

We present and characterize a novel mutation in KCNJ10. Unlike previously reported EAST/SeSAME patients, our patient was heterozygous, and contrary to previous studies, mimicking the heterozygous state by coexpression resulted in loss of channel function. We report in the same patient co-occurrence of a KCNT1 mutation resulting in a more severe phenotype. This study provides new insights into the phenotypic spectrum and to the genotype-phenotype correlations associated with EAST/SeSAME and MMFSI.
Keywords: Kir4.1, mutation, KCNJ10, seizures, genetics
Abstract
A 2-yr-old boy presented profound developmental delay, failure to thrive, ataxia, hypotonia, and tonic-clonic seizures that caused the death of the patient. Targeted and whole exome sequencing revealed two heterozygous missense variants: a novel mutation in the KCNJ10 gene that encodes for the inward-rectifying K+ channel Kir4.1 and another previously characterized mutation in KCNT1 that encodes for the Na+-activated K+ channel known as Slo2.2 or SLACK. The objectives of this study were to perform the clinical and genetic characterization of the proband and his family and to examine the functional consequence of the Kir4.1 mutation. The mutant and wild-type KCNJ10 constructs were generated and heterologously expressed in Xenopus laevis oocytes, and whole cell K+ currents were measured using the two-electrode voltage-clamp technique. The KCNJ10 mutation c.652C>T resulted in a p.L218F substitution at a highly conserved residue site. Wild-type KCNJ10 expression yielded robust Kir current, whereas currents from oocytes expressing the mutation were reduced, remarkably. Western Blot analysis revealed reduced protein expression by the mutation. Kir5.1 subunits display selective heteromultimerization with Kir4.1 constituting channels with unique kinetics. The effect of the mutation on Kir4.1/5.1 channel activity was twofold: a reduction in current amplitudes and an increase in the pH-dependent inhibition. We thus report a novel loss-of-function mutation in Kir4.1 found in a patient with a coexisting mutation in SLACK channels that results in a fatal disease.
NEW & NOTEWORTHY We present and characterize a novel mutation in KCNJ10. Unlike previously reported EAST/SeSAME patients, our patient was heterozygous, and contrary to previous studies, mimicking the heterozygous state by coexpression resulted in loss of channel function. We report in the same patient co-occurrence of a KCNT1 mutation resulting in a more severe phenotype. This study provides new insights into the phenotypic spectrum and to the genotype-phenotype correlations associated with EAST/SeSAME and MMFSI.
the k+ channel Kir4.1 is known to play a key role in controlling membrane potential, cellular excitability, and K+ fluxes. The primary subunit of the channel is composed of two putative membrane-spanning domains (TM1 and TM2) that are linked by an extracellular pore-forming region (H5), and of cytoplasmic amino and carboxyl terminals (Fig. 1E). A functional Kir channel is made up of four such subunits in a tetrameric complex (Yang et al. 1995). KCNJ10 may function as a homotetramer or it may form heterotetramers with the related subunit Kir5.1 (KCNJ16) (Pessia et al. 1996). Heteromeric assembly of Kir4.1 with Kir5.1 subunits confers functional and biophysical properties distinct from those of the homomeric Kir4.1 channel (Hibino et al. 2010). Specifically, coexpression of Kir4.1 with Kir5.1 subunits produces channels with stronger inward rectification and increased intracellular pH (pHi) sensitivity compared with the Kir4.1 homomer (Pessia et al. 2001; Tucker et al. 2000).
Fig. 1.

Detecting and analyzing the mutation. A: pedigree of the patient’s family indicating heterozygous state for the KCNJ10 mutation in both mother and proband. Squares represent males and circles females; arrow points to our affected proband. Detection of mutation was done by sequencing. B and C: electropherograms of KCNJ10 exons for the WT control (B) and the L218F mutation (C) in our patient. D: protein sequence of KCNJ10 from various species aligned using MUSCLE 3.6. The GenBank accession numbers for KCNJ10 of each species are indicated. E: schematic topology model of the human KCNJ10 subunit according to UniProt (ID P78508; http://www.uniprot.org/). The L218F variant lies in the cytoplasmic COOH-terminal domain.
In humans, autosomal recessive mutations of KCNJ10 have been reported to cause EAST/SeSAME syndrome, characterized by epilepsy, ataxia, sensorineural deafness, mental retardation, electrolyte imbalance, and salt-wasting tubulopathy (Bockenhauer et al. 2009; Cross et al. 2013; Scholl et al. 2009). The phenotypes reported in EAST/SeSAME syndrome are the result of the loss of function of Kir4.1 in the cells and organs that express the channel. For example, the seizures observed in EAST/SeSAME patients could be attributed to the predominant distribution of Kir4.1 homomer and Kir4.1/5.1 heteromers in glial cells of the central nervous system (Hibino et al. 2010; Sicca et al. 2011). These channels and gap junctions are involved in mediating clearing of excess K+ that accumulates as a result of synaptic excitation and that would otherwise cause continuous neuronal depolarization (Bedner and Steinhäuser 2013; Olsen and Sontheimer 2008). Loss of function of the Kir4.1 channel in the brain or spinal cord induces astrocyte depolarization, reduces astrocytic K+ and glutamate uptake, and results in a reduced seizure threshold (Djukic et al. 2007; Haj-Yasein et al. 2011). Conditional astrocytic Kir 4.1 knockout mice had smaller brains and displayed severe ataxia, seizures, and early lethality (Djukic et al. 2007). A delay in K+ clearance after synaptic activation was reported in the hippocampal slices of Kir 4.1 knockout mice (Haj-Yasein et al. 2011). Electrophysiological studies revealed that astrocytic Kir4.1 channels upregulate hippocampal spontaneous excitatory synaptic currents (Djukic et al. 2007) and downregulate long-term potentiation (Janigro et al. 1997) and short-term synaptic plasticity (in response to both prolonged repetitive stimulation and posttetanic potentiation), reflecting decreases in presynaptic glutamate release (Sibille et al. 2014). Thus Kir4.1 channels play a vital role in the modulation of synaptic strength.
In the ear, Kir4.1 channels expressed in the stria vascularis allow for the secretion of K+ into the endolymph, contributing significantly to the endocochlear potential (Nin et al. 2008), the disruption of which is the cause of deafness in EAST/SeSAME patients. Yet another example is the renal salt-wasting phenotype of the EAST/SeSAME syndrome that is due to the crucial role played by Kir4.1 and Kir5.1 in facilitating salt reabsorption in the distal convoluted tubule of the kidney (Tucker et al. 2000).
In this study we report, in a 2-yr-old Saudi boy, two monoallelic mutations: a novel mutation in the KCNJ10 gene and another mutation, previously discovered (Ishii et al. 2013; Kim et al. 2014; Møller et al. 2015; Ohba et al. 2015) and characterized (Kim et al. 2014; Rizzo et al. 2016), in the KCNT1 gene. The latter gene, also known as Slo2.2 or SLACK, encodes for the Na+-activated K+ channel subunit that contributes to hyperpolarization after a single action potential or following a repetitive firing of action potentials. When expressed in a heterologous expression system, the p.G288S missense mutation, also carried by our patient, resulted in larger whole cell K+ currents compared with wild-type KCNT1 currents (Kim et al. 2014; Rizzo et al. 2016), albeit a smaller unitary conductance has been reported (Kim et al. 2014). Seven of the eight patients reported to carry the mutation were diagnosed with malignant migrating focal seizures in infancy (MMFSI; OMIM no. 614959), an early-onset epileptic encephalopathy characterized by migrating multifocal seizures. Patients had symptoms that included severe psychomotor developmental delays, aphasia, and intellectual disability. One patient with the same p.G288S mutation was, however, diagnosed with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), a familial focal epilepsy syndrome quite distinct from MMFSI (Kim et al. 2014).
In this study, we focused investigations on the novel and uncharacterized KCNJ10 mutation. We examined the functional consequences of this KCNJ10 mutation by expressing it in Xenopus laevis oocytes and found a loss-of-function mechanism for both Kir4.1 and Kir4.1/5.1 channels. Moreover, we report an increased sensitivity of the mutant Kir4.1/5.1 channels to intracellular acidification.
MATERIALS AND METHODS
Genomic DNA sequencing.
Studies involving the proband and his family were approved by the Office of Research Affairs at King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia, and complied with the Helsinki Declaration. Informed consent for participation and publishing was obtained from the proband’s parents. A Comprehensive Epilepsy Panel including genes causing Mendelian forms of epilepsy was commercially performed (GeneDx, Gaithersburg, MD). The test methodology is accessible through https://www.genedx.com/wp-content/uploads/crmdocs/infosheetepilepsycomp.pdf. DNA was extracted from patient and family blood specimens. PCR was used to amplify exons plus additional flanking noncoding sequences. Cycle sequencing was carried out using the ABI Big Dye Terminator v.3.0 kit. Products were resolved by electrophoresis on an ABI 3730xl capillary sequencer. Sequencing was performed in both forward and reverse directions. Patient and family member sequences were aligned and compared with reference sequences and were interpreted as per the American College of Medical Genetics and Genomics guidelines (Richards et al. 2015).
Production of KCNJ10 constructs.
The primer pair L218F (forward: 5′-GCC AGG TGA CAG GAA AAC TGT TTC AGA CCC ACC AAA CCA AGG-3′ and reverse: 5′- CCT TGG TTT GGT GGG TCT GAA ACA GTT TTC CTG TCA CCT GGC-3′) was used to introduce the mutation L218F into the human Kir4.1 coding region using a PCR-based strategy. Human Kir4.1 cDNA was subcloned into a pTLB oocyte expression vector. The mutation p.L218F was introduced by site-directed mutagenesis performed using the QuikChange protocol (Stratagene, La Jolla, CA) and was verified by automated sequencing. In vitro transcription was done using the mMESSAGE mMACHINE SP6 transcription kit (Thermo Fisher Scientific). The transcribed cRNA concentration was quantified by electrophoresis with ethidium bromide staining and spectrophotometric analysis.
Heterologous expression of KCNJ10.
Wild-type and mutant channels were expressed in X. laevis oocytes as described in D’Adamo et al. (2015). Animal handling was in accordance with international standards of animal care, the Italian Animal Welfare Act approved by the local Veterinary Service Authority, and the NIH Guide for the Care and Use of Laboratory Animals. The X. laevis frogs were deeply anesthetized with an aerated solution containing 3-aminobenzoic acid ethyl ester methanesulfonate salt (5 mM) and sodium bicarbonate (60 mM), pH 7.3. Stage V–VI oocytes were isolated, digested with collagenase, injected with cRNA, and incubated at 16°C in ND96 solution containing (in mM) 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, and 5 HEPES, pH 7.4, with 50 μg/ml gentamicin. Frogs underwent no more than two surgeries, separated by at least 3 wk.
Electrophysiology.
Two-electrode voltage-clamp recordings (TEVC) were performed as previously described (Tucker et al. 2000). Briefly, whole cell currents were recorded using TEVC on oocytes at ∼22°C. Recordings were made on days 1–5 after cRNA injection with the use of a GeneClamp 500 amplifier (Axon Instruments) interfaced to a personal computer with an ITC-16 interface (Instrutech). The amplitude of currents measured from all cells increased substantially from day 1, peaked at day 3, and gradually decreased to reach close to day 1 levels on day 5. All currents and means reported are from recordings obtained on days 3 and 4 after injection. Microelectrodes were pulled to a tip resistance of <1 MΩ and backfilled with 3 M KCl. The extracellular solution contained (in mM) 90 KCl, 3 MgCl2, and 10 HEPES, pH 7.4. Desired intracellular pH (pHi) was achieved using a potassium acetate buffering system as previously described (Pessia et al. 2001). These experiments are based on previously established pHi changes in oocytes obtained using external acetate-buffered solutions (Tsai et al. 1995). Oocyte membrane potentials were held at −10 mV and stepped to various test potentials to elicit whole cell currents. Recordings were filtered at 2 kHz and acquired at 5 kHz with Pulse software and were analyzed with either PulseFit (HEKA, Lambrecht, Germany) or Origin 8 (OriginLab, Northampton, MA) software. Leak and capacitative currents were subtracted using a P/4 protocol.
Homology modeling.
The three-dimensional (3D) structure of heterotetramer Kir4.1 WT/L218F was built through comparative modeling using the Šali and Blundell (1993) software Modeller. The X-ray structure of the Kir3.1-prokaryotic Kir channel chimera (PDB ID: 2QKS) was used as a template (Nishida et al. 2007). Sequence alignment of the target sequence vs. the template was generated using ClustalX and further refined using Muscle (Edgar 2004); the calculated percentage of identity on the aligned sequences was 36.7%, whereas the similarity was 66.3%; only residues 25–349 of the Kir4.1 primary structure could be aligned with the corresponding segments of the X-ray template. Twenty homology models were generated and scored against the minimum number of constraint violations. Among them, the five lowest energy models were selected and analyzed using Procheck (Laskowski et al. 1996). The final model was chosen according to the highest percentage of residues in the allowed region of the Ramachandran plot (>90%). The L218F mutant was generated by substituting the leucine-218 side chain with that of the phenylalanine one using the VMD program (Humphrey et al. 1996). The final model was further minimized to reduce steric hindrance with neighboring atoms using the GROMACS4 algorithm and the GROMOS96 forcefield (Hess et al. 2008).
Immunoblots of oocyte lysates.
Xenopus laevis oocytes were recovered in phosphate-buffered saline (PBS; 5 µl per oocyte) supplemented with 0. 1mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Sigma) 72 h after injection with cRNAs and were lysed by passage through a 26-gauge needle 10 times. The lysates were centrifuged twice at 200 g for 5 min at 4°C to remove yolk and cellular debris. The supernatant proteins were resolved on a 12% polyacrylamide gel in reducing conditions and transferred to a nitrocellulose membrane that was blocked overnight in 3% nonfat dry milk in TBS-T (Tris-buffered saline-0.05% Tween 20). The blots were incubated for 1 h with the specific primary goat polyclonal Kir4.1 (1:500) in 3% BSA solution (Abcam, Cambridge, UK). After several washes in TBST (0.05% Tween 20), the membranes were incubated in 3% nonfat dry milk TBS-T (0.05% Tween 20) containing goat anti-rabbit antibody coupled to horseradish peroxidase (1:4,000; Santa Cruz) for 1 h. Immunoreactivity was visualized using enhanced chemiluminescence (GE Healthcare Europe, Diegen, Belgium). Images were acquired using the VersaDoc 1000 imaging system, and individual band densities were integrated by using Quantity One software (Bio-Rad).
Statistical analysis.
Statistical analysis was performed using the software programs Prism 6 (GraphPad Software, San Diego, CA) and Origin 8 (OriginLab). All data are means ± SE. Observed differences were evaluated by two-tailed unpaired Student’s t-test and were considered significant if P < 0.05. All means are averages obtained from independent experiments performed from oocytes collected from three different frogs. The number (n) of oocytes used in the experiments is indicated where appropriate.
RESULTS
Clinical findings.
A Saudi Arabian boy, a product of full-term normal pregnancy and delivery and born to nonconsanguineous parents (as determined by questionnaire), developed at the age of 3 mo seizures with up-rolling of eyes and fixed gaze that was recurrent and sometimes associated with oral cyanosis. The boy had no family history of seizure disorders. Parents and siblings, 5 boys and 2 girls, were all healthy (Fig. 1A). The initial attacks took place during sleep and usually lasted less than a minute. However, the frequency of seizures increased and by the age of 4 mo occurred 20–40 times a day. More complex seizures appeared, including smacking of the mouth with tonic-clonic movements of the upper and lower limbs, blinking of the eyes, and opening of the mouth. He was started on phenobarbital and levetiracetam. Trials of pyridoxine, folinic acid, biotin, clonazepam, and thiamin were not beneficial. At 4 mo, his weight was 5.7 kg (10th percentile), length 62 cm (25th percentile), and occipital-frontal circumference (OFC) 39.5 cm (3rd percentile). The brain magnetic resonance imaging (MRI) showed proper myelination and no structural abnormalities. The magnetic resonance spectroscopy (MRS; Fig. 2C) showed decreased ratios of both N-acetylaspartate/creatine (1.22; normal ratio being 2) (Filippi et al. 2002) and choline/creatine (0.84; normal ratio being 1.8) (Filippi et al. 2002). He was nondysmorphic but assumed a froglike position with hypotonia. The BAER (brain stem auditory evoked response) performed at the age of 4 mo showed normal auditory evoked potential responses in both ears. EEG (Fig. 2, A and B) showed that the awake background was disorganized, consisting of high-voltage, 300–400 μV, slow delta activity intermixed with multifocal epileptic discharges. Photic stimulation did not aggravate the discharges. This was consistent with hypsarrhythmia.
Fig. 2.
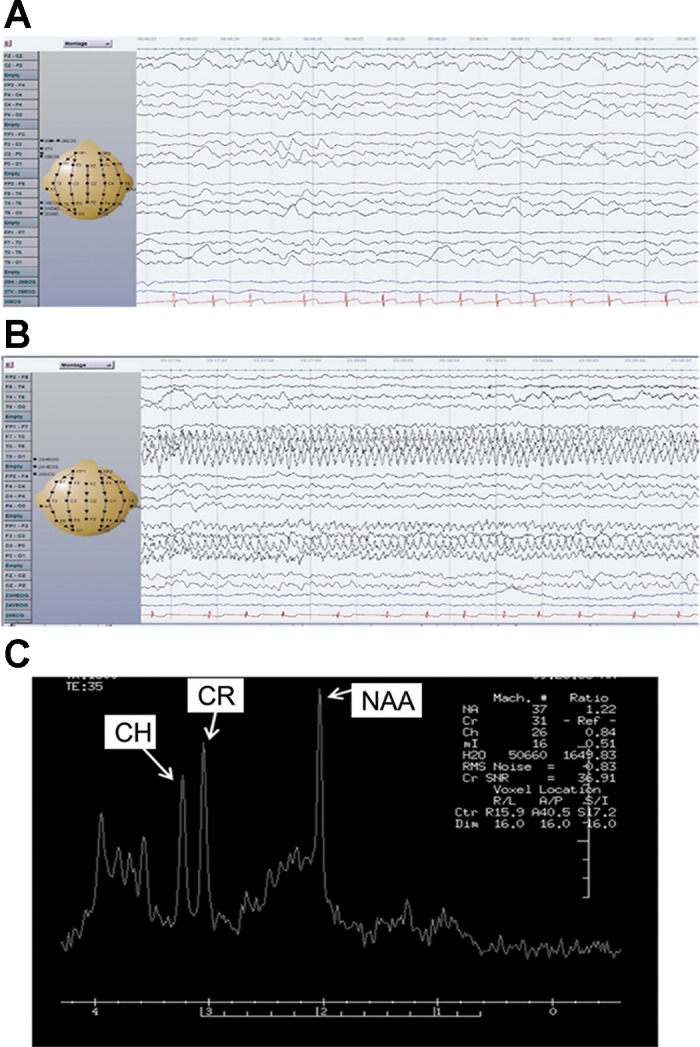
Clinical findings. A: continuous generalized slow background EEG. B: an ictal event characterized by rhythmic sharp waves and spikes involving left temporal and central parietal regions associated with subtle right eye deviation. C: magnetic resonance spectroscopy of the patient showing abnormally low levels of N-acetylaspartate (NAA) and choline (CH). CR, creatine.
On follow-up at the age of 21 mo, the boy showed signs of profound developmental delay. He was not cooing, fixing, or following. He had no head control and was not able to roll over or reach objects. He was also progressively failing to thrive and showed emaciation due to feeding intolerance and persistent vomiting. His barium swallow study did not show gastroesophageal reflux, but there was gut hypomotility on barium meal and small bowel follow-through. Attempts to augment nutrition, utilizing nasogastric feeding and medications (domperidone and omeprazole), had been tried with minimal success. His weight was 5.6 kg (−8 SD), length 69 cm (−5 SD), and OFC 39.5 cm (−6.5 SD). At 2 yr, the plasma electrolytes were as follows (normal ranges in parentheses and all units in mM): plasma potassium 4.7 (3.5–5), sodium 138 (135–147), magnesium 0.9 (0.7–1), chloride 105 (98–111), calcium 2.24 (2.1–2.26), phosphate 1.21 (1.36–2.26), CO2 20 (22–31), urea 2 (2.25–6.5), and creatinine 0.018 (0.017–0.04). The patient recently expired secondary to his underlying epileptic encephalopathy disease.
Identifying the mutation.
To identify genetic variants, possibly associated with the phenotype, the screening of a comprehensive epilepsy gene panel was performed (70 genes, sequencing and deletion/duplication analysis). This analysis revealed a monoallelic missense variant in the KCNJ10 coding region c.652C>T that resulted in a substitution of a leucine with a phenylalanine at the 218th amino acid of the Kir4.1 protein (p.L218F; Fig. 1, B and C). To our knowledge, this variant has not been published as a mutation or as a benign polymorphism. Protein homology analysis revealed that the residue L218 is strictly conserved among species (Fig. 1D) and lies in the COOH-terminal domain of the channel (Fig. 1E). Family screenings identified the KCNJ10 mutation in the proband’s mother, who was heterozygous yet asymptomatic (Fig. 1A). The mother being unaffected may be the result of reduced penetrance of the mutation, a phenomenon not uncommon in channelopathies (Cooper et al. 2013). Sequencing of KCNT1 coding region identified the monoallelic mutation c.862G>A (p.G288S) in the proband that was not found in any other family member. Whole exome sequencing confirmed the presence of the mutations in both KCNJ10 and KCNT1 genes.
3D homology model.
On the basis of crystal structure data, we generated a 3D homology model of a Kir4.1 channel and performed in silico mutagenesis. The analysis of this modeling indicated that the L218F mutation is located within the intracellular domain of the channel at the intersubunit interface (Fig. 3).
Fig. 3.
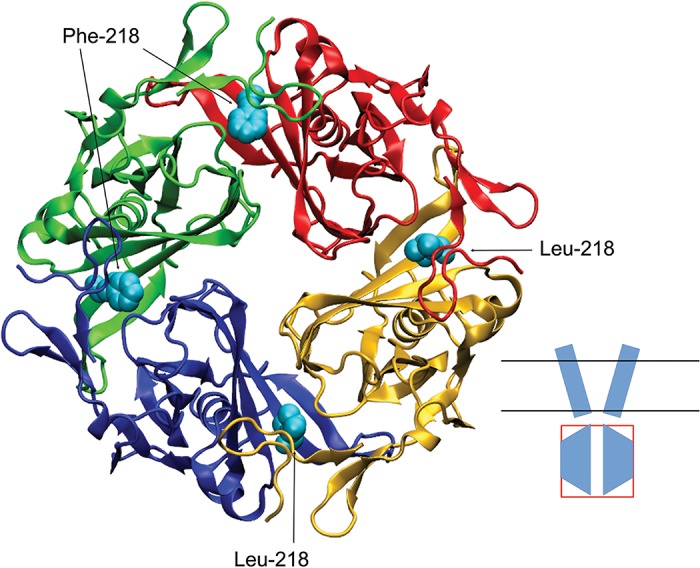
Three-dimensional homology model of the intracellular domain of Kir4.1 WT/L218F channel. Model indicates the structural difference of a larger phenylalanine at site 218 in the red and green subunits compared with that of a leucine, shown in the blue and yellow subunits. Inset shows a cartoon of the channel in which the intracellular domain is boxed.
L218F effect on protein levels and K+ currents.
To assess the pathogenic relevance of the newly identified KCNJ10 variant, we first performed Western blot (WB) analysis from Xenopus oocytes injected with either wild-type (WT) or L218F cRNA. The total membrane fractions were prepared and probed with the anti-Kir4.1 antibody (Fig. 4). The WT and L218F proteins were detected by the antibody as proteins of ~40 kDa, in close accord with its predicted molecular mass. These bands were not detected from mock-injected oocytes. However, the immunoreactive fainter band detected from oocytes injected with L218F clearly indicated that the mutant protein expressed by the cells was greatly reduced compared with control oocytes (Fig. 4). Densitometric analysis of Kir4.1 bands and normalization to the corresponding actin value indicated that the mutation reduced channel expression by ~65% (Fig. 4).
Fig. 4.
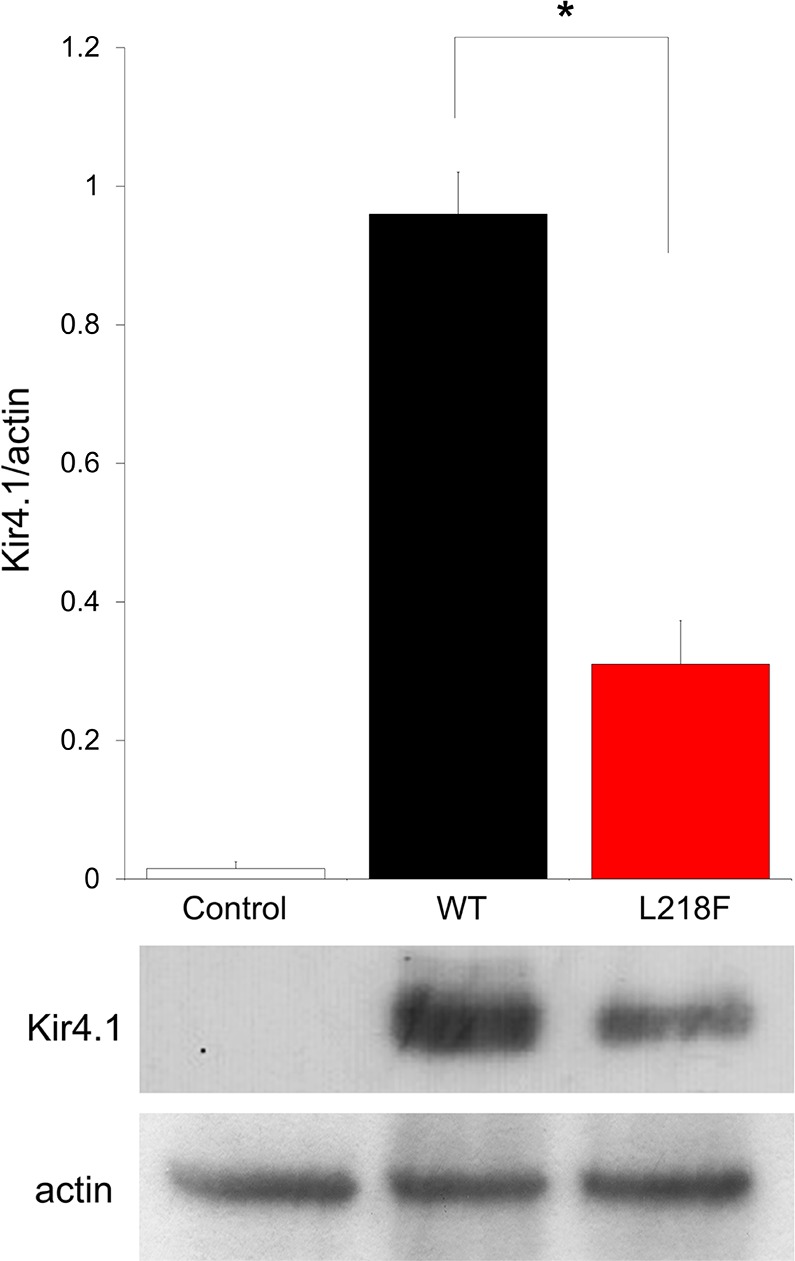
Recombinant Kir4.1 expression in Xenopus oocytes. Shown is densitometric analysis (top) of recombinant Kir4.1 bands (bottom) derived from total membrane protein extracts from mock-injected oocytes (left), WT cRNA-injected oocytes (middle), or L218F cRNA-injected oocytes (right), detected with anti-Kir4.1 antibody and normalized to the corresponding actin value, which was used as loading control. One representative immunoblot of oocyte lysates of 3 is shown. Data are means ± SE of 3 independent experiments (*P < 0.01 vs. WT).
Expression of channels with the L218F yielded currents with macroscopic kinetics similar to WT, exhibiting typical instantaneous currents upon hyperpolarization steps (Fig. 5A). However, the currents measured at both positive and negative test potentials showed smaller amplitudes for L218F than that of the WT when equal amounts of WT or mutated cRNAs were expressed (n = 14; Fig. 5B). For example (Fig. 5C), the steady-state mean current recorded at a test potential of −120 mV from oocytes injected with 0.5 ng of WT Kir4.1 (−56.50 ± 2.62 µA) was significantly different from the mean current recorded from oocytes injected with 0.5 ng of L218F (−22.71 ± 1.41 µA ; n = 14; P < 0.0001). To mimic the heterozygous state of the disease, WT and L218F mutant were coinjected in a 1:1 ratio and yielded current amplitudes that were intermediate between those of the WT and L218F and were significantly different (Fig. 5, A and B). The coinjection of 0.25 ng of WT and 0.25 ng of L218F mutant RNA resulted in a mean current of −41.60 ± 3 µA at −120 mV (n = 14).
Fig. 5.

A: Kir4.1 currents recorded from Xenopus oocytes injected with WT (0.5 ng) or mutant L218F (0.5 ng) or coinjected with both mutant L218F and WT (0.25 ng each) cRNA. Sample current families were recorded by two-electrode voltage clamp and evoked by voltage commands from a holding potential of −10 mV in −10-mV increments from +50 to −120 mV for 400 ms. Horizontal red dashed lines indicate 0 current. B: average steady-state current-voltage (I-V) relationships for WT Kir4.1, mutant L218F Kir4.1, and both WT + L218F Kir4.1 (blue). The current amplitudes measured from oocytes injected with mutant and those coinjected with both mutant and WT were smaller than that of the WT. C: bar graph showing average current amplitude recorded at −120 mV. I-V data points and bar graphs are results from 3 independent experiments performed using different batches of oocytes and recorded on day 3 or 4 after injection. Data are means ± SE (n = 14). Statistical significance of the differences was calculated using unpaired Student’s t-test. *P < 0.001; **P < 0.0001.
To promote proper assembly of Kir4.1 subunits with Kir5.1, both cRNAs were coinjected in a 1:20 ratio. Consistent with previously reported functional properties of Kir4.1/Kir5.1 channels (Pessia et al. 1996; Xu et al. 2000), the resulting macroscopic currents exhibited an instantaneous component followed by a time-dependent slow activation (Fig. 6). L218F mutation resulted in decreased heteromeric current amplitudes, recorded at both positive and negative test potentials, compared with those obtained from channels comprising WT Kir4.1 and Kir5.1 subunits (Fig. 6, A and B). For example (Fig. 6C), the mean current recorded at a test potential of −120 mV from oocytes injected with WT Kir4.1/Kir5.1 (−36.02 ± 4.77 µA, n = 8) was significantly different from the mean current recorded from oocytes injected with L218F Kir4.1/Kir5.1 (−17.69 ± 2.37 µA, n = 8; P < 0.005).
Fig. 6.

Effects of L218F mutation on heteromeric Kir4.1/Kir5.1 channel function. A: representative current families from oocytes expressing WT Kir4.1/Kir5.1 and L218F Kir4.1/Kir5.1 channels. Currents were evoked by voltage commands as described in Fig. 5, and all displayed the time-dependent “relaxation” typical of Kir4.1/Kir5.1 current. Horizontal red dashed lines indicate 0 current level. B: average steady-state I-V relationships for WT Kir41/5.1 and L218F Kir4.1/5.1. The current amplitudes measured from oocytes injected with mutant cRNA were smaller than those measured from oocytes injected with WT cRNA. C: bar graph showing that at −120 mV, the mean L218F Kir4.1/Kir5.1 current is significantly smaller than the mean WT Kir4.1/Kir5.1 current (n = 8; *P < 0.005). I-V data and bar graphs are representative of independent experiments performed using 2 different batches of oocytes (n = 8). These results indicate that L218F mutation reduced heteromeric current amplitudes. D–F: effect of L218F mutation on the pH sensitivity of Kir4.1/Kir5.1 channels. D: representative currents recorded from oocytes expressing WT Kir4.1/5.1 and L218 Kir4.1/5.1 cRNA. Currents were recorded during the perfusion of a membrane-permeable potassium acetate buffer that reduced the oocyte pHi from 7.2 to 6.4 and at a test potential of −120 mV. E: heteromeric current inhibition with time. WT cRNA-injected oocytes had a time course curve significantly different from that of the mutant cRNA-injected oocytes (n = 10; P < 0.0001). Current amplitudes, recorded at a test potential of −120 mV, were normalized to the peak current before the pHi was changed. The 2 curves were fitted with the equation Y = plateau + (Y0 − plateau)·exp [−K·(X − X0)]. F: bar graph showing residual current after 10 min of potassium acetate buffer perfusion. Residual L218F Kir4.1/Kir5.1 current was significantly less than that of the WT (n = 9; *P = 0.01). Results are from 3 different batches of oocytes.
Compared with homomeric Kir4.1, the heteromeric Kir4.1/Kir5.1 channels displayed an increased sensitivity to inhibition by intracellular H+ (pHi) (Pessia et al. 1996). Using a well-established potassium acetate buffering system known to modify pHi of oocytes (Choe et al. 1997; Pessia et al. 2001; Sicca et al. 2011; Tucker et al. 2000), we examined the mutation’s effects on pHi sensitivity of the heteromeric Kir4.1/Kir5.1 channels and found it to be increased with the mutation (Fig. 6D). Figure 6E shows the time course of inhibition of Kir4.1/Kir5.1 currents during the potassium acetate buffer application. The oocytes with the mutant channel expression had less residual current (14 ± 3%, n = 9) than oocytes with the WT (42 ± 8%, n = 9, P = 0.01) when pHi was lowered from 7.2 to 6.4 (Fig. 6F).
DISCUSSION
In this article we present a novel mutation in the Kir4.1 channel subunit, specifically at amino acid 218 located in the COOH-terminal domain of the subunit. Leucine at position 218 is conserved in homologs across species, which indicates its importance for normal channel function. Moreover, this residue is adjacent to the highly conserved Walker type A sequence (210–217), which binds nucleotide phosphates (Fig. 7). This particular sequence is necessary for ATP binding and is required for sustaining the channel’s activity (Takumi et al. 1995). It is possible that the difference in side chain structure, with leucine being aliphatic vs. the aromatic phenylalanine, leads to altered nucleotide binding and hydrolysis. In fact, alterations in channel sensitivity to nucleotides have been previously reported to occur as a consequence of a leucine substitution for a phenylalanine in an ATP-sensitive potassium (KATP) channel subunit (Cartier et al. 2003). The nearby glutamine at position 212 was recently found critical for normal inward rectification (Méndez-González et al. 2016), thereby putting more weight on the importance of this particular part of the channel. We also report, in the same patient, a de novo c.862G>A mutation in the KCNT1 gene that leads to a glycine-to-serine substitution at the highly conserved residue position 288, located at the 5th transmembrane domain of the protein. Patients harboring this mutation have been diagnosed with MMFSI and with ADNFLE (Kim et al. 2014; Rizzo et al. 2016). The association of the different phenotypes with the same p.G288S mutation may be the result of additional genetic variants such as the KCNJ10 identified in this study. Accordingly, KCNJ10 may be regarded as another contributing gene associated with MMFSI that broadens the spectrum of this seizure disorder. Presumably, the G288S substitution introduces a hydrogen bond that alters the pore structure and traps K+ ions, thereby impairing channel function (Ishii et al. 2013). Whereas this impairment was shown to decrease unitary conductance of the channel, the whole cell current was found to be enhanced (Kim et al. 2014; Rizzo et al. 2016). The possible explanation suggested for this is a change in channel-channel interaction that leads to an increased cooperative gating mechanism between individual mutant channels (Rizzo et al. 2016).
Fig. 7.

3D homology model of Kir4.1 showing the Walker type A sequence (A, stick; B and C, spheres) and the localization of the Leu-218 (B) and Phe-218 (C) residues.
Notably, KCNJ10 variants have been associated with either tonic-clonic seizures (Bockenhauer et al. 2009) or epileptic spasms (Sicca et al. 2011, 2016). Epileptic spasms associated with the hypsarrhythmia EEG pattern seen in our proband constitute the basis for the diagnosis of West syndrome. Lee et al. (2012) reported a patient who first presented with MMPSI (malignant migrating partial seizures of infancy) and later evolved to West syndrome. Unfortunately, genetic analysis of this patient was not performed. It could be worth investigating the presence of compound heterozygous mutations in both KCNJ10/KCNT1 genes.
We explored the functional properties of the L218F KCNJ10 mutation by expression in X. laevis oocytes and found that the mutation results in a decrease in whole cell Kir4.1 currents that can be explained, at least in part, by a reduction in protein expression. Coexpression of mutant and WT channels still showed a significant reduction in channel function, consistent with the presence of clinical findings in our heterozygote patient.
Although the KCNJ16 (Kir5.1) channel does not produce functional channels on its own, it coassembles selectively with Kir4.1 subunits to form distinct heteromeric channels. Kir4.1 becomes more sensitive to pHi in the physiologic range when coexpressed with Kir5.1 (Pessia et al. 2001). The pH-sensing mechanism, however, remains undetermined. We have previously proposed (Casamassima et al. 2003) that pH sensitivity and heteromeric interaction is regulated by residues, which include H190, at the proximal COOH terminus of Kir4.1. The L218F mutation observed in this study is also at the COOH-terminal domain of the channel. Rapid increases or decreases in H+ in the central nervous system that arise from electrical activity affect the function of both neurons and glia, a mechanism of which probably involves pH-sensitive ion channels such as Kir4.1/Kir5.1. These channels serve as important determinants of neuronal and astrocytic Pco2/pH sensitivity, as observed in Kir5.1 knockout mice (D’Adamo et al. 2011). pHi-sensitive Kir4.1/Kir5.1 provide a link between pHi and resting membrane potential. In astrocytes, Kir4.1/Kir5.1 channels have been proposed to play a K+ buffering role in response to changes in pHi (Hibino et al. 2010). In these cells, the electrogenic Na+- cotransporter is especially prevalent and is responsible for depolarization-induced intracellular alkalinization and membrane hyperpolarization (Chesler 2003). An increase in extracellular K+ concentration, a consequence of synaptic excitation, depolarizes the astrocyte membrane and accelerates the transporter resulting in a rapid rise in astrocyte pHi due to its uptake of one Na+ and two or three . The resulting intracellular alkalinization would enhance Kir4.1/Kir5.1 channel activity, facilitating K+ uptake. Coexpression of L218F Kir4.1 with Kir5.1 produced heteromeric L218F Kir4.1/Kir5.1 channels that showed decreased whole cell currents compared with the WT Kir4.1/Kir5.1 currents, confirming an effect on these heteromeric channels by the mutation. As previously described (Tucker et al. 2000), in this study acidic shifts of pHi reduced WT Kir4.1/Kir5.1 channel activity. The pHi-induced inhibition was greater in the oocytes expressing the L218F mutation. Increased pHi sensitivity of mutated Kir4.1/5.1 channels has been reported in previous studies involving patients with EAST/SeSAME syndrome (Williams et al. 2010). These results suggest that physiological conditions that lead to acidification, such as intense synaptic activity or physical stress, could further reduce mutated Kir4.1/Kir5.1 currents, leading to impaired glial cell K+ buffering that aggravates epilepsy and worsens symptoms. Thus pH homeostasis of patients with KCNJ10 mutations should be considered and may be of importance for the treatment given to patients with this disease.
Previously discovered KCNJ10 mutations resulting in EAST/SeSAME symptoms were all found in homozygous (Bockenhauer et al. 2009; Freudenthal et al. 2011; Parrock et al. 2013; Reichold et al. 2010; Scholl et al. 2009, 2012) and compound heterozygous patients (Scholl et al. 2009). Our patient was heterozygous for the KCNJ10 mutation. A possible explanation is the presence of a digenic mechanism where the appearance of symptoms resulting from one mutation may depend on the presence of another mutation on a different gene (Cooper et al. 2013). The co-occurrence of KCNT1 with the KCNJ10 mutation may have resulted in the augmentation of EAST/SeSAME phenotype. Such incidence, whereby a clinical phenotype of an autosomal recessive disease appears in double heterozygote probands, has been previously reported (e.g., da Silva-Costa et al. 2011; Liu et al. 2017; Yigit et al. 2015). However, unlike the results we report in this case, in studies that characterized EAST/SeSAME syndrome mutations, mimicking the heterozygous state by coexpressing mutant and wild-type channels all resulted in wild-type-like currents (Tang et al. 2010), which explains the lack of symptoms observed in heterozygous relatives of affected individuals and its presence in our proband. That in our study current decrease and changes in channel kinetics were detected with both Kir4.1 WT/L218F and L218F Kir4.1/Kir5.1 coexpressions could be due to the specific characteristics of the mutation involved, ours perhaps being more severe and located in a critical region for the channel protein function. Indeed, functional consequences and severity of symptoms differ according to mutation site and amino acids involved; some have more drastic effects on membrane potential and cell viability than others (Méndez-González et al. 2016).
Although the two names of this disease (EAST and SeSAME) are in fact abbreviated assemblages of the symptoms caused by KCNJ10 mutations, there is nevertheless a phenotypic heterogeneity in the presentation of these symptoms. For example, the A167V mutation resulted in hearing impairment and tubulopathy yet did not present the hallmark of the disease, epileptic seizures (Parrock et al. 2013). Conversely, hearing loss can be absent in the presence of cognitive delay, seizures, and severe ataxia (Scholl et al. 2012). It is unclear whether our 2-yr-old subject had hearing problems or whether it had not been recognized or well developed. Hearing in many affected EAST/SeSAME children appears normal in the first years of life but worsen with age, resulting in clinically significant defects at age 5–8 yr (Scholl et al. 2012). As for electrolyte abnormalities, data from five unrelated families revealed that electrolytes are typically normal at birth and during the first several years of life, becoming abnormal after 5 yr of age (Scholl et al. 2012). On the other hand, as reported for EAST/SeSAME syndrome patients, our proband had motor delay with evidence of ataxia apparent from the first year of life. Moreover, decreased levels of N-acetylaspartate and choline indicated in the MRS analysis of our patient point to neuronal loss and/or compromised neuronal metabolism.
In conclusion, our data demonstrate the presence of digenic heterozygous mutations in KCNJ10/KCNT1 identified in a patient with severe seizure and global developmental deficits. In summary, we report a novel loss-of-function mutation in KCNJ10 that alters the function of both Kir4.1 and Kir4.1/Kir5.1 channels. This and the coexisting p.G288S missense SLACK mutation could synergize to produce a more severe phenotype as observed in our proband. These findings provide further insights into the phenotypic spectrum and genotype-phenotype correlations associated with MMFSI and support the inclusion of KCNJ10 in a gene panel approach in the diagnosis of patients with MMFSI and severe developmental delay disorders.
GRANTS
This project was funded by Cassa di Risparmio di Perugia.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.P. and M.C.D. conceived and designed research; S.M.H., A.G., M.C., C.B., and L.M. performed experiments; S.M.H., A.B., and R.R. analyzed data; S.M.H., O.D., A.A.-S., M.P., M.A.-O., and M.C.D. interpreted results of experiments; S.M.H., A.G., R.R., and M.A.-O. prepared figures; S.M.H. drafted manuscript; S.M.H., O.D., M.P., and M.C.D. edited and revised manuscript; S.M.H., A.B., A.G., O.D., M.C., R.R., A.A.-S., C.B., L.M., M.P., M.A.-O., and M.C.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Elisa Marchetti for technical assistance.
REFERENCES
- Bedner P, Steinhäuser C. Altered Kir and gap junction channels in temporal lobe epilepsy. Neurochem Int 63: 682–687, 2013. doi: 10.1016/j.neuint.2013.01.011. [DOI] [PubMed] [Google Scholar]
- Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, Tobin J, Lieberer E, Sterner C, Landoure G, Arora R, Sirimanna T, Thompson D, Cross JH, van’t Hoff W, Al Masri O, Tullus K, Yeung S, Anikster Y, Klootwijk E, Hubank M, Dillon MJ, Heitzmann D, Arcos-Burgos M, Knepper MA, Dobbie A, Gahl WA, Warth R, Sheridan E, Kleta R. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier EA, Shen S, Shyng SL. Modulation of the trafficking efficiency and functional properties of ATP-sensitive potassium channels through a single amino acid in the sulfonylurea receptor. J Biol Chem 278: 7081–7090, 2003. doi: 10.1074/jbc.M211395200. [DOI] [PubMed] [Google Scholar]
- Casamassima M, D’Adamo MC, Pessia M, Tucker SJ. Identification of a heteromeric interaction that influences the rectification, gating, and pH sensitivity of Kir4.1/Kir5.1 potassium channels. J Biol Chem 278: 43533–43540, 2003. doi: 10.1074/jbc.M306596200. [DOI] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev 83: 1183–1221, 2003. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Choe H, Zhou H, Palmer LG, Sackin H. A conserved cytoplasmic region of ROMK modulates pH sensitivity, conductance, and gating. Am J Physiol Renal Physiol 273: F516–F529, 1997. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet 132: 1077–1130, 2013. doi: 10.1007/s00439-013-1331-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross JH, Arora R, Heckemann RA, Gunny R, Chong K, Carr L, Baldeweg T, Differ AM, Lench N, Varadkar S, Sirimanna T, Wassmer E, Hulton SA, Ognjanovic M, Ramesh V, Feather S, Kleta R, Hammers A, Bockenhauer D. Neurological features of epilepsy, ataxia, sensorineural deafness, tubulopathy syndrome. Dev Med Child Neurol 55: 846–856, 2013. doi: 10.1111/dmcn.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamo MC, Gallenmüller C, Servettini I, Hartl E, Tucker SJ, Arning L, Biskup S, Grottesi A, Guglielmi L, Imbrici P, Bernasconi P, Di Giovanni G, Franciolini F, Catacuzzeno L, Pessia M, Klopstock T. Novel phenotype associated with a mutation in the KCNA1(Kv1.1) gene. Front Physiol 5: 525, 2015. doi: 10.3389/fphys.2014.00525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Adamo MC, Shang L, Imbrici P, Brown SD, Pessia M, Tucker SJ. Genetic inactivation of Kcnj16 identifies Kir5.1 as an important determinant of neuronal PCO2/pH sensitivity. J Biol Chem 286: 192–198, 2011. doi: 10.1074/jbc.M110.189290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva-Costa SM, Martins FT, Pereira T, Pomilio MC, Marques-de-Faria AP, Sartorato EL. Searching for digenic inheritance in deaf Brazilian individuals using the multiplex ligation-dependent probe amplification technique. Genet Test Mol Biomarkers 15: 849–853, 2011. doi: 10.1089/gtmb.2011.0034. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci 27: 11354–11365, 2007. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797, 2004. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippi CG, Uluğ AM, Deck MD, Zimmerman RD, Heier LA. Developmental delay in children: assessment with proton MR spectroscopy. AJNR Am J Neuroradiol 23: 882–888, 2002. [PMC free article] [PubMed] [Google Scholar]
- Freudenthal B, Kulaveerasingam D, Lingappa L, Shah MA, Brueton L, Wassmer E, Ognjanovic M, Dorison N, Reichold M, Bockenhauer D, Kleta R, Zdebik AA. KCNJ10 mutations disrupt function in patients with EAST syndrome. Nephron, Physiol 119: 40–48, 2011. doi: 10.1159/000330250. [DOI] [PubMed] [Google Scholar]
- Haj-Yasein NN, Jensen V, Vindedal GF, Gundersen GA, Klungland A, Ottersen OP, Hvalby O, Nagelhus EA. Evidence that compromised K+ spatial buffering contributes to the epileptogenic effect of mutations in the human Kir4.1 gene (KCNJ10). Glia 59: 1635–1642, 2011. doi: 10.1002/glia.21205. [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447, 2008. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 90: 291–366, 2010. doi: 10.1152/physrev.00021.2009. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 14: 33–38, 1996. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Ishii A, Shioda M, Okumura A, Kidokoro H, Sakauchi M, Shimada S, Shimizu T, Osawa M, Hirose S, Yamamoto T. A recurrent KCNT1 mutation in two sporadic cases with malignant migrating partial seizures in infancy. Gene 531: 467–471, 2013. doi: 10.1016/j.gene.2013.08.096. [DOI] [PubMed] [Google Scholar]
- Janigro D, Gasparini S, D’Ambrosio R, McKhann G 2nd, DiFrancesco D. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci 17: 2813–2824, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, Kaczmarek LK. Human Slack potassium channel mutations increase positive cooperativity between individual channels. Cell Reports 9: 1661–1672, 2014. doi: 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8: 477–486, 1996. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lee EH, Yum MS, Jeong MH, Lee KY, Ko TS. A case of malignant migrating partial seizures in infancy as a continuum of infantile epileptic encephalopathy. Brain Dev 34: 768–772, 2012. doi: 10.1016/j.braindev.2011.11.011. [DOI] [PubMed] [Google Scholar]
- Liu YP, Bosch DG, Siemiatkowska AM, Rendtorff ND, Boonstra FN, Möller C, Tranebjærg L, Katsanis N, Cremers FP. Putative digenic inheritance of heterozygous RP1L1 and C2orf71 null mutations in syndromic retinal dystrophy. Ophthalmic Genet 30: 127–132, 2017. doi: 10.3109/13816810.2016.1151898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez-González MP, Kucheryavykh YV, Zayas-Santiago A, Vélez-Carrasco W, Maldonado-Martínez G, Cubano LA, Nichols CG, Skatchkov SN, Eaton MJ. Novel KCNJ10 gene variations compromise function of inwardly rectifying potassium channel 4.1. J Biol Chem 291: 7716–7726, 2016. doi: 10.1074/jbc.M115.679910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, van Kempen MJ, Klinkenberg S, Andrews I, Kelley K, Ronen GM, Callen D, McMahon JM, Yendle SC, Carvill GL, Mefford HC, Nabbout R, Poduri A, Striano P, Baglietto MG, Zara F, Smith NJ, Pridmore C, Gardella E, Nikanorova M, Dahl HA, Gellert P, Scheffer IE, Gunning B, Kragh-Olsen B, Dibbens LM. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia 56: e114–e120, 2015. doi: 10.1111/epi.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nin F, Hibino H, Doi K, Suzuki T, Hisa Y, Kurachi Y. The endocochlear potential depends on two K+ diffusion potentials and an electrical barrier in the stria vascularis of the inner ear. Proc Natl Acad Sci USA 105: 1751–1756, 2008. doi: 10.1073/pnas.0711463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Cadene M, Chait BT, MacKinnon R. Crystal structure of a Kir3.1-prokaryotic Kir channel chimera. EMBO J 26: 4005–4015, 2007. doi: 10.1038/sj.emboj.7601828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Kato M, Takahashi N, Osaka H, Shiihara T, Tohyama J, Nabatame S, Azuma J, Fujii Y, Hara M, Tsurusawa R, Inoue T, Ogata R, Watanabe Y, Togashi N, Kodera H, Nakashima M, Tsurusaki Y, Miyake N, Tanaka F, Saitsu H, Matsumoto N. De novo KCNT1 mutations in early-onset epileptic encephalopathy. Epilepsia 56: e121–e128, 2015. doi: 10.1111/epi.13072. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Functional implications for Kir4.1 channels in glial biology: from K+ buffering to cell differentiation. J Neurochem 107: 589–601, 2008. doi: 10.1111/j.1471-4159.2008.05615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrock S, Hussain S, Issler N, Differ AM, Lench N, Guarino S, Oosterveld MJ, Keijzer-Veen M, Brilstra E, van Wieringen H, Konijnenberg AY, Amin-Rasip S, Dumitriu S, Klootwijk E, Knoers N, Bockenhauer D, Kleta R, Zdebik AA. KCNJ10 mutations display differential sensitivity to heteromerisation with KCNJ16. Nephron Physiol 123: 7–14, 2013. doi: 10.1159/000356353. [DOI] [PubMed] [Google Scholar]
- Pessia M, Imbrici P, D’Adamo MC, Salvatore L, Tucker SJ. Differential pH sensitivity of Kir4.1 and Kir4.2 potassium channels and their modulation by heteropolymerisation with Kir5.1. J Physiol 532: 359–367, 2001. doi: 10.1111/j.1469-7793.2001.0359f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia M, Tucker SJ, Lee K, Bond CT, Adelman JP. Subunit positional effects revealed by novel heteromeric inwardly rectifying K+ channels. EMBO J 15: 2980–2987, 1996. [PMC free article] [PubMed] [Google Scholar]
- Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA 107: 14490–14495, 2010. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424, 2015. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo F, Ambrosino P, Guacci A, Chetta M, Marchese G, Rocco T, Soldovieri MV, Manocchio L, Mosca I, Casara G, Vecchi M, Taglialatela M, Coppola G, Weisz A. Characterization of two de novo KCNT1 mutations in children with malignant migrating partial seizures in infancy. Mol Cell Neurosci 72: 54–63, 2016. doi: 10.1016/j.mcn.2016.01.004. [DOI] [PubMed] [Google Scholar]
- Šali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815, 1993. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106: 5842–5847, 2009. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl UI, Dave HB, Lu M, Farhi A, Nelson-Williams C, Listman JA, Lifton RP. SeSAME/EAST syndrome–phenotypic variability and delayed activity of the distal convoluted tubule. Pediatr Nephrol 27: 2081–2090, 2012. doi: 10.1007/s00467-012-2219-4. [DOI] [PubMed] [Google Scholar]
- Sibille J, Pannasch U, Rouach N. Astroglial potassium clearance contributes to short-term plasticity of synaptically evoked currents at the tripartite synapse. J Physiol 592: 87–102, 2014. doi: 10.1113/jphysiol.2013.261735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F, Ambrosini E, Marchese M, Sforna L, Servettini I, Valvo G, Brignone MS, Lanciotti A, Moro F, Grottesi A, Catacuzzeno L, Baldini S, Hasan S, D’Adamo MC, Franciolini F, Molinari P, Santorelli FM, Pessia M. Gain-of-function defects of astrocytic Kir4.1 channels in children with autism spectrum disorders and epilepsy. Sci Rep 6: 34325, 2016. doi: 10.1038/srep34325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicca F, Imbrici P, D’Adamo MC, Moro F, Bonatti F, Brovedani P, Grottesi A, Guerrini R, Masi G, Santorelli FM, Pessia M. Autism with seizures and intellectual disability: possible causative role of gain-of-function of the inwardly-rectifying K+ channel Kir4.1. Neurobiol Dis 43: 239–247, 2011. doi: 10.1016/j.nbd.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T, Kiyama H, Sohmiya K, Nakanishi S, Kurachi Y. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem 270: 16339–16346, 1995. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- Tang X, Hang D, Sand A, Kofuji P. Variable loss of Kir4.1 channel function in SeSAME syndrome mutations. Biochem Biophys Res Commun 399: 537–541, 2010. doi: 10.1016/j.bbrc.2010.07.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai TD, Shuck ME, Thompson DP, Bienkowski MJ, Lee KS. Intracellular H+ inhibits a cloned rat kidney outer medulla K+ channel expressed in Xenopus oocytes. Am J Physiol Cell Physiol 268: C1173–C1178, 1995. [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Imbrici P, Salvatore L, D’Adamo MC, Pessia M. pH dependence of the inwardly rectifying potassium channel, Kir5.1, and localization in renal tubular epithelia. J Biol Chem 275: 16404–16407, 2000. doi: 10.1074/jbc.C000127200. [DOI] [PubMed] [Google Scholar]
- Williams DM, Lopes CM, Rosenhouse-Dantsker A, Connelly HL, Matavel A, O-Uchi J, McBeath E, Gray DA. Molecular basis of decreased Kir4.1 function in SeSAME/EAST syndrome. J Am Soc Nephrol 21: 2117–2129, 2010. doi: 10.1681/ASN.2009121227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Cui N, Yang Z, Qu Z, Jiang C. Modulation of kir4.1 and kir5.1 by hypercapnia and intracellular acidosis. J Physiol 524: 725–735, 2000. doi: 10.1111/j.1469-7793.2000.00725.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Determination of the subunit stoichiometry of an inwardly rectifying potassium channel. Neuron 15: 1441–1447, 1995. doi: 10.1016/0896-6273(95)90021-7. [DOI] [PubMed] [Google Scholar]
- Yigit G, Brown KE, Kayserili H, Pohl E, Caliebe A, Zahnleiter D, Rosser E, Bögershausen N, Uyguner ZO, Altunoglu U, Nürnberg G, Nürnberg P, Rauch A, Li Y, Thiel CT, Wollnik B. Mutations in CDK5RAP2 cause Seckel syndrome. Mol Genet Genomic Med 3: 467–480, 2015. doi: 10.1002/mgg3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]