

Abstract
Vitamin A based bisretinoid accumulation is a major focus in the study of macular degeneration. Whether specific endogenous lysosomal proteins can bind A2E, a pronounced bisretinoid in lipofuscin granules in retinal pigment epithelial cells, and interfere with enzymatic or photoinduced oxidation of such, has not been explored. Herein, using fluorescence and electronic absorption spectroscopy and mass spectrometry, we demonstrate that Saposin B, a critical protein in the degradation of sulfatides and “flushing” of lipids, can bind A2E, preventing its H2O2-dependent enzymatic oxidation by horseradish peroxidase and photooxidation by blue light (λ=450–460 nm).
Keywords: bisretinoids, enzymatic oxidation, macular degeneration, photooxidation, saposin B
Putting a lid on it
Degrading bisretenoids, understanding their cellular photochemistry and tracking their effects on cellular processes are critical in the study of age-related macular degeneration (AMD) development and treatment. Herein, we demonstrate that the vital cellular co-factor Saposin B binds A2E, a common bisretenoid found in granules in AMD patients, and prevents enzymatic and photooxidation/ degradation.
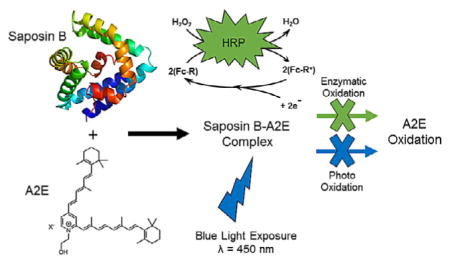
The bisretinoid 2-[(1E,3E,5E,7E)-2,5-dimethyl-8-(2,6,6-trimethyl-cyclohex-1-en-1-yl)octa-1,3,5,7-tetraen-1-yl]-1-(2-hydroxyethyl)-4-[(1E,3E,5E)-4-methyl-6-(2,6,6-trimethylcyclohex-1-en-1-yl)hexa-1,3,5-trien-1-yl]pyridinium (A2E; Figure 1) is a major fluorescent component of lipofuscin granules that accumulate in the lysosomes of cells of the retinal pigment epithelium (RPE) and is implicated in disease processes in age-related macular degeneration (AMD), recessive Stargardt disease (SD), and Best vitelli-form macular dystrophy.[1] A2E forms non-enzymatically in photoreceptors adjacent to RPE cells and is transferred (along with other components) through phagocytosis of outer-segment membrane by the RPE. All healthy eyes accumulate bisretenoids in RPE, while all other outer-membrane components phagocytosed are degraded. Thus, bisretenoid accumulation is not the result of active inhibition of lysosomal activity, differentiating any associated disease process from lysosomal storage diseases (LSD). A2E has been extensively investigated[2] and is the focus of therapeutic approaches that strive to reverse its accumulation in RPE.[3, 4] In 2011, a proof-of-concept study by Sparrow et al. demonstrated the feasibility of using an “enzyme-replacement” approach to A2E degradation by introducing horseradish peroxidase (HRP) into a human RPE cell line (ARPE-19), reducing A2E levels by 75 %.[5]
Figure 1.
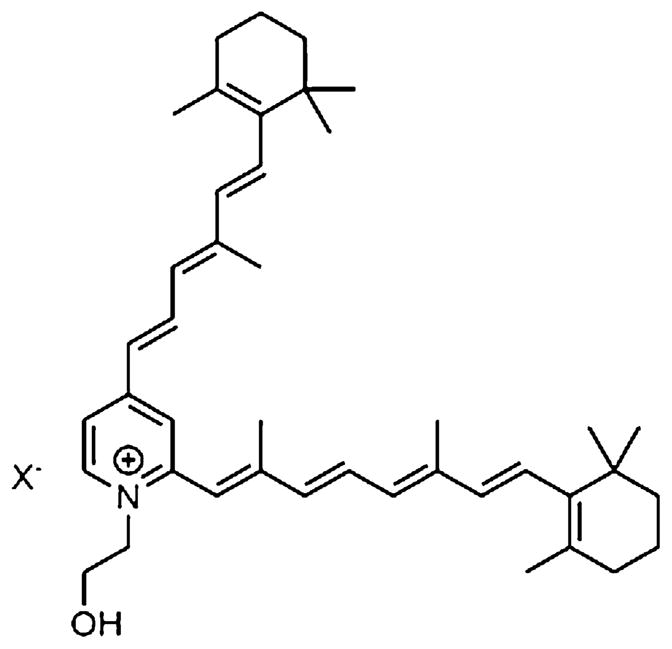
Structure of the bisretinoid A2E.
Although a major focus on A2E has been its role in oxidative stress,[6,7] few studies have demonstrated that A2E can directly interact with endogenous proteins. Work by Moiseyev et al. has demonstrated that A2E can inhibit non-palmitoylated, soluble, cytoplasmic retinoid isomerohydrolase (RPE65 isomerohydrolase) by direct binding (KD = 250 nM).[8] Yanagi et al. have demonstrated A2E to be an endogenous ligand for retinoic acid receptor (RAR), inducing sustained activation of RAR target genes.[9]
The phenotypic outcomes of such sustained activation includes neovascularization about the RPE.[9] Interestingly, Sparrow et al. have shown that conditions that promote A2E aggregation (i.e. nonpolar environments/microdomains) within the lysosome have also been shown to promote photooxidation.[10] Sparrow et al. have postulated that interactions between A2E and components of the lysosomal milieu, which they describe as possibly including polar and hydrophobic side-chains of proteins (without known examples), may serve to hold a fraction of A2E, available for redistribution amongst lysosomal microdomains after photobleaching.[10]
We have been interested in the multisubstrate specificity of human lysosomal Saposin B (SapB) and its implications in drug toxicity and/or disease progression.[11, 12] SapB is an intralysosomal, non-enzymatic proteinaceous co-factor that binds and presents 3-O-sulfogalactosylceramide (sulfatide) to the active site of arylsulfatase A (ASA) for desulfation to galactosylceramide.[13] The lack of functional SapB (or ASA) results in a buildup of sulfatide and the fatal LSD metachromatic leukodystrophy (MLD).[13] Interestingly, progressive RPE degeneration has been reported in patients with MLD.[14] In addition, Barres et al. have shown that sulfatide (but not galactosylceramide) can inhibit optic nerve growth, including the regrowth of damaged optic nerve.[15] SapB has also been shown to “flush” bound ligand, such as coenzyme Q10 (CoQ10), in human urine.[16] Given this understanding, and taking into account the intralysosomal nature of A2E accumulation, we hypothesized that SapB would bind A2E. Herein, we present evidence, using electronic absorption and fluorescence spectroscopies along with mass spectrometry, that SapB binds A2E (KD = 25 μM) with an observed binding stoichiometry of 2:1 SapB:A2E, and that such binding protects A2E from both enzymatic and photooxidation.
A spectrophotometric titration of SapB with A2E was first carried out. A2E[17] and SapB[18] were prepared and purified according to literature reports. To determine the A2E/protein binding stoichiometry, a fluorescence titration (monitoring the emission of tyrosine in SapB at λ=308 nm) was carried out by incrementally adding small amounts of A2E to a solution of SapB (Figure 2). The quenching of the fluorescence intensity at 308 nm exhibited a discontinuity at about 0.5:1 A2E/protein, suggesting the formation of a protein–A2E complex with one A2E ligand bound per SapB dimer. A double logarithmic plot of versus log[Q] confirmed a stoichiometry of approximately 1 A2E per SapB dimer (Figure 2, inset A) and provided a binding affinity (KA) of about 4 × 104 M−1.
Figure 2.

Spectrophotometric titration of SapB with A2E. Inset A: Double logarithmic plot of A2E quenching effect on the fluorescence of SapB. Inset B: Titration curves. Conditions: 40 μM SapB, 2 μL injections of 2 mM A2E (0.13 mole of A2E per mole of SapB), 50 mM phosphate buffer, pH 5.5 and 25 °C.
A2E has maximum absorbance bands around λ=340 nm and 440 nm and an emission spectrum centered around λ= 600 nm (with an excitation wavelength λexc = 440 nm; see Figure S1A in the Supporting Information). When excited at 278 nm, SapB has an intense fluorescence emission at 308 nm whereas A2E has very weak or residual fluorescence, in comparison. When excited at 308 nm, A2E has a very weak emission spectrum as shown in Figure S1B. Thus, the spectrophotometric titration of SapB with A2E reported in Figure 2 is following the quenching of the emission of SapB upon A2E binding, with little to no interference (‘inner-filter“ effect) from A2E fluorescence. Furthermore, the fact that the fluorescence emission of A2E at 605 nm shows a break at 0.5:1 A2E/SapB (Figure 3) is in support of the fluorescence data at 308 nm suggesting that the quenching is due to A2E binding and to A2E quenching the emission intensity of SapB. We note that SapB has no fluorescence at 605 nm when the excitation wavelength was set to 440 nm. Moreover, when A2E is titrated into SapB and the absorbance followed between 300 and 410 nm, a linear relationship is obtained suggesting, once again, that the observed break at 308 nm in Figure 2 is due to binding of A2E to SapB with little to no effect due to A2E (see Figure S2).
Figure 3.

Spectrophotometric titrations of SapB with A2E in the presence of CoQ10. Inset: An example fluorimetric titration. Conditions: 20 μM SapB, 1 μL injections of 1.9 mM A2E in methanol (0.13:1 A2E/SapB per injection), 50 mM phosphate buffer, pH 5.5 and 25 °C. The (SapB-CoQ10)complex were initially prepared at 2:1 protein:ligand ratio.
We have reported in an earlier study the binding properties of SapB to CoQ10 (a bona fide substrate).[18] To determine whether A2E binds to the same binding site on the protein as CoQ10, a series of fluorescence titration experiments (following the emission spectra of A2E at 605 nm) in the presence and absence of CoQ10 were performed. Figure 3 shows a near-identical fluorescence titration pattern when A2E is titrated into a SapB protein solution alone or a solution of SapB pre-complexed with CoQ10 (SapB:CoQ10)complex. These data suggest the hitherto unknown presence of a second binding site on SapB, in this case for A2E. The similarity of the titration curves in Figure 3 also suggests that the A2E binding site on SapB is not altered by the presence of CoQ10 and the affinity of SapB to A2E has not significantly changed, in support of a non-cooperative and distinct binding site for CoQ10 and A2E on SapB. This is further corroborated with the experimentally calculated dissociation constant for A2E binding to the SapB:CoQ10 complex (KD ≈ 52 μM, determined using rectangular hyperbola/nonlinear regression),[19, 20] a value similar to that for A2E binding to SapB alone (reported herein at ≈25 μM).
Since HRP is reported to oxidize the bisretinoid A2E,[5] we thought to examine whether SapB can protect A2E from oxidative transformations by HRP. When H2O2 is added to a solution of A2E containing HRP, a decrease in the absorbance value of A2E at 340 nm is observed (Figure 4) supporting that A2E acts as a reducing substrate for the HRP/H2O2 system, as expected.[5]
Figure 4.

Kinetic curves following the sequential additions of A2E, CoQ10, HRP and H2O2 to SapB as indicated on each curve. Conditions: 12 μM SapB, 6 μM A2E, CoQ10 or HRP, 1 μL of 3 % H2O2, 10 μL tween (1% v/v), 50 mM phosphate buffer, pH 5.5 and 25 °C. See also Figure S3.
When the experiment was repeated in the presence of A2E pre-complexed with SapB (2:1 SapB:A2E ratio; blue line), the absorbance change was very similar to, but less than, that of a control mixture of HRP and H2O2 (Figure 4; green line), supporting a protective role of SapB for A2E. Given we observe a curve for the (SapB-A2E)complex (blue line, Figure 4) that displays less change in relative absorption at 340 nm than the HRP/H2O2 control, we thought to check if the minor decrease in relative absorbance for the (SapB-A2E)complex was not due to partial A2E degradation, but rather SapB interaction with HRP mitigating degradation of the HRP enzyme by H2O2. Figure S4 shows the oxidative decay of A2E fluorescence at 605 nm by HRP/H2O2 in the absence and presence of SapB. Over the course of 400 min, A2E exhibited no fluorescence decay in the presence of SapB supporting a protective role of SapB for HRP, perhaps via protein–protein association. To confirm this latter hypothesis, a fluorescence quenching experiment following incremental additions of HRP to SapB was performed (Figure S5). The results demonstrate a relatively strong interaction between SapB and HRP with a binding constant of 1 × 106 M−1 and a stoichiometry of 1:1.
Having established the presence of two different binding sites on SapB (one for A2E and one for CoQ10), we also tested the ability of SapB to protect A2E in the presence of CoQ10 (Figure 4, Figure S3). The addition sequence (SapB-A2E)complex + CoQ10 + HRP + H2O2 (Figure S3) or the sequence (SapB-CoQ10)complex + A2E + HRP + H2O2 (Figure S3) showed absorbance values similar to the sequence (SapB +A2E)complex + HRP+ H2O2, a result consistent with the fluorescence data of Figure S5 and in strong support of a protective role of SapB for A2E. By contrast, a control experiment consisting of the addition sequence A2E + CoQ10 + HRP + H2O2 exhibited a similar absorbance curve to that in the absence of CoQ10 (Figure S3), supporting A2E oxidation.
We screened A2E photostability to blue light (450–460 nm 12W blue LED), whether in its free form or complexed to SapB (2:1 SapB:A2E; see the Supporting Information for details) in 50 mM phosphate buffer with 1 % methanol or 1 % DMSO or 0.1 mM SDS (the latter two being known facilitators of blue-light-induced A2E photooxidation).[10] The decline in absorbance is indicative of A2E oxidation (Figure 5), a result confirmed by electrospray mass spectrometry (Figures S6–S8), which shows significant oxidative reduction of A2E when bound to SapB over a period of 30 min.
Figure 5.

Effect of 450–460 nm blue light on 40 μM A2E, free or bound to SapB (80 μM) followed over 30 min at 444 nm in 50 mM phosphate buffer with either 1 % methanol (not indicated), 0.1 mM SDS (open and filled triangles) or 1 % DMSO (open and filled circles). Experimental slopes are as follow: A2E (0.0068 SEM ±0.0003, R2 =0.984); SapB-A2E (0.0026 SEM ±0.0002, R2 =0.952); A2E 1% DMSO (0.0170 SEM ±0.0009, R2 =0.982); SapB-A2E 1 % DMSO (0.0030 SEM ±0.0006, R2 =0.784); A2E 0.1 mM SDS (0.0170 SEM ±0.0009, R2 =0.982); SapB-A2E 0.1 mM SDS (0.0030 SEM ±0.0006, R2 =0.784).
Based on the invariable slopes for all experiments with A2E bound to SapB (Figure 5), it appears there is a constant protection factor at work, one not significantly affected by the external solvent polarity/environment, and counter to such environmental effects observed for oxidation of “free” A2E with blue light.[10]
The fact that SapB might play a role in mitigating photooxidation and then providing a source of subsequent fluorescence upon re-equilibrium between (SapB-A2E)complex and A2E levels is an interesting possibility, and in line with the Sparrow hypothesis.[10] The feasibility of A2E redistribution within the cell associated with SapB is made more relevant again in light of recent studies by Yamamoto et al. showing that SapB could regulate CoQ10 movement/levels within HepG2 cells.[21]
A2E has been reported to not interfere with ASA specific activity, but the experiments were conducted without time-dependent inhibition assays, accounting for equilibrium rates of the binding partners involved or knowledge of A2E interaction with the activator (i.e. SapB).[22] Subsequent studies have demonstrated that a delay in activity of ASA is likely that results in gradual, long-term accumulation of lipids.[23] Louis and Fluharty demonstrated that activator-dependent hydrolysis of myelin cerebroside sulfate by ASA could be affected in terms of slower hydrolysis rates by competition for activator by “other lipoidal constituents”, as yet still unknown.[24] Based on the data herein, it is likely that SapB binding of A2E would result in competition between A2E and ASA for activator, even temporarily, which would be a significant problem for a circadian process.[25]
In summary, we demonstrate that A2E binding by the lysosomal protein SapB prevents A2E oxidation by HRP or blue light. Such binding may then complicate attempts to produce an enzyme replacement therapy for A2E degradation/photooxidation and/or play a role in the “transport” or movement of A2E inside the cell (and possibly out of the cell). It would be of interest to assay the urine of patients with AMD or SD for the presence of (SapB-A2E)complex, especially since (SapB-CoQ10)complex has been observed, therein.[16] Finally, the possibility of A2E competition for activator suggests a need to assay for sulfatide build-up in patients with AMD or SD.
Acknowledgments
This work is supported by funding from New York State through the Center for Advanced Systems and Engineering- a NYSTAR designated center at Syracuse University, the National Science Foundation (NSF IGERT), Ichor Therapeutics (Lafayette, NY, US) (R.P.D)), and, in-part by, the Cottrell College Science Award (ID # 7892) from Research Corporation, and the National Institute of General Medical Sciences of the National Institutes of Health Award R15GM104879 (F.B.A.).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supporting Information (also containing the Experimental Section) and the ORCID identification number(s) for the author(s) of this article can be found under: http://dx.doi.org/10.1002/cptc.201700039.
References
- 1.Kiser PD, Palczewski K. Annu Rev Vis Sci. 2016;2:197–234. doi: 10.1146/annurev-vision-111815-114407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jarrett SG, Boulton ME. Mol Aspects Med. 2012;33:399–417. doi: 10.1016/j.mam.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sparrow JR. Proc Natl Acad Sci USA. 2016;113:4564–4569. doi: 10.1073/pnas.1600474113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ueda K, Zhao J, Kim HJ, Sparrow JR. Proc Natl Acad Sci USA. 2016;113:6904–6909. doi: 10.1073/pnas.1524774113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Y, Zhou J, Fishkin N, Rittman BE, Sparrow JR. J Am Chem Soc. 2011;133:849–857. doi: 10.1021/ja107195u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sparrow JR, Zhou J, Ben-Shabat S, Vollmer H, Itagaki Y, Nakanishi K. Invest Ophthalmol Visual Sci. 2002;43:1222–1227. [PubMed] [Google Scholar]
- 7.Rozanowska M, Jarvis-Evans J, Korytowski W, Boulton ME, Burke JM, Sarna T. J Biol Chem. 1995;270:18825–18830. doi: 10.1074/jbc.270.32.18825. [DOI] [PubMed] [Google Scholar]
- 8.Moiseyev G, Nikolaeva O, Chen Y, Farjo K, Ma Takahashi J-X. Proc Natl Acad Sci USA. 2010;107:17551–17556. doi: 10.1073/pnas.1008769107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iriyama A, Fujiki R, Takahashi H, Tamaki Y, Takezawa S, Takeyama K, Jang WD, Kato S, Yanagi Y. J Biol Chem. 2008;283:11947–11953. doi: 10.1074/jbc.M708989200. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Ueda K, Kim HJ, Sparrow JR. PLoS One. 2015;10:e138081. doi: 10.1371/journal.pone.0138081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huta BP, Roberts AM, Waters ES, Yu VY, Doyle RP, Mehlenbacher MR, Bou-Abdallah F. MedChemComm. 2014;5:787–791. [Google Scholar]
- 12.Huta BP, Mehlenbacher MR, Nie Y, Lai X, Zubieta C, Doyle RP, Bou-Abdallah F. ChemMedChem. 2016;11:277–282. doi: 10.1002/cmdc.201500494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kishimoto Y, Hiraiwa M, O’Brien JS. J Lipid Res. 1992;33:1255–1267. [PubMed] [Google Scholar]
- 14.Weiter JJ, Feingold M, Kolodny EH, Raghaven SS. Am J Ophthalmol. 1980;90:768–772. doi: 10.1016/s0002-9394(14)75191-8. [DOI] [PubMed] [Google Scholar]
- 15.Winzeler AM, Mandemakers WJ, Sun MZ, Stafford M, Phillips CT, Barres BA. J Neurosci. 2011;31:6481–6492. doi: 10.1523/JNEUROSCI.3004-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin G, Kubo H, Kashiba M, Horinouchi R, Hasegawa M, Suzuki M, Sagawa T, Oizumi M, Fujisawa A, Tsukamoto H, Yoshimura S, Yamamoto Y. J Clin Biochem Nutr. 2008;42:167–174. doi: 10.3164/jcbn.2008024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow JR. Proc Natl Acad Sci USA. 1998;95:14609–14613. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixson DD, Yu VY, Doyle RP. Anal Biochem. 2011;419:145–152. doi: 10.1016/j.ab.2011.08.042. [DOI] [PubMed] [Google Scholar]
- 19.Pollard TD. Mol Biol Cell. 2010;21:4061–4067. doi: 10.1091/mbc.E10-08-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrella EC, Machesky LM, Kaiser DA, Pollard TD. Biochemistry. 1996;35:16535–16543. doi: 10.1021/bi961498d. [DOI] [PubMed] [Google Scholar]
- 21.Kashiba M, Oizumi M, Suzuki M, Sawamura Y, Nagashima K, Yoshimura S, Yamamoto Y. J Clin Biochem Nutr. 2014;55:85–89. doi: 10.3164/jcbn.13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bergmann M, Schutt F, Holz FG, Kopitz J. Exp Eye Res. 2001;72:191–195. doi: 10.1006/exer.2000.0949. [DOI] [PubMed] [Google Scholar]
- 23.Holz FG, Schutt F, Kopitz J, Eldred GE, Kruse FE, Volcker HE, Cantz M. Invest Ophthalmol Visual Sci. 1999;40:737–743. [PubMed] [Google Scholar]
- 24.Louis AI, Fluharty AL. Dev Neurosci. 1991;13:41–46. doi: 10.1159/000112139. [DOI] [PubMed] [Google Scholar]
- 25.Gooley JJ, Chem-Pin Chua E. J Genet Genomics. 2014;41:231–250. doi: 10.1016/j.jgg.2014.04.001. [DOI] [PubMed] [Google Scholar]