

SUMMARY
During obesity, adipose tissue macrophages (ATMs) adopt a ‘metabolically-activated’ (MMe) phenotype. However, the functions of MMe macrophages are poorly understood. Here we combine proteomic and functional methods to demonstrate that in addition to potentiating inflammation, MMe macrophages also promote dead adipocyte clearance through lysosomal exocytosis. We identify NADPH-oxidase-2 (NOX2) as a driver of the inflammatory and adipocyte-clearing properties of MMe macrophages, and show that compared to wild-type, Nox2−/− mice exhibit a time-dependent metabolic phenotype during diet-induced obesity. After 8-weeks of high-fat feeding, Nox2−/− mice exhibit attenuated ATM inflammation and mildly improved glucose tolerance. After 16-weeks of high-fat feeding, Nox2−/− mice develop severe insulin resistance, hepatosteatosis, and visceral lipoatrophy characterized by dead adipocyte accumulation and defective ATM lysosomal exocytosis, a phenotype reproduced in myeloid cell-specific Nox2−/− mice. Collectively, our findings suggest that MMe macrophages perform detrimental and beneficial functions, whose contribution to metabolic phenotypes during obesity is determined by disease progression.
eTOC
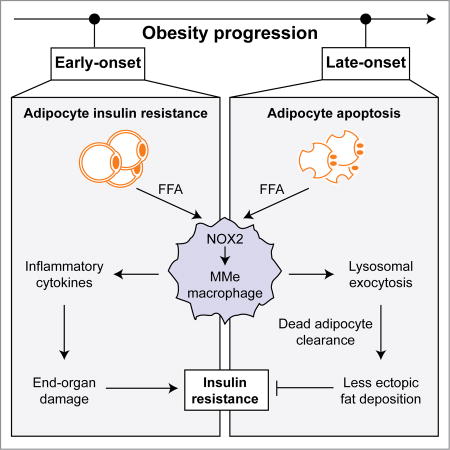
During obesity, adipose tissue macrophages are ‘metabolically-activated’ (MMe). Coats et al. show that MMe macrophages perform detrimental (potentiate inflammation) and beneficial (exocytose lysosomes to clear dead adipocytes) functions, controlled by NOX2. Nox2−/− mice exhibit improved/worsened metabolic phenotypes depending on high-fat-diet duration, highlighting the dynamic contributions of MMe macrophages in obesity.
INTRODUCTION
During obesity, macrophages accumulate in visceral adipose tissue where they promote chronic low-grade inflammation (Weisberg et al., 2003, Xu et al., 2003). It is well accepted that this inflammation is causally associated with insulin resistance in mice. Inhibiting several pathways that drive inflammatory signaling and/or production in macrophages improves insulin sensitivity during obesity (Han et al., 2013, Patsouris et al., 2008, Saberi et al., 2009, Wei et al., 2016).
In addition to producing inflammatory cytokines, ATMs have also been postulated to perform beneficial functions during diet-induced obesity (Fitzgibbons and Czech, 2016). They internalize excess free fatty acids (FFAs) released by insulin resistant adipocytes, thereby buffering metabolic tissues from the damage caused by ectopic accumulation of saturated FFAs. They also clear dead adipoctyes that accumulate during prolonged obesity when adipose tissue expands enough to produce hypoxia (Strissel et al., 2007, Sun et al., 2011). The clearance of these dead adipocytes promotes adipocyte turnover and maintains adipose tissue health during nutrient excess.
The diverse functions of macrophages, such as those described above, have often been conceptualized through an M1 and M2 paradigm (Gordon and Taylor, 2005). The M1 phenotype is caused by Th1 mediators such as LPS and IFNγ and is characterized by increased production of pro-inflammatory cytokines, while the M2 phenotype is driven by Th2 mediators (eg. IL4), which activates expression of immunosuppressive factors that promote macrophage clearance of dead cells and tissue remodeling (Odegaard et al., 2007). Although this M1/M2 paradigm has been a useful construct for understanding macrophage heterogeneity, recent studies suggest that it cannot adequately describe the functions of ATMs during obesity.
Studies from Ferrante’s group showed that macrophages from obese mice induce lysosomal and lipid metabolism pathways (Xu et al., 2013), raising the possibility that these pathways might help macrophages to clear dead adipocytes and their large FFA reservoirs. Moreover, studies from our laboratory, showed that saturated FFAs produce a pro-inflammatory, ‘metabolically-activated’ (MMe) macrophage phenotype that is mechanistically distinct from M1 or M2 activation (Kratz et al., 2014). Although we showed that MMe macrophages accumulate in visceral and subcutaneous adipose tissue of obese humans and mice, their roles in regulating macrophage inflammation, function, and insulin sensitivity have not been explored.
Here we combine proteomic and functional analyses with genetic models and dietary interventions to investigate the roles of MMe macrophages in diet-induced obesity (DIO). We show that metabolic activation of macrophages induces inflammatory cytokine production (a detrimental function) and lysosomal exocytosis to dead adipocytes (a beneficial function). We identify NADPH-oxidase-2 (NOX2) as a key regulator of the inflammatory cytokine expression and lysosomal exocytosis in MMe macrophages, and show that ablating Nox2 both improves and worsens the metabolic phenotype during DIO depending on the duration of high-fat feeding. Collectively, our findings underscore the dynamic functions of ATMs during DIO, and demonstrate the importance of the MMe phenotype in regulating these functions and their metabolic consequences.
RESULTS
Metabolic activation induces macrophage inflammation and lysosomal exocytosis in vitro and in vivo
We previously showed that saturated free fatty acids (FFAs) produce macrophage inflammation via a ‘metabolically activated’ (MMe) pathway that is mechanistically distinct from the classically activated M1 phenotype induced by bacterial challenge (Kratz et al., 2014). Although we showed that these MMe macrophages are present in adipose tissue of obese humans and mice, their functions in diet-induced obesity and insulin resistance are poorly understood.
To interrogate how metabolic activation might alter macrophage function, we produced pro-inflammatory MMe macrophages by treating murine bone marrow-derived macrophages (BMDMs) with glucose, insulin, and palmitate (Fig. 1A) and performed plasma membrane proteomics. We focused on plasma membrane proteins because they mediate a variety of important macrophage functions including phagocytosis, cell migration, and antigen presentation.
Fig. 1. MMe macrophages overexpress cytokines and accumulate cell surface lysosomal membrane proteins in vitro and in vivo.

Panels A-D: BMDMs (M0) were metabolically activated (MMe) or classically activated (M1). Panel A: Inflammatory cytokine expression levels. Panel B. Plasma membrane proteomics analysis with the t-test and G-test identifies proteins induced (yellow) and suppressed (blue) on the cell surface of MMe macrophages (relative to M0). Panel C: Gene ontology analysis of plasma membrane proteins elevated on the cell surface of MMe macrophages (relative to M0). Relative abundances of proteins are presented as a heatmap. Panel D: Flow cytometric quantification of cell surface LAMP1 and LAMP2. Panels E–F: BMDMs (M0) were treated with conditioned media collected from 3T3-L1 adipocytes (live or apoptotic) or apoptotic neutrophils (PMN). Panel E: Inflammatory cytokine expression levels. Panel F: Flow cytometric quantification of cell surface LAMP1 and LAMP2. Panels G–J: Analysis of ATMs from C57BL/6 mice fed a low-fat (LFD) or high-fat diet HFD for 16 weeks. Panel G: ATMs were isolated from the stromal vascular fraction (SVC) using anti-CD11b-coupled magnetic beads. ATM purity and recover were confirmed by staining for CD11b in the purified ATMs, SVC, and flow through (FT). Panel H: ATM purity was assessed by staining for CD11c and F4/80. Panel I: Inflammatory cytokine expression levels. Panel J: Heatmap of the relative abundance of cell surface lysosomal membrane proteins. Panel K: A comparison of cell surface levels of LAMP2, NPC1, and ATP6v1a in vitro and in vivo; proteins were quantified by mass spectrometry. Results are mean ± SEM; n=3–6, *, p<0.05 Student’s t-test; ##p<0.05 Student’s t-test and G>1.5 G-test. See also Fig. S1 and Table S1.
Mass spectrometric analysis of the plasma membrane identified 664 proteins in M0 (unstimulated) and MMe macrophages. Statistical analysis with the t-test and G-test identified 176 proteins that were differentially abundant (FDR<0.05) in MMe versus M0 macrophages (Fig. 1B). Gene ontology analysis of proteins induced in MMe macrophages identified enrichments in several annotations expected for a cell surface proteome (i.e. plasma membrane, membrane raft, extracellular matrix; Fig. 1C), suggesting that our methods for isolating plasma membrane proteins were robust. Moreover, we observed increased levels of MMe-associated proteins (PLIN2, P62) but not M1-associated proteins (CD40, STAT1) that we previously identified in proteomic analyses of human MMe and M1 macrophages (Kratz et al., 2014) (Fig. S1).
Unexpectedly, plasma membrane proteins induced in MMe macrophages were enriched in lysosomal membrane proteins (p=10−6; Fig. 1C, Table S1), and this enrichment was specific to the MMe phenotype since they were not observed in M1 macrophages (Fig. 1C). To confirm these observations using an alternative approach, we quantified cell surface LAMP1 and LAMP2 levels on M0, MMe, and M1 macrophages by flow cytometry. Consistent with our proteomics data, we found that LAMP1 and LAMP2 were induced on the cell surface of MMe, but not M1, macrophages (Fig. 1D, Fig. S1). Because lysosomal membrane proteins appear on the cell surface during lysosomal exocytosis, when lysosomal and plasma membranes fuse (Rodriguez et al., 1997), these findings suggest that metabolic activation induces macrophages to exocytose their lysosomes.
Previous studies showed that lysosomal exocytosis by macrophages is required for them to clear dead adipocytes (Haka et al., 2016). Since macrophages (~20µm diameter) are much smaller than adipocytes (~120µm diameter), they cannot clear dead adipocytes through a traditional phagocytosis mechanism. Instead, the macrophages form a tight attachment (a lysosomal synapse) on the dead adipocyte. They acidify the contact region using the plasma membrane proton pumping V-ATPase and secrete lysosomal contents into the lysosomal synapse. The lysosomal lipases in the acidic milieu liberate FFAs that are subsequently internalized by macrophages, leading to foam cell formation.
Since FFAs are required for metabolic activation, and dead adipocytes are an abundant reservoir of FFA, we hypothesized that dead adipocytes could induce the inflammation and lysosomal exocytosis associated with the MMe phenotype. To test this hypothesis, we treated unstimulated BMDMs with media conditioned by live or apoptotic 3T3-L1 adipocytes and measured inflammatory cytokine expression by qRT-PCR and quantified lysosomal exocytosis by staining for cell surface LAMP1 and LAMP2.
We found that media from apoptotic adipocytes coordinately induced the expression of Il1β and Il6 (Fig. 1E) and the appearance of cell surface LAMP1 and LAMP2 on macrophages (Fig. 1F). The effects on cytokines and lysosomal exocytosis were specific to apoptotic adipocytes since media from live 3T3-L1 adipocytes or apoptotic neutrophils (a dead cell without FFA) were incapable of inducing these changes (Figs. 1E–F). Thus, dead adipocytes release factors that induce the inflammation and lysosomal exocytosis characteristic of MMe macrophages.
Our previous studies showed that ATMs from obese mice adopt an MMe phenotype (Kratz et al., 2014). We therefore reasoned that ATMs from obese mice would also show coordinated induction of cytokine expression and lysosomal exocytosis. To test this, we fed C57BL/6 mice a low-fat diet or 45% high-fat diet for 16 weeks, isolated ATMs (Fig. 1G), confirmed their purity based on CD11c and F4/80 staining (98.1%; Fig. 1H), and assessed cytokine expression by qRT-PCR and cell surface lysosomal membrane protein levels by mass spectrometry.
Consistent with previous findings, ATMs from obese mice were characterized by elevated Tnfα, Il6, and Il1β expression (Fig. 1I). Moreover, plasma membrane proteins elevated in ATMs from obese mice (relative to lean mice) were significantly enriched for lysosomal membrane proteins (p=10−7). Indeed, 10 lysosomal membrane proteins were induced on the cell surface of ATMs from obese mice (Fig. 1J–K, Table S1), 6 of which (LAMP2, ATP6V1B2, ATP6V1A, AHNAK, NPC1, ANXA2) were also induced on the cell surface of MMe macrophages in vitro.
TLR2, NOX2, and MYD88 coordinately regulate inflammation and lysosomal exocytosis in MMe macrophages
What signaling pathways regulate cytokine expression and lysosomal exocytosis in MMe macrophages? To begin to answer this question, we searched the literature for proteins known to mediate macrophage responses to FFAs, the driver of the MMe phenotype. We focused on toll-like receptor 2 (TLR2), NADPH oxidase 2 (NOX2), MYD88, and TLR4 because previous studies showed that these proteins are required for palmitate-induced inflammation and/or ER stress in macrophages (Kratz et al., 2014, Robblee et al., 2016, Seimon et al., 2010).
We first investigated the effects of these knockouts on inflammatory cytokine expression. We found that ablating Nox2 (Gp91 subunit), Tlr2, Myd88, or Tlr4 attenuated Il6 and Il1β levels in MMe macrophages (Fig. 2A, Fig. S2). Interestingly, the anti-inflammatory effect of ablating Nox2 or Tlr2 was specific to MMe macrophages, since cytokine expression in M1 macrophages was unaffected (Fig. 2A). In contrast, ablating Tlr4 or Myd88, an adaptor protein used by most TLRs to activate NF-κB (Akira et al., 2001), lowered inflammatory cytokine expression in both MMe and M1 macrophages (Fig. 2A, Fig. S2). Together, these studies identified TLR2, NOX2, MYD88, and TLR4 as regulators of inflammatory cytokine expression in MMe macrophages, and further demonstrated that distinct signaling pathways regulate inflammatory cytokine expression in MMe and M1 macrophages.
Fig. 2. TLR2, NOX2, and MYD88 coordinately regulate inflammatory cytokine expression and lysosomal exocytosis in MMe macrophages.

Panel A: Inflammatory cytokine expression in metabolically activated (MMe) and classically activated macrophages (M1) BMDMs made from wild-type (wt), TLR2-deficient (Tlr2−/−), NOX2-deficient (Gp91−/−), or MYD88-deficient (Myd88−/−) mice. Levels are expressed relative to M0 for each genotype. Panels B–D: Quantification of macrophage lysosomal exocytosis and lipid uptake from live or apoptotic 3T3-L1 adipocytes. Panel B: Schematic of the assay: (i) macrophage lysosomal exocytosis was quantified as the capture of previously endocytosed biotin-fluorescein-dextran (green) on the cell surface of streptavidin-labeled apoptotic adipocytes, (ii) macrophage lipid uptake was quantified as the amount of LipidTOX signal (red) inside Alexa444-CtB-labeled macrophages (green). Scale bar = 20 µm Panel C: Quantification of lysosomal exocytosis and lipid uptake by wt, Tlr2−/−, Gp91−/−, or Myd88−/− BMDMs. Panel D: Relationship between macrophage lipid uptake and lysosomal exocytosis. Panel E: Relative uptake of palmitate from the media by wt, Tlr2−/−, Gp91−/−, or Myd88−/− BMDMs. Panel F: Lysosomal exocytosis by Nox2−/− macrophages pre-treated with TNFα. Results are mean ± SEM; n=3–12, *, p<0.05 Student’s t-test. See also Fig. S2.
We further investigated the effects of these knockouts on lysosomal exocytosis. To this end, we used a more physiologically relevant co-culture assay (Haka et al., 2016) that quantifies i) the delivery of lysosomal contents from macrophages to apoptotic 3T3-L1 adipocytes, and ii) the subsequent accumulation of adipocyte-derived lipids in the macrophage (Fig. 2B). As a control, macrophages were co-incubated with live adipocytes, demonstrating the specificity of these processes to apoptotic adipocytes (Fig. 2B).
We found that lysosomal exocytosis to apoptotic adipocytes was significantly impaired in Nox2−/−, Tlr2−/−, and Myd88−/− macrophages (Fig. 2C). Moreover, Nox2−/−, Tlr2−/−, and Myd88−/− macrophages also accumulated fewer lipids from apoptotic adipocytes (Fig. 2C), perhaps due to their inability to deliver lysosomal lipases to mobilize FFAs from the adipocyte lipid droplet. Consistent with this interpretation, the extent of macrophage lipid accumulation was strongly and positively correlated (R2=0.94, p<0.0001) to the amount of lysosomal exocytosis (Fig. 2D). Moreover, Nox2−/−, Tlr2−/−, and Myd88−/− macrophages were not defective in their ability to internalize palmitate (Fig. 2E), one of the major FFAs liberated during lipolysis from adipocytes.
Interestingly, lysosomal exocytosis to apoptotic adipocytes was not impaired in Tlr4−/− macrophages (Fig. S2), even though this knockout suppressed inflammatory cytokine expression. These findings suggest that the defective lysosomal exocytosis observed in Tlr2−/−, Nox2−/−, and Myd88−/− macrophages was not reliant upon the suppression of inflammatory cytokines. Consistent with this hypothesis, supplementing Nox2−/− macrophages with TNFα, a key cytokine produced by ATMs, could not rescue the defect in lysosomal exocytosis (Fig. 2F).
ATM inflammation precedes visceral fat adipocyte death during DIO
Our in vitro findings suggest that MMe macrophages might play complex roles during DIO. On the one hand, MMe macrophages overexpress inflammatory cytokines, which could promote insulin resistance (Chawla et al., 2011, Olefsky and Glass, 2010). On the other hand, MMe macrophages exocytose their lysosomes and clear dead adipocytes, which could limit insulin resistance by maintaining adipose tissue health and protecting against ectopic lipid deposition in the liver (Shulman, 2014, Strissel et al., 2007, Sun et al., 2011). Since these potentially detrimental and beneficial properties of MMe macrophages are coordinately regulated by TLR2, NOX2, and MYD88, we reasoned that interpreting the metabolic phenotypes of mice deficient in one or more of these proteins might be challenging.
To help facilitate interpretation, we first determined whether ATM inflammation and adipocyte death (and hence the need for dead adipocyte clearance) occurred at different time points during DIO. Previous studies showed that ATM inflammation might be an early and persistent event, while adipocyte death may predominantly occur after prolonged high-fat feeding when adipose tissue becomes hypoxic (Fig. 3A) (Strissel et al., 2007). However, these studies were performed in mice fed a 60% HFD, and our studies used the 45% HFD. To better understand the timing of these events and to establish reproducible conditions to investigate the effects of our knockouts on these processes in vivo, we studied adipose tissue biology in wild-type C57BL/6 mice fed a low-fat diet or 45% high-fat diet for 8 weeks and 16 weeks (Fig. 3B).
Fig. 3. ATM inflammation precedes visceral fat adipocyte death during DIO.

Wild-type C57BL/6 mice were fed a low-fat diet (LFD) or 45% high-fat diet (HFD) for 8 or 16 weeks. Panel A: A model for the timing of ATM inflammation and epididymal fat death during DIO. Panel B: Body weight gain. Panel C: mRNA expression of inflammatory cytokines and lipid metabolism genes in purified ATMs. Panel D: Hif1α mRNA levels in epididymal fat (a marker of hypoxia). Panel E: Caspase-3 (CASP3) activity levels in epididymal fat. Panel F: Epididymal fat was stained with antibodies against PLIN2 (red; identifies adipocytes) and MAC2 (green; identifies macrophages), and DAPI (blue; identifies nuclei). Macrophages in crown-like structures (CLS; arrow) were quantified by microscopy and standardized per unit area. Panel G: H&E staining of liver and quantification of liver triglyceride (TG) levels. Results are mean ± SEM; n=5–15, *, p<0.05 Student’s t-test. See also Figs. S3–S4.
Eight weeks of high-fat feeding was sufficient to induce the MMe phenotype in ATMs characterized by increased inflammatory cytokines (Tnfα, Il6, and Il1β) and genes involved in lipid metabolism (Plin2, Abca1, Cd36) (Fig. 3C). Despite the presence of ATM inflammation, several lines of evidence indicated that epididymal fat health in obese mice was not significantly compromised at this time point. First, adipose tissue levels of Hif1α were not elevated (Fig. 3D), suggesting that hypoxia, a key driver of adipocyte death (Wang et al., 1995), had not yet occurred. Second, adipose tissue caspase-3 activity, a marker of apoptosis (Nicholson et al., 1995), was not elevated (Fig. 3E). Third, the number of crown-like structures, an indicator of adipocyte death (Murano et al., 2008), was unchanged (Fig. 3F, Fig. S3). Finally, ectopic fat deposition in the liver, which has been associated with adipocyte death in visceral fat (Strissel et al., 2007), was undetectable (Fig. 3G, Fig. S4).
Although ATMs still over-expressed the cytokines and lipid metabolism genes characteristic of MMe macrophages following 16 weeks of high-fat feeding (Fig. 3C), the state of epididymal fat health was substantially different at this time point. Adipose tissue Hif1α levels were elevated (Fig. 3D), suggesting that the tissue was hypoxic. Moreover, adipose tissue capsase-3 activity and crown-like structures were markedly increased (Figs. 3E–F, Fig. S3), indicating the presence of apoptotic adipocytes. Consistent with the deterioration of epididymal fat health and increased FFA release, the MMe phenotype was more pronounced at this time point, particularly with respect to the lipid metabolism genes (Fig. 3C). Moreover, ectopic fat deposition in the liver was now detectable in obese mice (Fig. 3G, Fig. S4), and this deposition could not be explained by changes in genes involved in triglyceride synthesis (Fas1, Acc1, and Srebp1) or fatty acid metabolism (Pparα and Cpt1α) in the liver (Fig. S4).
These findings suggest that while metabolically activated ATMs are present both at 8 weeks and 16 weeks of 45% high-fat feeding, adipocyte death, and hence the requirement for macrophages to clear dead adipocytes, may only be an important consideration after 16 weeks of high-fat feeding.
Nox2−/− mice have improved ATM inflammation and glucose tolerance after 8 weeks of high-fat feeding
Our identification of NOX2, TLR2, and MYD88 as drivers of cytokine expression and lysosomal exocytosis in MMe macrophages provided an opportunity to inhibit these processes in vivo and assess effects on obesity-associated insulin resistance. We focused our initial studies on Nox2−/− mice because NOX2 is predominantly expressed by phagocytic cells, including macrophages and neutrophils (Bedard and Krause, 2007).
We began by interrogating the metabolic phenotypes of Nox2−/− mice following 8 weeks of high-fat feeding. Since ATM inflammation was present at this time point, but adipocyte apoptosis had not yet occurred (see Fig. 3), we hypothesized that the anti-inflammatory effect of Nox2−/− would predominate and protection from metabolic derailments during DIO would be observed at this early time point. This hypothesis makes several predictions.
First, ablating Nox2 should attenuate ATM inflammation. Although Nox2−/− mice fed the HFD were heavier than wild-type mice (Fig. 4A), ATMs isolated from visceral fat were substantially less inflamed, as evidenced by decreased expression of Tnfα, Il6, and Il1β (Fig. 4B). Because NOX2 drives inflammatory cytokine expression in MMe but not M1 macrophages (see Fig. 2), these findings reinforce the notion that ATMs are inflamed via a metabolically activated pathway in vivo (Kratz et al., 2014).
Fig. 4. Nox2−/− mice have improved ATM inflammation and glucose tolerance after 8 weeks of HFD.

Wild-type (wt) and Gp91−/− mice were fed a low-fat diet (LFD) or 45% high-fat diet (HFD) for 8 weeks. Panel A: Body weight gain. Panel B: Inflammatory cytokine expression in purified ATMs. Panel C: Hif1α mRNA levels in epididymal fat (a marker of hypoxia). Panel D: Epididymal fat was stained with antibodies against PLIN2 (red; identifies adipocytes) and MAC2 (green; identifies macrophages), and DAPI (blue; identifies nuclei). Macrophages in crown-like structures (CLS; arrow) were quantified by microscopy and standardized per unit area. Panel E: Caspase-3 (CASP3) activity levels in epididymal fat. Panel F: H&E staining of liver and quantification of liver triglyceride (TG) levels. Panel G: Fasting blood glucose levels and serum insulin levels. Panels H–I: Glucose tolerance and insulin tolerance tests in wt and Gp91−/− mice fed the HFD. Glucose tolerance and insulin tolerance were assessed by area under the curve (AUC) analysis. Results are mean ± SEM; n=5–15, *, p<0.05 Student’s t-test. See also Figs. S3–S5.
Second, ablating Nox2 should not increase the number of dead adipocytes. Indeed, Nox2−/− mice showed no differences in adipocyte death indicators; Hif1α levels, caspase-3 activity and the number crown-like structures were unchanged, and ectopic fat accumulation in the liver was only minimally and insignificantly increased in Nox2−/− mice relative to wild-type mice fed the HFD (Figs. 4C–F, Figs. S3–S4). Moreover, none of these adipocyte death indicators were elevated in obese relative to lean mice irrespective of genotype (Figs. 4C–F), which is consistent with our findings that 8 weeks of 45% HFD is insufficient to induce adipocyte death (see Fig. 3).
Third, this hypothesis predicts that Nox2−/− mice should demonstrate improved metabolic parameters. Indeed, Nox2−/− mice fed the HFD had lower fasting glucose levels (Fig. 4G) and improved glucose tolerance (Fig. 4H). However, fasting insulin levels and insulin tolerance remained unchanged (Figs. 4G, 4I), perhaps due to the increased body weight, the mild hepatosteatosis, and the increased number of ATMs in Nox2−/− mice (Fig. S5), all of which would limit the anti-inflammatory effect of the knockout.
Taken together, these findings provide further evidence that ATMs are inflamed via an MMe pathway in vivo, and further suggest that inhibiting inflammatory cytokine expression in MMe macrophages (by ablating Nox2) protects against metabolic dysfunction following 8 weeks of high-fat feeding.
Nox2−/− mice develop severe visceral lipoatrophy, hepatosteatosis, and insulin resistance following 16 weeks of HFD
Next, we investigated the metabolic phenotypes of Nox2−/− mice fed a HFD for 16 weeks. Since ATM inflammation and adipocyte death were both present at this time point (see Fig. 3), and ablating Nox2 attenuated metabolic activation and lysosome secretion in response to dead adipocytes (see Fig. 2), we hypothesized that Nox2−/− mice might exhibit a more complex phenotype at this later time point. To explore this, we characterized the adipose tissue, liver, and metabolic parameters of Nox2−/− mice fed the HFD for 16 weeks.
The protection observed in Nox2−/− mice following 8 weeks of HFD was reversed after prolonged high-fat feeding. Nox2−/− mice fed a HFD for 16 weeks were more obese, hyperglycemic, hyperinsulinemic, and insulin resistant than wild-type controls (Figs. 5A–D). Nox2−/− mice retained glucose tolerance, but they required higher levels of insulin production to maintain glucose homeostasis (Fig. 5C).
Fig. 5. Nox2−/− mice develop severe visceral lipoatrophy, hepatosteatosis, and insulin resistance following 16 weeks of HFD.

Wild-type (wt) and Gp91−/− mice were fed a low-fat diet (LFD) or 45% high-fat diet (HFD) for 16 weeks. Panel A: Body weight gain. Panel B: Fasting blood glucose levels and serum insulin levels. Panels C–D: Glucose tolerance test (GTT) and insulin tolerance test (ITT) in wt and Gp91−/− mice fed the HFD. Insulin levels during the GTT are shown in the inset. Glucose tolerance and insulin tolerance were assessed by area under the curve (AUC) analysis. Panel E: Epididymal fat was stained with antibodies against PLIN2 (red; identifies adipocytes) and MAC2 (green; identifies macrophages), and DAPI (blue; identifies nuclei). Macrophages in crown-like structures (CLS; arrow) were quantified by microscopy and standardized per unit area. Panel F: Caspase-3 (CASP3) activity levels in epididymal fat. Panel G: Epididymal fat mass following in wt and Gp91−/− mice following 8 weeks and 16 weeks of HFD. Panel H: Inflammatory cytokine expression in purified ATMs. Panels I–J: H&E staining of liver and quantification of liver triglyceride (TG) levels. Panel K: Liver weight. Panel L: Hepatic expression of genes involved in TG synthesis and metabolism in mice fed the HFD. Panels M–N: Linear regression analysis of body weight or epididymal fat caspase-3 activity versus liver TGs and fasting glucose levels. Results are mean ± SEM; n=5–15, *, p<0.05 Student’s t-test. See also Figs. S3–S6.
An examination of the epididymal fat in Nox2−/− mice fed the HFD for 16 weeks revealed marked increases in the number of crown-like structures (Fig. 5E, Fig. S3) and caspase-3 activity (Fig. 5F) relative to control mice, suggesting a higher accumulation of dead adipocytes. The extent of this adipocyte death was quite substantial given that Nox2−/− mice fed the HFD had less epididymal fat mass than wild-type mice (Fig. 5G), despite being more obese (Fig. 5A). In contrast, Nox2−/− mice had more epididymal fat than wild-type mice following 8 weeks of HFD (Fig. 5G). These findings suggest that Nox2−/− mice are capable of expanding their epididymal fat mass during early DIO, but that they cannot sustain it during prolonged exposure to the HFD.
Despite the increased number of dead adipocytes in Nox2−/− mice, which should induce macrophage inflammation (see Fig. 1), ATM expression of Tnfα, Il6, and Il1β was similar to that of wild-type mice (Fig. 5H). Thus, it is likely that the anti-inflammatory effect of ablating Nox2 was still present at 16 weeks, but masked by the hyper-inflammatory environment produced by the dead adipocytes.
Consistent with the deterioration of epididymal fat health and mass at 16 weeks of HFD, Nox2−/− mice displayed severe hepatosteatosis, characterized by increased hepatic triglyceride mass and liver weight (Figs. 5I–K, Fig. S4), This hepatosteatosis could not be explained by changes in the expression of liver genes involved in triglyceride synthesis (Fas1, Acc1, and Srebp1) or fatty acid metabolism (Pparα and Cpt1α) (Fig. 5L).
Together, these studies showed that Nox2−/− mice develop severe late-onset adipocyte death, hepatosteatosis, and insulin resistance. Since Nox2−/− mice are more obese than their wild-type counterparts, we explored the possibility that these late-onset effects were due to excess body mass. To test this, we fed wild-type mice a HFD until their average body weight matched Nox2−/− mice at 16 weeks (‘wild-type match’) and quantified key aspects of the metabolic phenotype.
We found that ‘wild-type match’ mice could not mimic the late onset dead adipocyte accumulation, hepatosteatosis, and insulin resistance observed in Nox2−/− mice (Figs. S3–S4, S6), suggesting that these metabolic abnormalities were not driven by the increased body weight of Nox2−/− mice fed the HFD. Indeed, liver triglycerides and fasting glucose levels were not correlated to body weight in wild type and Nox2−/− mice fed the HFD (Figs. 5M–N). In contrast, these parameters were significantly and positively correlated to the extent of adipocyte death in epididymal fat, as determined by caspase-3 activity (Figs. 5M–N). Thus, the inability of Nox2−/− mice to sustain their epididymal fat may help to explain the late-onset hepatosteatosis and metabolic defects observed in these mice.
Dead adipocyte accumulation is associated with defective lysosomal exocytosis by Nox2−/− ATMs
In principle, the number of dead adipocytes observed in vivo is determined both by the rate of death and the rate of clearance (Fig. 6A); the latter process, which is mediated by ATMs, has not been the subject of intensive study. Since Nox2−/− macrophages exhibited defective lysosomal exocytosis and clearance of apoptotic adipocytes in vitro (see Figs. 1,2), we reasoned that defective dead adipocyte clearance by ATMs might help to explain the late-onset accumulation of dead adipocytes. We used three approaches to test this hypothesis.
Fig. 6. ATMs from obese Nox2−/− mice exhibit lower levels of lysosomal membrane proteins on the cell surface.

Wild-type (wt) and Gp91−/− mice were fed a low-fat diet (LFD) or 45% high-fat diet (HFD) for 16 weeks. Panel A: A model for the determinants of dead adipocyte number in vivo. Panel B: Hif1α mRNA levels in epididymal fat (a marker of hypoxia). Panel C: Inflammatory cytokine expression levels in epididymal fat. Panels D–E: ATMs were purified from wt and Gp91−/− mice and interrogated by plasma membrane proteomics. Panel D: Heatmap of the relative abundance of cell surface lysosomal membrane proteins. Panel E: Examples of several lysosomal membrane proteins on the cell surface of ATMs; proteins were quantified by mass spectrometry. Results are mean ± SEM; n=2–5, #, p<0.05, G-test. See also Table S1.
First, we investigated whether drivers of adipocyte death were elevated in Nox2−/− mice. Adipocyte apoptosis can be caused by hypoxia and/or inflammation (Sun et al., 2011), both of which increase as adipose tissue expands during obesity. Neither of these mechanisms are a likely cause of dead adipocyte accumulation in Nox2−/− mice since adipose tissue levels of Hif1α (hypoxia marker) and Tnfα and Il1β (pro-inflammatory markers) were unchanged after 16 weeks of high-fat feeding compared to wild type mice (Figs. 6B–C). Moreover, ATM inflammation (see Fig. 5) and ATM number (Fig. S5) were not significantly different between wild type and Nox2−/− mice fed the HFD.
Second, we determined whether ATMs from Nox2−/− mice had lower cell surface levels of lysosomal membrane proteins, the hallmark of lysosomal exocytosis (Rodriguez et al., 1997). We isolated ATMs from HFD-fed wild-type and Nox2−/− mice and interrogated them by plasma membrane proteomics. This analysis identified 8 lysosomal membrane proteins on the cell surface of wild-type ATMs, all of which were lower on the cell surface of ATMs from Nox2−/− mice (Fig. 6D–E, Table S1). Importantly, the decreased levels of lysosomal membrane proteins on Nox2−/− ATMs were likely underestimated given that Nox2−/− mice had significantly more dead adipocytes, which should stimulate lysosomal exocytosis (see Figs. 1,2). These findings suggest that ATMs from Nox2−/− mice exhibit impaired lysosomal exocytosis in vivo.
Third, we determined whether ablating Nox2 in myeloid cells specifically could reproduce the late-onset accumulation of dead adipocytes and associated metabolic derailments. We crossed Nox2fl/fl mice with LysM-Cre+/− mice to generate myeloid cell specific Nox2-deficient mice (Nox2fl/flLysM-Cre+/−) and littermate controls (Nox2fl/flLysM-Cre−/−). After validating Nox2 knockout in ATMs (Fig. S7), we examined the metabolic phenotypes of these mice following prolonged high-fat feeding.
Myeloid-cell specific ablation of Nox2 mimicked the late-onset accumulation of dead adipocytes in the absence of increased hypoxia or adipose tissue inflammation (Figs. 7A–D, Fig. S3), hepatosteatosis and increased liver weight (Fig. 7E–F, Fig. S4), obesity (Fig. 7G), hyperglycemia (Fig. 7H), and hyperinsulinemia (Fig. 7I) phenotypes of the whole animal knockout. Of note, the stronger increase in the number of crown-like structures in the LysM-Cre model relative to the global knockout was due to the presence of fewer crown-like structures in LysM-Cre−/− control mice, which likely reflects the variability in the appearance of crown-like structures in vivo.
Fig. 7. Ablating Nox2 in myeloid cells is sufficient to produce late-onset lipoatrophy, hepatosteatosis, and insulin resistance.

A comparison of the late-onset phenotypes in whole animal NOX2-deficient (global KO) and myeloid cell-deficient (LysM-Cre KO) fed the HFD. All parameters are plotted relative to the appropriate wild-type control (CTRL). Panel A: Epididymal fat was stained with antibodies against PLIN2 (red; identifies adipocytes) and MAC2 (green; identifies macrophages), and DAPI (blue; identifies nuclei). Macrophages in crown-like structures (CLS; arrow) were quantified by microscopy and standardized per unit area. Panel B: Relative caspase-3 (CASP-3) activity in epididymal fat. Panel C: Relative epididymal fat mass. Panel D: Relative levels of Hif1α (a hypoxia marker) and Tnfα and Il1β (inflammatory markers) in epididymal fat. Panel E: H&E staining of the liver. Panel F: Relative liver weight. Panel G: Relative body weight gain. Panels H–I: Relative fasting glucose and insulin levels. Data for the global control and KO mice are re-presented in modified form for comparative purposes. Results are mean ± SEM; n=5–10, *, p<0.05, Student’s t-test. See also Figs. S3–S4,S7.
These findings suggest that the accumulation of dead adipocytes and the associated metabolic dysfunction are driven by the loss of Nox2 in myeloid cells, and not due to unanticipated effect in adipocytes.
DISCUSSION
Accumulating evidence suggests that ATMs perform both detrimental and beneficial functions during obesity. They produce inflammatory cytokines that promote insulin resistance (Chawla et al., 2011, Olefsky and Glass, 2010). They also might protect metabolic tissues from the deleterious effects of excess FFAs and contribute to adipose tissue homeostasis by clearing dead adipocytes. Traditionally, these diverse functions have been attributed to distinct ATM populations; the detrimental functions have been associated with M1-like macrophages, which may predominate during early DIO (Lumeng et al., 2007), while the beneficial functions have been ascribed to M2-like macrophages, which may accumulate during prolonged DIO (Shaul et al., 2010).
Here we provide evidence that inflammatory cytokine production and dead adipocyte clearance are functional properties of a single MMe macrophage phenotype, that is present during early and late DIO. We further show that inflammatory signaling through NOX2, TLR2, and MYD88 coordinately regulates both the detrimental and beneficial functions of MMe macrophages. Accordingly, ablating Nox2 produces a complex metabolic phenotype determined by the duration of high-fat feeding, which in turn highlights the relative importance of macrophage inflammation and dead adipocyte clearance during the progression of obesity.
After 8 weeks of HFD, wild-type mice exhibited ATM inflammation in the absence of adipose tissue hypoxia and adipocyte death, indicating a low need for dead adipocyte clearance at this time point. Accordingly, the anti-inflammatory benefit of ablating Nox2 was observed without exposing the costs associated with impaired dead adipocyte clearance. Nox2−/− mice had attenuated ATM inflammation and exhibited improvements in fasting glucose and glucose tolerance, even though mice were more obese, had more ATMs in epididymal fat, and were trending to hepatosteatosis – all of which should exacerbate metabolic dysfunction during DIO. Since ablating Nox2 inhibits inflammation in MMe but not M1 macrophages, our findings further underscore the importance of metabolic activation in producing ATM inflammation in obesity.
In contrast, after 16 weeks of HFD, we found that dead adipocytes accumulated in epididymal fat of wild type mice, and this accumulation was significantly exacerbated in Nox2−/− mice, suggesting that the costs associated with impaired dead adipocyte clearance were evident at this time point.
Three lines of evidence suggest that this late-onset dead adipocyte accumulation in Nox2−/− mice was due to impaired clearance by ATMs. First, we showed that inflammation and hypoxia, the main drivers of adipocyte death, were not increased in Nox2−/− mice relative to wild type mice. Second, we showed that ablating Nox2 interfered with dead adipocyte clearance through lysosomal exocytosis in vitro and further showed that Nox2−/− ATMs had impaired lysosomal exocytosis in vivo. Third, we showed that mice lacking Nox2 in myeloid cells specifically reproduced this late onset phenotype. Although neutrophils are also targeted by the LysM-Cre system (Clausen et al., 1999), ATMs are the most abundant cell type in adipose tissue and form the adipocyte-clearing crown-like structures during DIO. It is therefore unlikely that neutrophils significantly contribute to the impaired adipocyte clearance observed in vivo. However, since neutrophils promote insulin resistance during DIO (Talukdar et al., 2012), we cannot ignore their potential contribution to the metabolic phenotypes observed in the Nox2−/− mice. Collectively, these findings implicate metabolically activated ATMs and their ability to exocytose their lysosomes to dead adipocytes as a key determinant of adipose tissue remodeling and health during prolonged obesity.
The late-onset dead adipocyte accumulation was so extensive in Nox2−/− mice, that they had less epididymal fat mass than their wild type counterparts despite being more obese, suggesting that visceral fat could no longer store excess dietary nutrients. This phenotype shares some similarities with lipodystrophy, where the inability to store nutrients in adipose tissue results in ectopic fat accumulation in the liver and hepatosteatosis, which in turn drives insulin resistance (Fiorenza et al., 2011). Indeed, Nox2−/− mice fed the high-fat diet for 16 weeks exhibited severe hepatosteatosis, which was likely responsible for the hyperglycemia, hyperinsulinemia, and insulin resistance at this time point (Cohen et al., 2011, Shulman, 2014).
However, unlike lipodystrophy, Nox2−/− mice did not exhibit inherent defects in adipose tissue development or expansion. Epididymal fat in Nox2−/− mice fed the LFD was normal at both time points. Furthermore, epididymal fat mass was actually higher in Nox2−/− mice fed the HFD for 8 weeks relative to wild type mice. Instead, Nox2−/− mice had a diminished capacity to maintain their epididymal fat during prolonged nutrient excess, when adipose tissue hypoxia and adipocyte death were apparent, and ATM-mediated dead adipocyte clearance was required.
The clearance of dead adipocytes poses two unique challenges for macrophages. First, because adipocytes are so large, macrophages cannot phagocytose them. Instead, their clearance relies on the formation of extracellular lysosomal compartments between many macrophages and one adipocyte (Haka et al., 2016), resulting in the formation of crown-like structures. Second, adipocytes are predominantly comprised of triglycerides. Thus, during clearance, macrophages are exposed to FFAs such as palmitate, which induce macrophage inflammation (Kratz et al., 2014). For this reason, clearance of dead adipocytes, unlike the clearance of other dead cells (Fadok et al., 1998), is an inflammation propagating event. Indeed crown-like structures have been positively correlated with inflammation in adipose tissue of obese humans and mice (Lumeng et al., 2007).
From this perspective, the MMe macrophage is well positioned to handle the unique challenges imposed by dead adipocytes. FFAs released by dead adipocytes promote the MMe macrophage phenotype and its associated upregulation of lysosomal exocytosis, lipid metabolism, and inflammation. Lysosomal exocytosis may drive FFA liberation to help clear the TG droplet, which may be further enabled by the induction of lysosomal genes recently reported in ATMs from morbidly obese mice (Xu et al., 2013). Increased expression of lipid metabolism genes (ie. ABCA1, CD36, PLIN2) could help macrophages to productively handle this excess fat, while inflammation could facilitate recruitment of additional macrophages to the crown-like structure to assist in clearance.
Of course, prolonged macrophage inflammation also carries its consequences. Cytokines such as TNFα promote damage to metabolic organs and drive insulin resistance during obesity (Hotamisligil et al., 1993) and our findings during early DIO provide further support for this widely accepted paradigm.
On the other hand, recent studies showed that the metabolic consequences of this inflammation might not be as straightforward as this paradigm suggests. For example, thermoneutral housing accelerated the onset of adipose tissue inflammation but did not worsen the metabolic phenotype (Tian et al., 2016). Similarly, ablating TNFα expression by adipocytes exacerbated obesity-associated insulin resistance and hepatosteatosis (Wernstedt Asterholm et al., 2014).
Our findings reinforce the emerging complex relationship between inflammation, macrophages, and metabolic phenotypes during DIO. Indeed, we observed contradictory metabolic phenotypes with the same anti-inflammatory perturbation (Nox2−/−) over two time points during DIO. Interestingly, previous studies with Nox2−/− mice fed the HFD reported similar contradictory findings. One study reported improvements in fasting glucose, glucose tolerance, and brain injury in Nox2−/− mice fed a 60% HFD for 14 weeks (Pepping et al., 2013). Another study showed increased adiposity due to hyperphagia, and worsened insulin resistance and hepatosteatosis in Nox2−/− fed a 60% HFD for 18 weeks (Costford et al., 2014).
Although we cannot rule out the contribution of other NOX2-driven mechanisms, the contradictory findings that we and others report with Nox2−/− mice may be conceptualized by integrating the beneficial and detrimental functions of ATMs through a single MMe phenotype, whose impact on adipose tissue biology and metabolic phenotypes is determined by the duration of high-fat feeding. Future experiments will be required to determine whether this paradigm holds true in other anti-inflammatory settings.
EXPERIMENTAL PROCEDURES
Mice
All animal studies were approved by the University of Chicago IACUC (ACUP#72209). Wild-type, Gp91−/−, Tlr2−/−, Tlr4−/−, and Myd88−/− male mice on the C57BL/6 background are from Jackson Labs. For DIO studies, wild-type, Gp91−/−, Tlr2−/−, and Tlr4−/− mice were placed on a low-fat or high-fat diet (45% fat, Research Diets Inc. D12451) at 8 weeks of age for up to 16 weeks.
Nox2fl/fl mice
Nox2fl/fl mice were described recently (Sag et al., 2017) and were crossed with LysM-cre knock in mice (Jackson Laboratories 004781) to generate LysM-cre+/− Nox2fl/fl and litter mate control Nox2fl/fl mice. See supplemental methods for genotyping details.
Differentiation and activation of bone marrow-derived macrophages
Murine bone marrow-derived macrophages (BMDMs) were differentiated from bone marrow stem cells as previously described (Kratz et al., 2014). For M1 activation, BMDMs were treated with LPS (5ng/mL) and IFNγ (12ng/mL) for 24h. For MMe activation, macrophages were treated with a combination of glucose (30 mM), insulin (10 nM), and palmitate (0.4mM) for 24h. Macrophages were also treated with media conditioned by live 3T3-L1 adipocytes, apoptotic 3T3-L1 adipocytes, or apoptotic neutrophils for 24h.
Macrophage clearance of apoptotic adipocytes
Macrophage clearance of apoptotic adipocytes and subsequent accumulation of lipids were performed as previously described (Haka et al., 2016). See supplemental methods for additional details.
ATM isolation and analysis
ATMs were isolated using anti-CD11b antibody coupled to magnetic beads as previously described (Kratz et al., 2014) and purity was assessed by flow cytometry. ATMs were interrogated by qRT-PCR, plasma membrane proteomics, and flow cytometry. A workflow for flow cytometric analysis of ATMs is provided in the online supplement (Fig. S5).
Plasma membrane proteomics
Plasma membrane proteins of BMDMs or ATMs were isolated and analyzed by mass spectrometry as previously described (Becker et al., 2012). Proteins were quantified by spectral counting and statistical significance was assessed using a combination of the G-test (G-statistic) and t-test (p-value) with correction for the false discovery rate as previously described (Heinecke et al., 2010).
Plasma measurements
Mice were fasted for 3h and serum insulin levels were measured by ELISA (Millipore) and blood glucose levels were measured with a One Touch Ultra 2 glucometer (Lifescan). For GTT, mice were fasted for 16h, injected with 1 g/kg glucose, and blood was collected at several time points for the measurement of glucose and insulin levels. For ITT, mice were fasted for 6h, injected with 0.5 mU/g insulin, and blood was collected at several time points for the measurement of glucose.
Adipose tissue measurements
Epididymal fat was stained with antibodies against murine MAC2 (Cedarlane) and PLIN2 (ABCAM) and fluorescence images were acquired using an Olympus IX81 inverted wide field microscope. The number of crown-like structures in epididymal fat was by counting the number of adipocytes surrounded by MAC-2 positive signal per unit area. Between 4–6 mice were used per condition and 3–5 images were used per mouse. Epididymal fat caspase-3 activity was measured using the Apo-ONE kit (Promega) according to the manufacturer’s protocol.
Liver measurements
Livers were perfused with 30mL of PBS prior to excision, homogenized, and triglycerides were measured using a triglyceride quantification kit (Abcam) according to the manufacturer’s protocol.
Statistics
Statistical significance was assessed using an unpaired, two-tailed, Student’s t-test. Replicate numbers for each experiment are indicated in the figure legends.
Supplementary Material
HIGHLIGHTS.
Inflammation and dead adipocyte clearance by MMe macrophages require NOX2
Nox2−/− improves the metabolic phenotype in early DIO, but worsens it in late DIO
Early improvements associate with suppressed ATM inflammation
Late worsening associates with lower ATM lysosomal exocytosis to dead adipocytes
Acknowledgments
This research was supported by grants from the National Institutes of Health (R01DK102960 (L.B.), R37DK27083 (F.R.M.), R01HL093324 (F.R.M.), R01DK055267 (C.J.R.), P30 DK020595 (DRTC Cell Biology Core, University of Chicago), T32DK087703 (support for B.R.C., S.F.), T32DK007074 (support for A.H.), the Bernice Goldblatt Endowment Fellowship, University of Chicago (support for C.C.), and the American Heart Association (10SDG3600027 (L.B.)).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
Conceptualization: All authors. Investigation: B.R.C., K.Q.S, V.C.B.L., G.Z., E.P., A.H., S.F., L.Z., B.H., A.S.H., C.C. Writing – Original Draft: L.B. Writing – Reviewing & Editing: All authors. Supervision: L.B., F.R.M. Funding Acquisition: L.B., F.R.M.
References
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–80. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Becker L, Liu NC, Averill MM, Yuan W, Pamir N, Peng Y, Irwin AD, Fu X, Bornfeldt KE, Heinecke JW. Unique proteomic signatures distinguish macrophages and dendritic cells. PLoS One. 2012;7:e33297. doi: 10.1371/journal.pone.0033297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11:738–49. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–77. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–23. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costford SR, Castro-Alves J, Chan KL, Bailey LJ, Woo M, Belsham DD, Brumell JH, Klip A. Mice lacking NOX2 are hyperphagic and store fat preferentially in the liver. Am J Physiol Endocrinol Metab. 2014;306:E1341–53. doi: 10.1152/ajpendo.00089.2014. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorenza CG, Chou SH, Mantzoros CS. Lipodystrophy: pathophysiology and advances in treatment. Nat Rev Endocrinol. 2011;7:137–50. doi: 10.1038/nrendo.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgibbons TP, Czech MP. Emerging evidence for beneficial macrophage functions in atherosclerosis and obesity-induced insulin resistance. J Mol Med (Berl) 2016;94:267–75. doi: 10.1007/s00109-016-1385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–64. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Haka AS, Barbosa-Lorenzi VC, Lee HJ, Falcone DJ, Hudis CA, Dannenberg AJ, Maxfield FR. Exocytosis of Macrophage Lysosomes Leads to Digestion of Apoptotic Adipocytes and Foam Cell Formation. J Lipid Res. 2016 doi: 10.1194/jlr.M064089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MS, Jung DY, Morel C, Lakhani SA, Kim JK, Flavell RA, Davis RJ. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science. 2013;339:218–22. doi: 10.1126/science.1227568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinecke NL, Pratt BS, Vaisar T, Becker L. PepC: proteomics software for identifying differentially expressed proteins based on spectral counting. Bioinformatics. 2010;26:1574–5. doi: 10.1093/bioinformatics/btq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–25. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23. doi: 10.2337/db06-1076. [DOI] [PubMed] [Google Scholar]
- Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, Cinti S. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res. 2008;49:1562–8. doi: 10.1194/jlr.M800019-JLR200. [DOI] [PubMed] [Google Scholar]
- Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG. Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab. 2008;8:301–9. doi: 10.1016/j.cmet.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepping JK, Freeman LR, Gupta S, Keller JN, Bruce-Keller AJ. NOX2 deficiency attenuates markers of adiposopathy and brain injury induced by high-fat diet. Am J Physiol Endocrinol Metab. 2013;304:E392–404. doi: 10.1152/ajpendo.00398.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robblee MM, Kim CC, Porter Abate J, Valdearcos M, Sandlund KL, Shenoy MK, Volmer R, Iwawaki T, Koliwad SK. Saturated Fatty Acids Engage an IRE1alpha-Dependent Pathway to Activate the NLRP3 Inflammasome in Myeloid Cells. Cell Rep. 2016;14:2611–23. doi: 10.1016/j.celrep.2016.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Webster P, Ortego J, Andrews NW. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol. 1997;137:93–104. doi: 10.1083/jcb.137.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi M, Woods NB, De Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–29. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sag CM, Schnelle M, Zhang J, Murdoch CE, Kossmann S, Protti A, Santos CX, Sawyer GJ, Zhang X, Mongue-Din H, Richards DA, Brewer AC, Prysyazhna O, Maier LS, Wenzel P, Eaton PJ, Shah AM. Distinct Regulatory Effects of Myeloid Cell and Endothelial Cell Nox2 on Blood Pressure. Circulation. 2017;135:2163–77. doi: 10.1161/CIRCULATIONAHA.116.023877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seimon TA, Nadolski MJ, Liao X, Magallon J, Nguyen M, Feric NT, Koschinsky ML, Harkewicz R, Witztum JL, Tsimikas S, Golenbock D, Moore KJ, Tabas I. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010;12:467–82. doi: 10.1016/j.cmet.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul ME, Bennett G, Strissel KJ, Greenberg AS, Obin MS. Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet--induced obesity in mice. Diabetes. 2010;59:1171–81. doi: 10.2337/db09-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–41. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, 2nd, Defuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–8. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, Mcnelis J, Lu M, Li P, Yan Q, Zhu Y, Ofrecio J, Lin M, Brenner MB, Olefsky JM. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med. 2012;18:1407–12. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian XY, Ganeshan K, Hong C, Nguyen KD, Qiu Y, Kim J, Tangirala RK, Tonotonoz P, Chawla A. Thermoneutral Housing Accelerates Metabolic Inflammation to Potentiate Atherosclerosis but Not Insulin Resistance. Cell Metab. 2016;23:165–78. doi: 10.1016/j.cmet.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–4. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, Ory DS, Semenkovich CF. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–298. doi: 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, Mccann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, Scherer PE. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014;20:103–18. doi: 10.1016/j.cmet.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–30. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, Van Eijk M, Serlie MJ, Ferrante AW., Jr Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab. 2013;18:816–30. doi: 10.1016/j.cmet.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.