 h and then lyophilized via vacuum centrifugation. The labeled control and biofuel treatment samples were reconstituted in buffer A (20 mM ammonium formate, pH = 10.0), pooled, and fractionated using Durashell-C18 column (250 × 4.6 mm, 5 μm particle size, 100 Å pore size, Agela) by HPLC system at a flow rate of 0.8 mL min−1. The HPLC gradient consisted of 95% buffer A for 15 min; 5–15% buffer B (20 mM ammonium formate, pH = 10.0, 80% v/v acetonitrile) for 25 min; 15–38% buffer B for 15 min; 38–90% buffer B for 1 min followed by 90% buffer B for 9 min and 95% buffer A for 5 min. The chromatograms were monitored by ultraviolet absorbance at 218 nm. The fractions were collected every 1 min after the 5th min and 48 fractions were obtained for each injection. The fractions were desalted with Sep-Pak Vac C18 cartridges (Waters, USA), vacuum centrifuged to dryness and reconstituted in buffer C (2% acetonitrile, 0.1% formic acid) for LC–MS/MS analysis.
LC–MS/MS proteomic analysis
The mass spectroscopy analysis was performed using a TripleTOF 5600 mass spectrometer coupled with online Eksigent nano LC system (AB Sciex, USA). For LC conditions, a nanobored C18 column (15 cm × 75 μm, 5 μm particle size) with a picofrit nanospray tip (New Objectives, USA) and loading pump with a constant flow rate of 2 μL min−1 were applied. The peptides were separated by a micro flow rate of 0.3 μL min−1 and the injection volume was 8 µL. LC gradient was adjusted according to different peptide fractions, and in general, 95–90% buffer C for 0.1 min; 10–25% buffer D (98% acetonitrile, 0.1% formic acid) for 60 min; 25–48% buffer D for 25 min; 48–80% buffer D for 1 min followed by 80% buffer D for 4 min and 95% buffer C for 10 min. The parameters of mass spectrometer were set as previously described [23]. In brief, positive ions were monitored with selected mass ranges of 350–1250 m/z for TOF MS scan and 100–1500 m/z for product ion scan. The peptides with + 2 to + 4 charge states were selected for MS/MS. The ten most abundant ions above a five count threshold in an accumulation period of 0.1 s and a dynamic exclusion period of 25 s were selected for MS/MS. The relative abundance of the proteins in the samples was calculated based on the peak areas of the iTRAQ reporter ions.
Proteomic data analysis
The MS data were processed using ProteinPilot 4.5 (AB Sciex) software. Peptide identification and protein summarization were performed using the Paragon and Pro Group algorithms implemented in ProteinPilot. The parameters were set as iTRAQ labeling at N-terminal and lysine residues, cysteine modification by methyl methanethiosulfonate and trypsin as a protease. Proteins identified with low false discovery rate (≤ 1%) were used for further analysis. For iTRAQ quantification, the peptides were automatically selected by the software to calculate the reporter peak area. The resulting dataset was auto bias-corrected to eliminate any variations caused by unequal mixing while combining different samples. The MS/MS data were searched against genome annotations of C. reinhardtii CC-503 deposited in NCBI database [81]. Proteins with 2.0-fold change between biofuel-treated and control samples and P values of statistical evaluation less than 0.05 were determined as differentially expressed proteins. KOG was analyzed using the WebMGA tool [82]. The UniProtKB identifiers of proteins were retrieved from the UniProtKB database [83], and the identified proteins were subjected to GO [84] and KEGG pathway [85] analyses. The fundamental functions of proteins and metabolic pathways were counted and analyzed. The enrichment of differentially regulated proteins in GO terms and metabolic pathways was carried out using the following formula: m/n > M/N, where N is the number of all proteins with GO or KEGG pathway annotation information; n is the number of the differentially regulated proteins with GO or KEGG pathway annotation information; M is the number of proteins with a given GO term or KEGG pathway annotation; and m is the number of the differentially regulated proteins with a given GO term or KEGG pathway annotation. The GO terms or KEGG pathways with P values < 0.05 as threshold in the Chi-square test were considered as enriched GO terms or KEGG pathways by the biofuel-responsive proteins.
qRT-PCR analysis
The algal cells were collected by centrifugation at 8000×g for 5 min at 4 °C. Approximately 10 mg of cell pellets were resuspended in 1 ml TRIzol reagent (Invitrogen) for RNA preparation according to manufacturers’ instructions, and the purified RNA was dissolved in 50 μL DEPC-treated water. Total RNA was quantified, and 1 μg of RNA was digested by RNase-free DNase followed by reverse transcription using a SuperScript VILO master mix for 1st strand cDNA synthesis (Invitrogen). Primers targeting C. reinhardtii genes were designed using the Primer Express v3.0 software. The gene name and primer sequences are listed in Additional file 8: Table S7. Real-time PCR analysis was performed on an ABI Vii7™ Real-time PCR System (applied biosystems) using Fast SYBR Green Master Mix kit (applied biosystems). The actin gene was amplified as internal controls. The relative expression level of target genes was calculated by the formula 2−ΔΔCt, where
Additional files
Additional file 1: Figure S1. Cell morphology observation under light microscope (100 ×). Scale bars of 50 μm were indicated.
Additional file 2: Table S1. Accession numbers of proteins detected in this study.
Additional file 3: Table S2. List of differentially regulated proteins in ethanol treatment.
Additional file 4: Table S3. List of differentially regulated proteins in butanol treatment.
Additional file 5: Table S4. GO annotation of proteins detected in this study.
Additional file 6: Table S5. GO enrichment analysis of differentially regulated proteins in ethanol treatment.
Additional file 7: Table S6. GO enrichment analysis of differentially regulated proteins in butanol treatment.
Additional file 8: Table S7. Primer pairs used in qRT-PCR analysis.
Authors’ contributions
YGJ carried out the iTRAQ-LC–MS/MS experiments and was a major contributor in writing the manuscript. HQ and APL conducted microalgae culture and sample collection. PX performed bioinformatics analyses to interpret the data. QS conducted the qRT-PCR experiment. JXW, ZLH, and APL designed the experiment and drafted the manuscript. All the authors read and approved the final manuscript.
Acknowledgements
The authors thank Ms. Lili Han and Xianxia Cao for their technical supports.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files.
Consent for publication
The authors have consented for publication.
Ethics approval and consent to participate
Not applicable.
Funding
This work was partially supported by the Natural Science Foundation of SZU (#827–000081), the National Natural Science Foundation of China (#31470431,#31500071,#31670116), the Guangdong Innovation Research Team Fund (2014ZT05S078), the Shenzhen Grant Plan for Science & Technology (JCYJ20120613112512654 and JCYJ20140418182819134), and the Nanshan District key lab for biopolymers and safety evaluation (#KC2014ZDZJ0001A), used for the design of the study, data collection, data analysis, interpretation of data, and in writing the manuscript, respectively.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- GO
gene ontology
- Hsps
heat shock proteins
- iTRAQ
isobaric tag for relative and absolute quantitation
- KOG
eukaryotic cluster of orthologous groups of proteins
- LC–MS/MS
liquid chromatography–tandem mass spectrometry
- qRT-PCR
real-time quantitative reverse transcription PCR
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1186/s13068-017-0931-9) contains supplementary material, which is available to authorized users.
Contributor Information
Yongguang Jiang, Email: jiangyg@szu.edu.cn.
Peng Xiao, Email: xiaopengycyz@163.com.
Qing Shao, Email: 549978093@qq.com.
Huan Qin, Email: 1936972014@qq.com.
Zhangli Hu, Email: huzl@szu.edu.cn.
Anping Lei, Email: bioaplei@szu.edu.cn.
Jiangxin Wang, Phone: +86 755 26958849, Email: jxwang@szu.edu.cn.
References
- 1.Chisti Y. Biodiesel from microalgae. Biotechnol Adv. 2007;25:294–306. doi: 10.1016/j.biotechadv.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Dutta K, Daverey A, Lin J-G. Evolution retrospective for alternative fuels: first to fourth generation. Renew Energy. 2014;69:114–122. doi: 10.1016/j.renene.2014.02.044. [DOI] [Google Scholar]
- 3.Brennan L, Owende P. Biofuels from microalgae—a review of technologies for production, processing, and extractions of biofuels and co-products. Renew Sustain Energy Rev. 2010;14:557–577. doi: 10.1016/j.rser.2009.10.009. [DOI] [Google Scholar]
- 4.Radakovits R, Jinkerson RE, Darzins A, Posewitz MC. Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell. 2010;9:486–501. doi: 10.1128/EC.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wijffels RH, Barbosa MJ. An outlook on microalgal biofuels. Science. 2010;329:796–799. doi: 10.1126/science.1189003. [DOI] [PubMed] [Google Scholar]
- 6.Qin S, Lin H, Jiang P. Advances in genetic engineering of marine algae. Biotechnol Adv. 2012;30:1602–1613. doi: 10.1016/j.biotechadv.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 7.Lü J, Sheahan C, Fu P. Metabolic engineering of algae for fourth generation biofuels production. Energy Environ Sci. 2011;4:2451–2466. doi: 10.1039/c0ee00593b. [DOI] [Google Scholar]
- 8.Michinaka Y, Shimauchi T, Aki T, Nakajima T, Kawamoto S, Shigeta S, Suzuki O, Ono K. Extracellular secretion of free fatty acids by disruption of a fatty acyl-CoA synthetase gene in Saccharomyces cerevisiae. J Biosci Bioeng. 2003;95:435–440. doi: 10.1016/S1389-1723(03)80041-5. [DOI] [PubMed] [Google Scholar]
- 9.Melis A, Happe T. Hydrogen production. Green algae as a source of energy. Plant Physiol. 2001;127:740–748. doi: 10.1104/pp.010498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metzger P, Largeau C. Botryococcus braunii: a rich source for hydrocarbons and related ether lipids. Appl Microbiol Biotechnol. 2005;66:486–496. doi: 10.1007/s00253-004-1779-z. [DOI] [PubMed] [Google Scholar]
- 11.Balat M, Balat H, Öz C. Progress in bioethanol processing. Prog Energy Combust Sci. 2008;34:551–573. doi: 10.1016/j.pecs.2007.11.001. [DOI] [Google Scholar]
- 12.García V, Päkkilä J, Ojamo H, Muurinen E, Keiski RL. Challenges in biobutanol production: how to improve the efficiency? Renew Sustain Energy Rev. 2011;15:964–980. doi: 10.1016/j.rser.2010.11.008. [DOI] [Google Scholar]
- 13.Pfromm PH, Amanor-Boadu V, Nelson R, Vadlani P, Madl R. Bio-butanol vs. bio-ethanol: a technical and economic assessment for corn and switchgrass fermented by yeast or Clostridium acetobutylicum. Biomass Bioenergy. 2010;34:515–524. doi: 10.1016/j.biombioe.2009.12.017. [DOI] [Google Scholar]
- 14.Deng M-D, Coleman JR. Ethanol synthesis by genetic engineering in cyanobacteria. Appl Microbiol Biotechnol. 1999;65:523–528. doi: 10.1128/aem.65.2.523-528.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dürre P. Biobutanol: an attractive biofuel. Biotechnol J. 2007;2:1525–1534. doi: 10.1002/biot.200700168. [DOI] [PubMed] [Google Scholar]
- 16.Steen EJ, Chan R, Prasad N, Myers S, Petzold CJ, Redding A, Ouellet M, Keasling JD. Metabolic engineering of Saccharomyces cerevisiae for the production of n-butanol. Microb Cell Fact. 2008;7:36. doi: 10.1186/1475-2859-7-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJY, Hanai T, Liao JC. Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng. 2008;10:305–311. doi: 10.1016/j.ymben.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Lan EI, Liao JC. Metabolic engineering of cyanobacteria for 1-butanol production from carbon dioxide. Metab Eng. 2011;13:353–363. doi: 10.1016/j.ymben.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Dunlop MJ. Engineering microbes for tolerance to next-generation biofuels. Biotechnol Biofuels. 2011;4:1. doi: 10.1186/1754-6834-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H-S, Kim N-R, Yang J, Choi W. Identification of novel genes responsible for ethanol and/or thermotolerance by transposon mutagenesis in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2011;91:1159–1172. doi: 10.1007/s00253-011-3298-z. [DOI] [PubMed] [Google Scholar]
- 21.Mao S, Luo Y, Zhang T, Li J, Bao G, Zhu Y, Chen Z, Zhang Y, Li Y, Ma Y. Proteome reference map and comparative proteomic analysis between a wild type Clostridium acetobutylicum DSM 1731 and its mutant with enhanced butanol tolerance and butanol yield. J Proteome Res. 2010;9:3046–3061. doi: 10.1021/pr9012078. [DOI] [PubMed] [Google Scholar]
- 22.Qiao J, Wang J, Chen L, Tian X, Huang S, Ren X, Zhang W. Quantitative iTRAQ LC-MS/MS proteomics reveals metabolic responses to biofuel ethanol in cyanobacterial Synechocystis sp. PCC 6803. J Proteome Res. 2012;11:5286–5300. doi: 10.1021/pr300504w. [DOI] [PubMed] [Google Scholar]
- 23.Tian X, Chen L, Wang J, Qiao J, Zhang W. Quantitative proteomics reveals dynamic responses of Synechocystis sp. PCC 6803 to next-generation biofuel butanol. J Proteom. 2013;78:326–345. doi: 10.1016/j.jprot.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 24.Dienst D, Georg J, Abts T, Jakorew L, Kuchmina E, Borner T, Wilde A, Duhring U, Enke H, Hess WR. Transcriptomic response to prolonged ethanol production in the cyanobacterium Synechocystis sp. PCC6803. Biotechnol Biofuels. 2014;7:21. doi: 10.1186/1754-6834-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anfelt J, Hallstrom B, Nielsen J, Uhlen M, Hudson EP. Using transcriptomics to improve butanol tolerance of Synechocystis sp. strain PCC 6803. Appl Environ Microbiol. 2013;79:7419–7427. doi: 10.1128/AEM.02694-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Chen L, Huang S, Liu J, Ren X, Tian X, Qiao J, Zhang W. RNA-seq based identification and mutant validation of gene targets related to ethanol resistance in cyanobacterial Synechocystis sp. PCC 6803. Biotechnol Biofuels. 2012;5:1. doi: 10.1186/1754-6834-5-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L, Wu L, Wang J, Zhang W. Butanol tolerance regulated by a two-component response regulator Slr 1037 in photosynthetic Synechocystis sp. PCC 6803. Biotechnol Biofuels. 2014;7:89. doi: 10.1186/1754-6834-7-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu H, Ren X, Wang J, Song Z, Shi M, Qiao J, Tian X, Liu J, Chen L, Zhang W. Integrated OMICS guided engineering of biofuel butanol-tolerance in photosynthetic Synechocystis sp. PCC 6803. Biotechnol Biofuels. 2013;6:106. doi: 10.1186/1754-6834-6-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Bae JY, Lee YM, Kwon H, Moon HY, Kang HA, Yee SB, Kim W, Choi W. Construction of Saccharomyces cerevisiae strains with enhanced ethanol tolerance by mutagenesis of the TATA-binding protein gene and identification of novel genes associated with ethanol tolerance. Biotechnol Bioeng. 2011;108:1776–1787. doi: 10.1002/bit.23141. [DOI] [PubMed] [Google Scholar]
- 30.Atsumi S, Wu TY, Machado IM, Huang WC, Chen PY, Pellegrini M, Liao JC. Evolution, genomic analysis, and reconstruction of isobutanol tolerance in Escherichia coli. Mol Syst Biol. 2010;6:449. doi: 10.1038/msb.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scranton MA, Ostrand JT, Fields FJ, Mayfield SP. Chlamydomonas as a model for biofuels and bioproducts production. Plant J. 2015;82:523–531. doi: 10.1111/tpj.12780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jinkerson RE, Jonikas MC. Molecular techniques to interrogate and edit the Chlamydomonas nuclear genome. Plant J. 2015;82:393–412. doi: 10.1111/tpj.12801. [DOI] [PubMed] [Google Scholar]
- 33.D-j Shi, C-l Wang, K-m Wang. Genome shuffling to improve thermotolerance, ethanol tolerance and ethanol productivity of Saccharomyces cerevisiae. J Ind Microbiol Biotechnol. 2009;36:139–147. doi: 10.1007/s10295-008-0481-z. [DOI] [PubMed] [Google Scholar]
- 34.Ruhl J, Schmid A, Blank LM. Selected Pseudomonas putida strains able to grow in the presence of high butanol concentrations. Appl Environ Microbiol. 2009;75:4653–4656. doi: 10.1128/AEM.00225-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyes LH, Almario MP, Kao KC. Genomic library screens for genes involved in n-butanol tolerance in Escherichia coli. PLoS ONE. 2011;6:e17678. doi: 10.1371/journal.pone.0017678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knoshaug EP, Zhang M. Butanol tolerance in a selection of microorganisms. Appl Biochem Biotechnol. 2009;153:13–20. doi: 10.1007/s12010-008-8460-4. [DOI] [PubMed] [Google Scholar]
- 37.Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Maréchal-Drouard L. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science. 2007;318:245–250. doi: 10.1126/science.1143609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wienkoop S, Weiß J, May P, Kempa S, Irgang S, Recuenco-Munoz L, Pietzke M, Schwemmer T, Rupprecht J, Egelhofer V. Targeted proteomics for Chlamydomonas reinhardtii combined with rapid subcellular protein fractionation, metabolomics and metabolic flux analyses. Mol BioSyst. 2010;6:1018–1031. doi: 10.1039/b920913a. [DOI] [PubMed] [Google Scholar]
- 39.Minty JJ, Lesnefsky AA, Lin F, Chen Y, Zaroff TA, Veloso AB, Xie B, McConnell CA, Ward RJ, Schwartz DR. Evolution combined with genomic study elucidates genetic bases of isobutanol tolerance in Escherichia coli. Microb Cell Fact. 2011;10:1. doi: 10.1186/1475-2859-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu J, Chen L, Wang J, Qiao J, Zhang W. Proteomic analysis reveals resistance mechanism against biofuel hexane in Synechocystis sp. PCC 6803. Biotechnol Biofuels. 2012;5:68. doi: 10.1186/1754-6834-5-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stanley D, Bandara A, Fraser S, Chambers PJ, Stanley GA. The ethanol stress response and ethanol tolerance of Saccharomyces cerevisiae. J Appl Microbiol. 2010;109:13–24. doi: 10.1111/j.1365-2672.2009.04657.x. [DOI] [PubMed] [Google Scholar]
- 42.Widiastuti H, Kim JY, Selvarasu S, Karimi IA, Kim H, Seo JS, Lee DY. Genome-scale modeling and in silico analysis of ethanologenic bacteria Zymomonas mobilis. Biotechnol Bioeng. 2011;108:655–665. doi: 10.1002/bit.22965. [DOI] [PubMed] [Google Scholar]
- 43.Ding J, Huang X, Zhang L, Zhao N, Yang D, Zhang K. Tolerance and stress response to ethanol in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2009;85:253–263. doi: 10.1007/s00253-009-2223-1. [DOI] [PubMed] [Google Scholar]
- 44.von Gromoff ED, Treier U, Beck CF. Three light-inducible heat shock genes of Chlamydomonas reinhardtii. Mol Cell Biol. 1989;9:3911–3918. doi: 10.1128/MCB.9.9.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson MD, Paavola CD, Lenvik TR, Gantt JS. Chlamydomonas transcripts encoding three divergent plastid chaperonins are heat-inducible. Plant Mol Biol. 1995;27:1031–1035. doi: 10.1007/BF00037029. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki K, Nakanishi H, Bower J, Yoder DW, Osteryoung KW, Miyagishima SY. Plastid chaperonin proteins Cpn60 alpha and Cpn60 beta are required for plastid division in Arabidopsis thaliana. BMC Plant Biol. 2009;9:38. doi: 10.1186/1471-2229-9-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rutherford BJ, Dahl RH, Price RE, Szmidt HL, Benke PI, Mukhopadhyay A, Keasling JD. Functional genomic study of exogenous n-butanol stress in Escherichia coli. Appl Environ Microbiol. 2010;76:1935–1945. doi: 10.1128/AEM.02323-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Du X, Takagi H. N-Acetyltransferase Mpr1 confers ethanol tolerance on Saccharomyces cerevisiae by reducing reactive oxygen species. Appl Microbiol Biotechnol. 2007;75:1343–1351. doi: 10.1007/s00253-007-0940-x. [DOI] [PubMed] [Google Scholar]
- 49.Asada K. Ascorbate peroxidase—a hydrogen peroxide-scavenging enzyme in plants. Physiol Plant. 1992;85:235–241. doi: 10.1111/j.1399-3054.1992.tb04728.x. [DOI] [Google Scholar]
- 50.Takeda T, Yoshimura K, Yoshii M, Kanahoshi H, Miyasaka H, Shigeoka S. Molecular characterization and physiological role of ascorbate peroxidase from halotolerant Chlamydomonas sp. W80 strain. Arch Biochem Biophys. 2000;376:82–90. doi: 10.1006/abbi.1999.1564. [DOI] [PubMed] [Google Scholar]
- 51.Eltayeb AE, Kawano N, Badawi GH, Kaminaka H, Sanekata T, Morishima I, Shibahara T, Inanaga S, Tanaka K. Enhanced tolerance to ozone and drought stresses in transgenic tobacco overexpressing dehydroascorbate reductase in cytosol. Physiol Plant. 2006;127:57–65. doi: 10.1111/j.1399-3054.2006.00624.x. [DOI] [Google Scholar]
- 52.Arenas FA, Covarrubias PC, Sandoval JM, Perez-Donoso JM, Imlay JA, Vasquez CC. The Escherichia coli BtuE protein functions as a resistance determinant against reactive oxygen species. PLoS ONE. 2011;6:e15979. doi: 10.1371/journal.pone.0015979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Olsson S, Puente-Sanchez F, Gomez MJ, Aguilera A. Transcriptional response to copper excess and identification of genes involved in heavy metal tolerance in the extremophilic microalga Chlamydomonas acidophila. Extremophiles. 2015;19:657–672. doi: 10.1007/s00792-015-0746-1. [DOI] [PubMed] [Google Scholar]
- 54.Möhlmann T, Tjaden J, Schwöppe C, Winkler HH, Kampfenkel K, Neuhaus HE. Occurrence of two plastidic ATP/ADP transporters in Arabidopsis thaliana L. Eur J Biochem. 1998;252:353–359. doi: 10.1046/j.1432-1327.1998.2520353.x. [DOI] [PubMed] [Google Scholar]
- 55.Hollenstein K, Dawson RJ, Locher KP. Structure and mechanism of ABC transporter proteins. Curr Opin Struct Biol. 2007;17:412–418. doi: 10.1016/j.sbi.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 56.Dettmer J, Hong-Hermesdorf A, Stierhof YD, Schumacher K. Vacuolar H+-ATPase activity is required for endocytic and secretory trafficking in Arabidopsis. Plant Cell. 2006;18:715–730. doi: 10.1105/tpc.105.037978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finbow ME, Harrison MA. The vacuolar H+-ATPase: a universal proton pump of eukaryotes. Biochem J. 1997;324(Pt 3):697–712. doi: 10.1042/bj3240697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cid C, Garcia-Descalzo L, Casado-Lafuente V, Amils R, Aguilera A. Proteomic analysis of the response of an acidophilic strain of Chlamydomonas sp. (Chlorophyta) to natural metal-rich water. Proteomics. 2010;10:2026–2036. doi: 10.1002/pmic.200900592. [DOI] [PubMed] [Google Scholar]
- 59.Zhang SY, Sun GX, Yin XX, Rensing C, Zhu YG. Biomethylation and volatilization of arsenic by the marine microalgae Ostreococcus tauri. Chemosphere. 2013;93:47–53. doi: 10.1016/j.chemosphere.2013.04.063. [DOI] [PubMed] [Google Scholar]
- 60.Hommel A, Hesse D, Volker W, Jaschke A, Moser M, Engel T, Bluher M, Zahn C, Chadt A, Ruschke K, et al. The ARF-like GTPase ARFRP1 is essential for lipid droplet growth and is involved in the regulation of lipolysis. Mol Cell Biol. 2010;30:1231–1242. doi: 10.1128/MCB.01269-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Voigt J, Stevanovic S, Schirle M, Fausel M, Maier J, Adam KH, Marquardt O. A 14-3-3 protein of Chlamydomonas reinhardtii associated with the endoplasmic reticulum: nucleotide sequence of the cDNA and the corresponding gene and derived amino acid sequence. Biochim Biophys Acta. 2004;1679:180–194. doi: 10.1016/j.bbaexp.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 62.Eitzinger N, Wagner V, Weisheit W, Geimer S, Boness D, Kreimer G, Mittag M. Proteomic analysis of a fraction with intact eyespots of Chlamydomonas reinhardtii and assignment of protein methylation. Front Plant Sci. 2015;6:1085. doi: 10.3389/fpls.2015.01085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blifernez O, Wobbe L, Niehaus K, Kruse O. Protein arginine methylation modulates light-harvesting antenna translation in Chlamydomonas reinhardtii. Plant J. 2011;65:119–130. doi: 10.1111/j.1365-313X.2010.04406.x. [DOI] [PubMed] [Google Scholar]
- 64.Crespo JL, Diaz-Troya S, Florencio FJ. Inhibition of target of rapamycin signaling by rapamycin in the unicellular green alga Chlamydomonas reinhardtii. Plant Physiol. 2005;139:1736–1749. doi: 10.1104/pp.105.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Imamura S, Kawase Y, Kobayashi I, Sone T, Era A, Miyagishima SY, Shimojima M, Ohta H, Tanaka K. Target of rapamycin (TOR) plays a critical role in triacylglycerol accumulation in microalgae. Plant Mol Biol. 2015;89:309–318. doi: 10.1007/s11103-015-0370-6. [DOI] [PubMed] [Google Scholar]
- 66.Chou ML, Fitzpatrick LM, Tu SL, Budziszewski G, Potter-Lewis S, Akita M, Levin JZ, Keegstra K, Li HM. Tic40, a membrane-anchored co-chaperone homolog in the chloroplast protein translocon. EMBO J. 2003;22:2970–2980. doi: 10.1093/emboj/cdg281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mao RF, Rubio V, Chen H, Bai L, Mansour OC, Shi ZZ. OLA1 protects cells in heat shock by stabilizing HSP70. Cell Death Dis. 2013;4:e491. doi: 10.1038/cddis.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Danilov RA, Ekelund NG. Effects of Cu2+, Ni2+, Pb2+, Zn2+ and pentachlorophenol on photosynthesis and motility in Chlamydomonas reinhardtii in short-term exposure experiments. BMC Ecol. 2001;1:1. doi: 10.1186/1472-6785-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allakhverdiev SI, Murata N. Salt stress inhibits photosystems II and I in cyanobacteria. Photosynth Res. 2008;98:529–539. doi: 10.1007/s11120-008-9334-x. [DOI] [PubMed] [Google Scholar]
- 70.Chekounova E, Voronetskaya V, Papenbrock J, Grimm B, Beck CF. Characterization of Chlamydomonas mutants defective in the H subunit of Mg-chelatase. Mol Genet Genom. 2001;266:363–373. doi: 10.1007/s004380100574. [DOI] [PubMed] [Google Scholar]
- 71.Chai C, Fang J, Liu Y, Tong H, Gong Y, Wang Y, Liu M, Wang Y, Qian Q, Cheng Z, et al. ZEBRA2, encoding a carotenoid isomerase, is involved in photoprotection in rice. Plant Mol Biol. 2011;75:211–221. doi: 10.1007/s11103-010-9719-z. [DOI] [PubMed] [Google Scholar]
- 72.Yang W, Wittkopp TM, Li X. Critical role of Chlamydomonas reinhardtii ferredoxin-5 in maintaining membrane structure and dark metabolism. Proc Nat Acad Sci USA. 2015;112:14978–14983. doi: 10.1073/pnas.1515240112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu C, Willmund F, Whitelegge JP, Hawat S, Knapp B, Lodha M, Schroda M. J-domain protein CDJ2 and HSP70B are a plastidic chaperone pair that interacts with vesicle-inducing protein in plastids 1. Mol Biol Cell. 2005;16:1165–1177. doi: 10.1091/mbc.E04-08-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang S, Shen G, Li Z, Golbeck JH, Bryant DA. Vipp1 is essential for the biogenesis of photosystem I but not thylakoid membranes in Synechococcus sp. PCC 7002. J Biol Chem. 2014;289:15904–15914. doi: 10.1074/jbc.M114.555631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matters GL, Beale SI. Structure and expression of the Chlamydomonas reinhardtii alad gene encoding the chlorophyll biosynthetic enzyme, delta-aminolevulinic acid dehydratase (porphobilinogen synthase) Plant Mol Biol. 1995;27:607–617. doi: 10.1007/BF00019326. [DOI] [PubMed] [Google Scholar]
- 76.Allen MD, Kropat J, Merchant SS. Regulation and localization of isoforms of the aerobic oxidative cyclase in Chlamydomonas reinhardtii. Photochem Photobiol. 2008;84:1336–1342. doi: 10.1111/j.1751-1097.2008.00440.x. [DOI] [PubMed] [Google Scholar]
- 77.Randolph-Anderson BL, Sato R, Johnson AM, Harris EH, Hauser CR, Oeda K, Ishige F, Nishio S, Gillham NW, Boynton JE. Isolation and characterization of a mutant protoporphyrinogen oxidase gene from Chlamydomonas reinhardtii conferring resistance to porphyric herbicides. Plant Mol Biol. 1998;38:839–859. doi: 10.1023/A:1006085026294. [DOI] [PubMed] [Google Scholar]
- 78.Mastrobuoni G, Irgang S, Pietzke M, Aßmus HE, Wenzel M, Schulze WX, Kempa S. Proteome dynamics and early salt stress response of the photosynthetic organism Chlamydomonas reinhardtii. BMC Genom. 2012;13:1. doi: 10.1186/1471-2164-13-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.van Lis R, Baffert C, Coute Y, Nitschke W, Atteia A. Chlamydomonas reinhardtii chloroplasts contain a homodimeric pyruvate:ferredoxin oxidoreductase that functions with FDX1. Plant Physiol. 2013;161:57–71. doi: 10.1104/pp.112.208181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gorman DS, Levine R. Cytochrome f and plastocyanin: their sequence in the photosynthetic electron transport chain of Chlamydomonas reinhardi. Proc Natl Acad Sci USA. 1965;54:1665–1669. doi: 10.1073/pnas.54.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Genome assembly and annotation report: Chlamydomonas reinhardtii.http://www.ncbi.nlm.nih.gov/genome/147. Accessed 10 Jun 2016.
- 82.Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genom. 2011;12:1–9. doi: 10.1186/1471-2164-12-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.The UniProt knowledgebase. http://www.uniprot.org/. Accessed 15 Jun 2016.
- 84.Gene Ontology Consortium Gene ontology consortium: going forward. Nucleic Acids Res. 2015;43:D1049–D1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Cell morphology observation under light microscope (100 ×). Scale bars of 50 μm were indicated.
Additional file 2: Table S1. Accession numbers of proteins detected in this study.
Additional file 3: Table S2. List of differentially regulated proteins in ethanol treatment.
Additional file 4: Table S3. List of differentially regulated proteins in butanol treatment.
Additional file 5: Table S4. GO annotation of proteins detected in this study.
Additional file 6: Table S5. GO enrichment analysis of differentially regulated proteins in ethanol treatment.
Additional file 7: Table S6. GO enrichment analysis of differentially regulated proteins in butanol treatment.
Additional file 8: Table S7. Primer pairs used in qRT-PCR analysis.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional information files.