

ABSTRACT
Porphyromonas gingivalis is a Gram-negative, anaerobic bacterium considered to be an important pathogen of periodontal disease that is also implicated in adverse pregnancy outcome (APO). Until recently, our understanding of the role of P. gingivalis in APO has been limited and sometimes contradictory. The purpose of this review is to provide an overview of past and current research on P. gingivalis that addresses some of the controversies concerning the role of this organism in the pathogenesis of APO.
KEYWORDS: Porphyromonas gingivalis, preeclampsia, preterm delivery, fetal growth restriction, defective deep placentation, uterine NK cells, decidual macrophages
Introduction
Porphyromonas gingivalis is a Gram-negative, anaerobic bacterium considered to be an important pathogen of periodontal disease [1]. As a periodontal pathogen, P. gingivalis may indirectly contribute to adverse pregnancy outcomes (APO) by facilitating the release of bacterial products or inflammatory mediators into the maternal circulation that reach the maternal-fetal interface [2]. P. gingivalis could also directly promote APO via invasion and injury to utero-placental tissues; this is supported by several studies that have detected P. gingivalis DNA/antigen in the placenta, amniotic fluid, umbilical cord, and neonatal nasogastric aspirates from complicated pregnancies [3–11]. However, the significance of P. gingivalis as a causative agent of APO is sometimes viewed with skepticism due to several confounding factors. For instance, periodontal disease in of itself is a poor predictor of preterm birth, preeclampsia, fetal growth restriction, or perinatal death [2,12]. Moreover, a consensus of interventional studies in which pregnant women received periodontal treatment did not show an overall significant reduction in the rate of preterm birth, fetal growth restriction, low birth weight, stillbirth, or miscarriage in these cohorts [2,12]. Also confounding is that P. gingivalis DNA or antigen has also been detected in placentas from healthy pregnancies, albeit at a lower microbial load and lower frequency than women with preeclampsia or preterm birth [3–7]. It is also difficult to extrapolate the contribution P. gingivalis to APO when its detection in utero-placental tissues is usually in association with other oral bacterial species [3–5], or when it is not detected in placental tissues from complicated pregnancies even when other oral bacterial species are present [13].
Experimental infection in various animal models confirms that invasion of the uterine compartment by P. gingivalis produces a diverse array of APO, including utero-placental pathology, enhanced expression of pro-T helper (TH)1 type cytokines (IFN-γ, IL-2, IL-12, and TNF-α), fetal growth restriction (FGR), and spontaneous preterm delivery [12–15]. Since these outcomes are manifestations of different pathologic mechanisms within the fetal compartment, it may seem somewhat perplexing that one organism could be responsible for all of these complications. For example, FGR can be a sequela of abnormal placentation or placental angiogenesis, and can accompany severe or early onset preeclampsia [14]. Inflammation associated with FGR or preeclampsia is usually maternal in origin [14]. On the other hand, preterm labor is a complication of overt inflammation that begins in the fetal sac and is usually initiated by activation of toll-like receptors or inflammasomes [15,16]. In this scenario, low birth weight is a result of prematurity rather than placental dysfunction that restricts proper growth of the fetus.
Recent studies regarding the pathogenic mechanisms of P. gingivalis provide new insights into the role of this organism in periodontal disease and/or APO. The purpose of this review is to provide an overview of past and current research on P. gingivalis that addresses some of the controversies concerning the role of this organism in the pathogenesis of APO. In addition to presenting new data generated by our research group, we searched PubMed for publications relevant to the topic of this review. Search terms for this review included: Porphyromonas gingivalis, periodontal disease, periodontitis, gingivitis, hormones and pregnancy, preeclampsia, preterm delivery, fetal growth restriction, defective deep placentation, uterine NK cells, and decidual macrophages.
The interaction between pregnancy-related hormones and oral health
The phenomenon of pregnancy-induced gingivitis is widely reported [17,18], and our current understanding of the link between oral disease and APO indicate that any associated risk is likely precipitated by early or preexisting oral disease. While hormonal changes have been shown to increase bleeding on probing and gingival inflammation in healthy women with normal plaque index and a healthy periodontium, this has been demonstrated to occur independently of pro-inflammatory cytokines IL-1β and TNF-α, which are involved in recruitment of neutrophils and monocytes [17]. These findings indicate that there is direct hormonal induction of gingivitis that does not involve a host response to oral plaque. This finding is significant in that it demonstrates that a clinical diagnosis of gingivitis during pregnancy is not necessarily indicative of oral disease. In a diseased state, pregnancy has been shown to exacerbate existing oral disease, even in the absence of a notable increase in oral plaque [19]. In normal uncomplicated pregnancies, susceptibility to pregnancy-induced gingivitis is reported to occur between 12 and 28 weeks of pregnancy [20]. These findings support the conclusion that should periodontal intervention reduce APO for individuals at risk, the intervention should be performed as soon as a woman knows she is pregnant or (preferably) before becoming pregnant.
In general, sex hormones are reported to increase vascular permeability and proliferation while inhibiting oral mucosal tissue repair, suggesting that sex hormone associated gingivitis (e.g. puberty, menstruation, pregnancy) alters the effectiveness of the epithelial barrier to the oral microbiota [17,18,21]. One study demonstrated that both Prevotella intermedia and P. gingivalis are able to reduce testosterone to 5-alpha dihydrotestosterone (DHT) and induce DHT synthesis by fibroblasts [22]. More recently, a study investigating the role of androgen regulation during placentation showed that the placenta uses both androgen receptors and histone lysine demethylases to mediate androgen signaling and epigenetic regulation of gene expression during placental development [23]. These authors suggested that abnormal androgen signaling might alter placental development. Notably, it has been demonstrated through multiple animal and human studies that androgens play a role in APOs [24,25]. In such a scenario, one may imagine that early placental colonization by microorganisms like P. gingivalis that are capable of interfering with androgen signaling may adversely impact placental development through this mechanism.
Female sex hormones may directly promote microbial growth, as they stimulate P. intermedia growth [26,27] and an increase in both P. intermedia and P. gingivalis in response to hormonal changes during pregnancy [17,19] has been reported. However, due to variable and sometimes conflicting reports in the literature, there is no strong consensus linking increased concentrations of female sex hormones and an increased number (microbial load) of specific periodontal pathogens. Rather, such studies highlight the complexity of the interactive relationships between bacterial constituents of the oral microbiota. Moreover, it has been suggested that sex hormones participate in signaling between pathogens and hosts; therefore, studies that reported sex-linked association of susceptibility to infectious disease may involve microbial responses (e.g. expression of virulence factors) other than just microbial proliferation [28]. Thus, it has been difficult to develop better criteria for identifying ‘at risk’ individuals, since it is a challenge to define the underlying mechanisms responsible for the reported associations between APO and periodontal disease, APO and certain oral community profiles, and/or APO and specific oral pathogens.
Periodontal disease, TH17/Treg cell imbalance, and APO
TH17 cells and IL-17 are reported to increase in periodontal lesions and exacerbate the P. gingivalis ligand-induced inflammatory process [29]. More recently, it has been demonstrated that P. gingivalis favors T helper cell polarization to a TH17 profile with generation of TH17-related cytokines [30] and that the P. gingivalis induced T cell differentiation shift is highly specific towards a TH17 cell phenotype in an IL-6 dependent manner [31]. Moreover, a periodontitis rat model study showed a variable site-specific TH17/Treg cell ratio in the oral tissues but detected a TH17 cell increase with a relative Treg cell decrease in peripheral blood, which was proposed to potentially impact development of systemic inflammatory diseases [32]. This hypothesis is supported by a recent study that found that P. gingivalis-induced periodontitis exacerbates arthritis in an IL-17RA (receptor A) dependent manner in an antigen-induced arthritis murine model [33]. The balance of TH17 cells relative to Treg cells is a central component to balancing the immune response such that pathogens are appropriately recognized, but inappropriate immune responses to self and harmless antigens are minimized [19]. Pregnancy requires recalibration of this balance to protect the mother from infection while accepting a semi-allogenic fetus. Multiple studies have shown that an excess of TH17 with a reduced Treg cell profile can lead to APO and is reviewed in detail by Figueiredo and Schumacher, 2016 [34]. Therefore, it is reasonable to hypothesize that a combination of preexisting periodontal disease and P. gingivalis placental colonization would increase the risk of APO in part by inducing a TH17/Treg cell imbalance.
Could P. gingivalis affect the placental microbiome?
The paradigm that the placenta is a sterile environment has been challenged by several reports that show bacterial DNA or live bacteria present in placentas obtained from normal pregnancies [3–5,35–38]. Interestingly, one study that used comparative 16s ribosomal DNA-based and whole-genome shotgun metagenomic analyses on 320 placental specimens reported that the placental microbiome most closely resembles supragingival plaque [35]. Subsequent studies do not necessarily describe the same microbial community composition within the placenta [36,39,40]. However, they do report a link between APO and placental dysbiosis, such as enrichment of Fusobacterium species in placentas from preterm and low-birth-weight pregnancies [36,40] or trace levels of Porphyromonas, Prevotella, Variovorax, and Dialister in placentas from preeclamptic women [39]. Given that P. gingivalis promotes oral dysbiosis [41,42], we propose that this may be an important mechanism through which P. gingivalis contributes to adverse pregnancy outcomes. In a pilot study conducted by our research group, we examined the composition of the oral microbiome of Sprague-Dawley (SD) rats that received repeated oral inoculations of sterile vehicle or 109 CFU of P. gingivalis strain A7A1-28 (Figure 1). Oral swabs were collected before the inoculation procedure was started, at the end of the 3-month inoculation phase, and at time of necropsy when animals were at gestation day 18. Metagenomic sequencing targeting seven of the nine hypervariable regions (V2, V3, V4, V6–7, V8, and V9) of the 16S bacterial rRNA gene was performed using the Ion 16S Metagenomics Kit (Thermo Fisher Scientific, Waltham, MA, USA). Our approach showed that Streptococcaceae and Micrococcaceae families collectively comprised about 80–95% of the oral microbiota of SD rats and consistently dominated the oral microbiota at each time point (Figure 1). Consistent with previous reports [41], we found that infection with P. gingivalis strain A7A1-28 induced oral dysbiosis that persisted during pregnancy, albeit with a different composition than before pregnancy. While a relatively minor but notable proportional population decrease for both Streptococcaceae and Micrococcaceae families was observed at the end of the 3-month A7A1-28 inoculation phase, this observation coincided with an increase in population proportion of other microbial families and, more importantly, a dramatic increase in diversity in the oral microbiome. While a downward shift in oral microbial diversity was observed at necropsy of the A7A1-28 infected and pregnant SD rats, they maintained a greater oral microbial diversity relative to control. Interestingly, the Pasteurellaceae family, which constituted less than 1% of the total population in both the control and 3-month A7A1-28 post-inoculation microbiota, shifted to a consensus population proportion of 5.47% at necropsy. This family was identified to primarily consist of Haemophilus parainfluenzae – an opportunistic pathogen associated with a wide range of infections, including intra-abdominal and genital infections, and a common colonizer of the mucosa [44]. Although the literature to date is relatively sparse, H. parainfluenzae is also a species of concern with respect to mother-to-infant infections and APO [44–47]. Our findings further support the hypothesis that P. gingivalis-induced dysbiosis results in an oral microbiome profile that permits colonization of potential pathogens and favors an increase in population proportion of resident opportunistic pathogens, increasing the overall risk of microbial infection-associated APOs.
Figure 1.
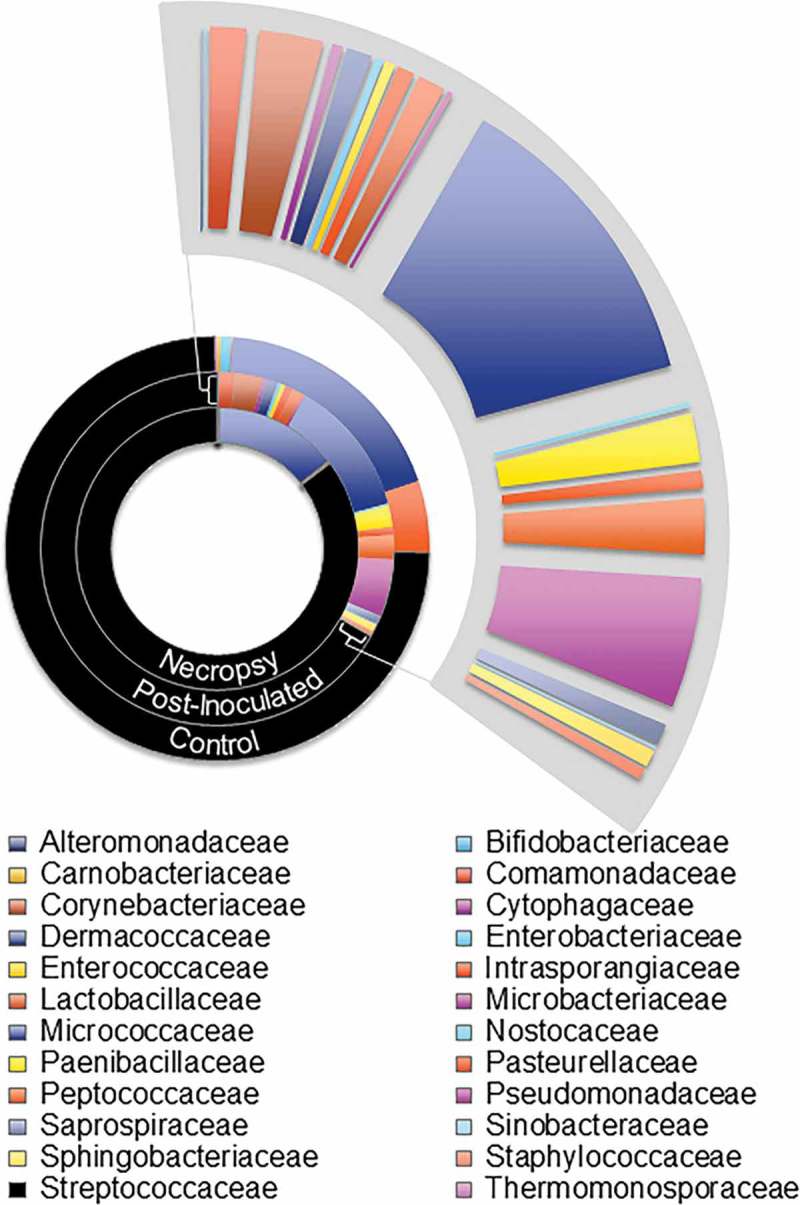
Consensus oral microbiome profiles of Sprague-Dawley (SD) female rats at the family taxonomic level. The consensus data for pre-inoculation baseline, post-P. gingivalis inoculated before breeding, and pregnant (gestation day 18) necropsy were used to generate this family-level comparison (results have not been published elsewhere).
Female rats received repeated oral inoculations with P. gingivalis strain A7A1-28 as previously described [43]. Oral samples were collected 7 days after the last inoculation (post-P. gingivalis group). Breeding was started 2 weeks after the last oral inoculum and dams were sacrificed at gestation day 18.
Hajishengallis et al. [41] demonstrated that P. gingivalis disrupts the composition of the oral community structure, facilitating the overgrowth of oral commensals that in turn enhance oral inflammation. A significant feature of this study is that it showed that the commensal oral microbiota must be present in order for P. gingivalis to induce periodontal disease. Nakajima et al. [42] have recently shown that P. gingivalis-mediated dysbiosis is not limited to the oral cavity. Specifically, oral inoculation of C57BL/6 mice with P. gingivalis increases the proportion of Bacteroidetes and decreases the proportion of Firmicutes within the ileum, with subsequent increased gut permeability and dissemination of gut bacteria into the liver. To date, it has not been determined whether P. gingivalis enhances intrauterine infections by other microbes, but there is a rationale for conducting such experiments. Monotypic infection with P. gingivalis in pregnant rodents produces lesions that disrupt the maternal-fetal barrier involving inflammation of uterine arteries and infiltration of the uterine submucosa by neutrophils [48]. These lesions could facilitate invasion of the placenta by other bacteria that gain entry into the uterus, since polymicrobial colonization/infections are more common at the maternal-fetal interface than monotypic infections [3–5,8,9,39].
The microbial community-wide dysbiotic effect of P. gingivalis is largely driven by this microbe’s ability to subvert host antimicrobial defenses without suppressing inflammation, which in turn facilitates the growth of bacterial species that can tolerate and exploit the inflammatory environment (reviewed by Zenobia and Hajishengallis [49]. Although the polymicrobial etiology of inflammatory diseases such as periodontal disease is associated with dysbiosis of the commensal microbiota, disease susceptibility may depend on an individual’s tolerance to dysbiosis in the context of intrinsic host factors such as genotype and immunological status, or extrinsic host factors such as diet, stress, and behavior. This is further predicated on the relative pathogenicity (e.g. virulence factors) of the constituent members of the microbiota. While there are several P. gingivalis virulence mechanisms that promote dysbiosis [49], the ability of P. gingivalis to synthesize different lipopolysaccharide (LPS) structures that either activate or antagonize TLR4, such as lipid A1 and 4′ phosphatases [50], is particularly relevant to the maternal-fetal interface. The uterus, placenta, and fetal membranes express TLRs 1–9 and dysregulation of TLR function in these tissues can promote APO [51,52]. In the oral cavity, modification of P. gingivalis lipid A is required for successful colonization and overgrowth of plaque bacteria [50]. This was confirmed in vivo with a ligature model of periodontitis in rabbits that were inoculated with P. gingivalis strain A7436 or lipid phosphatase mutants that were ‘locked’ into producing a TLR4 agonist or antagonist [50]. In this study, all A7436 lipid A mutants altered the composition of the oral microbiota, but only TLR4 antagonist and wild-type A7436 enhanced the growth of Fusobacterium nucleatum, which is implicated in preterm delivery [53,54] and it is often found in association with P. gingivalis in placenta, amniotic fluid, and nasogastric aspirates from preterm infants [4,8]. Furthermore, the placenta is a blood-rich tissue and red blood cells contain hemin. At least in vitro, hemin-rich medium promotes the growth of TLR4 antagonist forms of P. gingivalis [55], and thus the placental environment may be enriching for this lipid A structure. This notion is implied by results from a previous study that examined the impact of Campylobacter rectus and P. gingivalis strain A7436 coinfection in a rodent model of APO [56]. Specifically, Arce et al. found that coinfection with A7436 reduced C. rectus-induced expression of TLR4 in the placenta, but produced FGR that did not occur in animals singly infected with C. rectus [56]. P. gingivalis-induced FGR is indeed associated with an increased placental expression of TH1-type cytokines: IFN-γ, IL-2, and IL-12 with a concurrent reduction in TH2 type IL-10 and TGF-β2 [57]. It should be noted that FGR is a syndrome with multiple pathologic etiologies [14,58]. Underlying causes of FGR include inflammation from unknown causes or secondary to hematogenous infection, as well as underlying vasculopathies such as poorly remodeled uterine arteries or abnormal placental angiogenesis [58].
P. gingivalis as a pathogen of the placental bed
Uterine vascular changes during pregnancy
Although a thorough understanding of uterine changes during pregnancy is beyond the scope of this review, it is important to summarize some key features of this process in order to appreciate the impact P. gingivalis has on this system. Successful pregnancy depends on adequate remodeling of uterine spiral arteries from narrow lumen/high resistance vessels to dilated/low resistance vessels that lack their musculoelastic wall. Optimal transformation of the uterine arteries allows the placenta to receive an adequate blood supply without pulsatile turbulent flow that could damage the placenta. The actual process of spiral artery remodeling involves endothelial cell swelling, separation and dedifferentiation of vascular smooth muscle cells (VSMC), degradation of the extracellular matrix with deposition of fibrinoid material, and infiltration of the vessel wall with invading trophoblasts [59]. Effective spiral artery remodeling depends on a series of coordinated events between arterial endothelium, VSMC, uterine natural killer cells (uNK), macrophages (MΦ), and extravillous trophoblasts (EVT) [59]. Changes in endothelial cells and VSMC begin early during decidualization, which is driven by increasing concentrations of progesterone and estrogen [60].
During early pregnancy, uNK cells make up ~70% of leukocytes within the placental bed [61,62]. uNK cells have a distinctly different phenotype than circulating NK cells in that they are CD56bright/CD16− and express a repertoire of activating and inhibitory receptors (NKRs) that can fluctuate in response to EVT and invading pathogens [63]. Although the biological functions of uNK cells is still largely unknown, these cells appear to be important for spiral artery remodeling. Experimental ablation of uNK cells in pregnant rats reduces EVT invasion and induces vascular and perivascular necrosis with fibrosis surrounding the artery [64]. Human uNK cells induce disruption of VSMC and breakdown of the extracellular matrix in vitro, possibly making the arteries more penetrable to EVT cells [65]. Their role in vascular remodeling is also implied by their phenotype. UNK cells produce angiogenic factors (VEGF, placental growth factor, and angiopoietin 2), cytokines (GM-CSF, IFN-γ, CSF-1, and TNF-α), matrix metalloproteinases, and surface receptors (NK complex of lectin related genes and leukocyte receptor complex of immunoglobulin-related genes) that interact with MHC class I molecules uniquely expressed by EVT (HLA-G, HLA-E, and HLA-C) [62].
Macrophages comprise ~20% of the decidual leukocyte population [61]. Unlike uNK cells, which predominate during the first trimester of pregnancy, macrophages maintain their levels within the decidua throughout pregnancy [61]. However, macrophages perform similar tasks in that they also regulate the remodeling of the spiral arteries through release of various cytokines, matrix metalloproteinases, and angiogenic factors [66]. Decidual macrophage (dMΦ) populations are heterogeneous, but most tend towards M2 polarity phenotypes (M2a, M2b, and M2c) based on their expression of IL-10, TGF-β, indoleamine 2,3-dioxygenase (IDO), IL-6, and/or TNF-α [67], and their ability to suppress uNK cytotoxicity toward EVT [68]. dMΦ can minimize inflammation by phagocytosing cellular debris from apoptotic VSMC [67], but also have the capacity to revert to a pro-inflammatory phenotype, as seen in recurrent miscarriage, preeclampsia, and fetal growth restriction [67,69].
EVTs facilitate spiral artery remodeling by promoting apoptosis of arterial endothelium and VSMC [70]. Another important feature of EVTs is that they express unique MHC class I molecules (HLA-G, E, and C), which maintain maternal tolerance of the fetus via immune modulation of uNK cells, macrophages, and T cells [71]. Thus, anything that blocks EVT invasion into the uterus or inappropriately leads to EVT death could result in implantation failure or poor placentation.
It is important to note that EVTs are functionally distinct from villous trophoblasts. First, EVT are highly motile and capable of invading the uterus, whereas villous trophoblasts are not [70]. Second, EVTs are susceptible to invasion by intracellular bacteria [72], unlike villous trophoblasts, which are highly resistant to microbial invasion [73]. During the first trimester of pregnancy when EVTs form anchoring villi to attach the placenta to the decidua and the uterine wall, this susceptibility affords the opportunity for vertical transmission of pathogens to the fetus [72].
The impact of impaired spiral artery remodeling on pregnancy
Inadequate remodeling of the myometrial segments of the uterine spiral arteries is referred to as defective deep placentation (DDP) [74,75]. Studies of DDP are challenging because adequate specimens can only be obtained by invasive biopsy, hysterectomy, or from cadavers [75]. Despite this limitation, it has become clear that DDP underpins a spectrum of obstetric complications ranging from first- and second-trimester spontaneous abortion, preterm premature rupture of membranes (PPROM), and spontaneous preterm birth to intrauterine growth restriction (IUGR) and abruption placentae [75,76]. These complications are often referred to as the great obstetrical syndromes (GOS) [75]. Remarkably, each of these obstetric disorders has different clinicopathologic features within the placenta and/or fetal membranes that set them apart from each other [75,76]. The diversity of the GOS underscores the significance of DDP as a central pathologic mechanism that begins during the earliest weeks of pregnancy before any complications manifest. Thus, any effective strategy to prevent DDP would require early intervention, possibly even before pregnancy begins.
P. gingivalis as a vascular pathogen of the placental bed
Given the difficulty in obtaining human placental bed specimens, it is not surprising that there is no information to identify whether P. gingivalis colonizes the deep uterine vasculature or if this is linked to APO. However, there are some implications that support this notion. The inner third of the placental bed, the decidua, ‘peels off’ with the placenta when it is removed, so it is readily available for analysis. Studies that have sampled the decidua for the presence of P. gingivalis DNA reported that 70–92% of specimens from women with preeclampsia were positive [4,5]. Another study that screened a very preterm cohort (≤32 weeks’ gestation) consisting of spontaneous preterm birth, small for gestational age, preeclampsia, and preeclampsia with HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count) cases found the presence of P. gingivalis within the placenta significantly linked to shorter gestation lengths (OR 0.63 [95% CI: 0.48–0.85]; p = 0.002), regardless of the diagnosis of APO [6]. Although spontaneous preterm birth, small for gestational age, preeclampsia, and preeclampsia with HELLP syndrome have different pathologies within the placenta, DDP can be found in all these disorders [75]. Thus, DDP could be a common mechanism by which P. gingivalis could contribute to APO.
Both in vivo and in vitro infection models indicate that P. gingivalis can interfere with remodeling of the uterine spiral arteries. In a pregnant rat model of infection, Belanger et al. were the first to provide evidence that P. gingivalis induces metrial arteritis [48]. This lesion was most common in dams inoculated with P. gingivalis strain A7436. Subsequent studies by our research group have demonstrated that infection with fimbriae-expressing P. gingivalis strain A7436 or A7A1-28 does indeed interfere with the physiologic remodeling of the uterine spiral artery in rats (Figure 2). In this model, reduced spiral artery remodeling is characterized by retention of arterial endothelium and VSMCs, with a significant reduction in EVT density in the placental bed. In these tissues, P. gingivalis antigen is most commonly detected in stromal cells that are often associated with uNK cells and CD68+ macrophages (Figure 3), opening the possibility that P. gingivalis may be perturbing the function of these cells in a paracrine manner.
Figure 2.
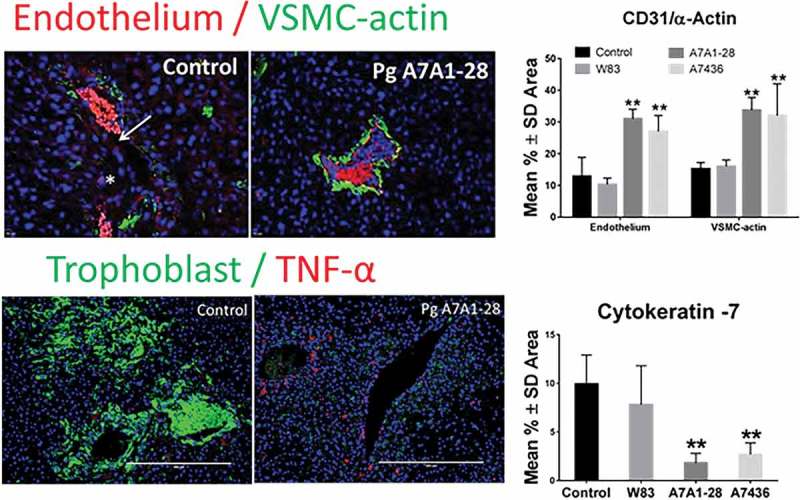
Infection with P. gingivalis strains A7A1-28 and A7436 but not W83 impairs remodeling of uterine spiral arteries in SD rats. Animals received repeated oral inoculations of sterile vehicle (control) or P. gingivalis as described in Figure 1. Endothelial cells were detected with rabbit polyclonal antibody to CD31 (Abbiotec™, San Diego, CA). VSMC were identified with anti-α-actin mouse clone 1A4 (Biorad Laboratories, Hercules, CA). EVT were detected with anti-cytokeratin 7 mouse monoclonal antibody clone LP1K (Abcam®, Cambridge, MA) and nuclei were stained with DAPI. Calibrated digital images were analyzed with Image J 1.50b analysis software (Rasband; National Institutes of Health, USA). Morphometry of spiral artery remodeling was performed with the particle analysis feature. Data were analyzed by one-way ANOVA and Tukey’s tests. **Values different from control and W83 p < 0.01 (n = 5). Unpublished data were presented at the International Federation of Placenta Associations meeting held in Portland, OR, September 2016.
Figure 3.
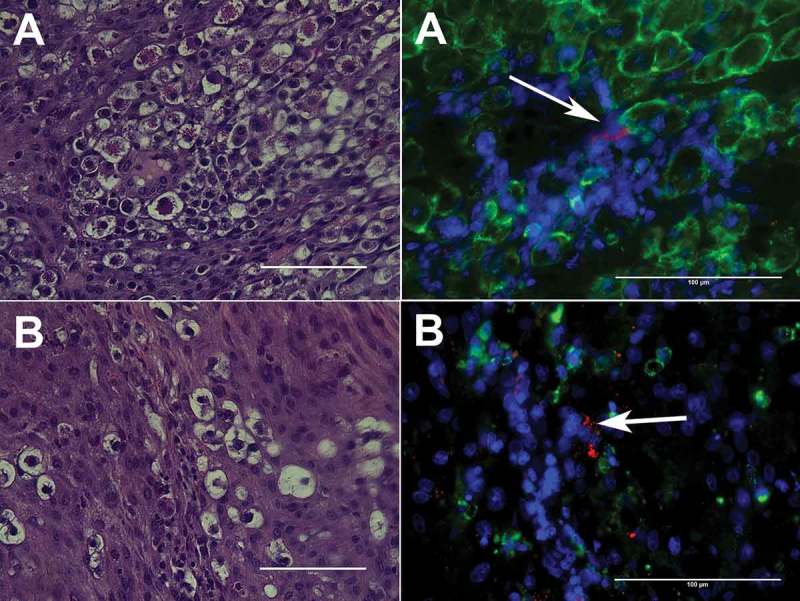
Representative images of P. gingivalis antigen in the metrial triangle of SD rats in association with uNK cells (A) or CD68+ decidual macrophages (B). P. gingivalis (red) was detected with a rabbit polyclonal antibody to whole cell W83 [6,77], uNK cells were identified with mouse monoclonal antibody clone ANK61 to NK cell activation structure, decidual macrophages were labeled with anti-CD68 mouse clone ED1antibody (Abcam®, Cambridge, MA), and nuclei (blue) were stained with DAPI (results have not been published elsewhere).
A preliminary study conducted by our group in nonhuman primates suggests that infection with P. gingivalis alters decidual NK cell populations in vivo. Pregnant rhesus macaques received a total of three intravenous inoculations of sterile vehicle or 105 CFU of P. gingivalis strain A7A1-28 that were administered a week apart during the first trimester of pregnancy. Animals were euthanized at gestation day 50 ± 3 days (latter part of the first trimester; term = gestation day 165). Decidual cells were isolated as previously described [78] and evaluated by flow cytometry (Figure 4). Data analysis was performed manually with FlowJo v10.3 (Flow Jo, LLC, Ashland, OR) and also with Bioconductor package Cytofkit [79] to generate unbiased, multidimensional t-distributed stochastic neighbor embedding (tSNE) [80] population cluster maps. With a panel designed to define NK cell subsets in the decidua (dNKs), we observed a decrease in the absolute number, percentage, and median fluorescence intensity of CD56bright dNK cells during P. gingivalis infection, with a concomitant increase in CD16+ NK cells. Both of these subsets had increased expression of cytotoxicity receptor NKp46 and activation marker NKG2D with infection; however, we did not observe significant differences in the expression of activation marker CD69, proliferation marker Ki-67, or inhibitory marker PD-1 in dNKs from our infected versus control cohorts.
Figure 4.
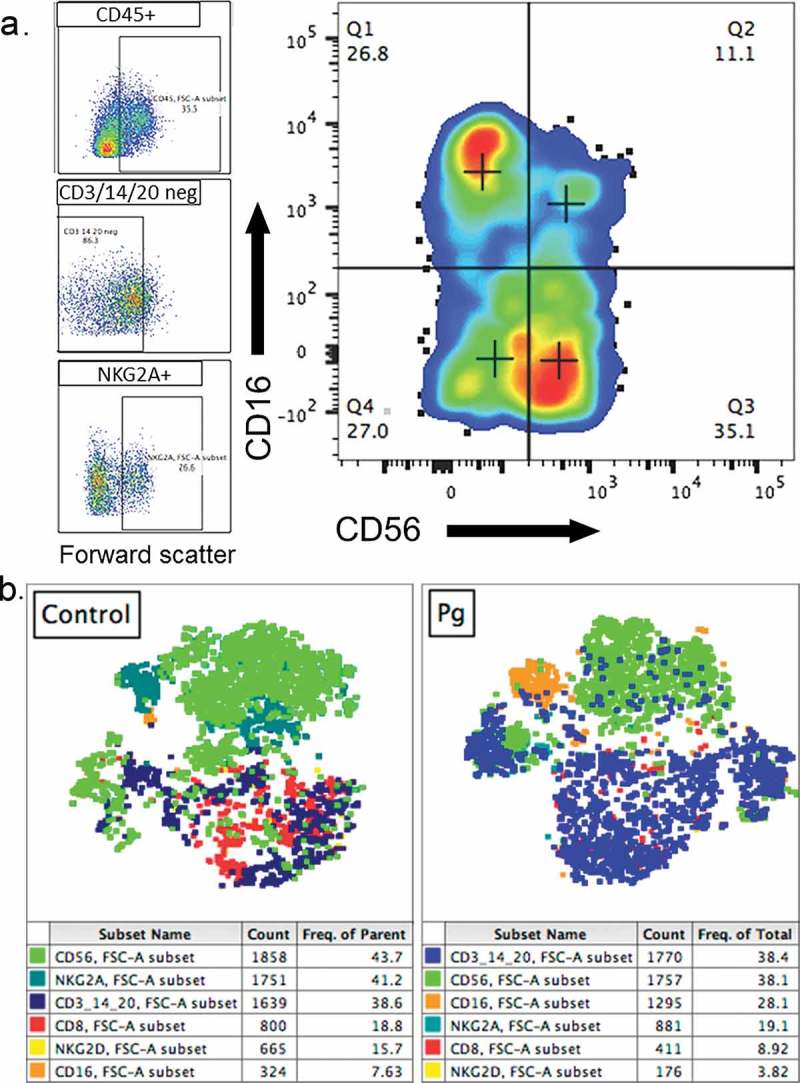
Representative manual gating strategy performed with FlowJo v10.3 software to identify dNKs defined as live, single cells that were CD45+/CD14–/CD20–/CD3–/NKG2A+ (A). Representative unbiased, multidimensional tSNE clustering analysis of first trimester decidual leukocytes conducted with Bioconductor package Cytofkit (B). tSNE clustering analysis revealed a greater proportion of CD3+/CD14+/CD20+ cells and lower proportion of NKG2A+ and CD56+ cells with P. gingivalis (Pg) infection compared with controls (bottom row). Each dot represents an individual cell, colored by marker expression as shown in the corresponding table below each graph (results have not been published elsewhere).
While CD16+ NK cells are the predominant NK cell type in peripheral blood, they have high cytotoxicity and are present at much lower levels in the decidua. In a healthy pregnancy, most dNKs are CD56bright cells that possess granules such as perforin and granzyme B but have low potential for degranulation and are mainly pro-angiogenic, cytokine secreting and immunomodulatory. Double-negative or CD56dim populations have been described as secreting low levels of IFN-γ and TNF-α and having a detrimental impact on pregnancy [63]. A number of studies have found that alterations in dNK subsets are higher in cases of unexplained recurrent miscarriage, infertility, and implantation failure [81–84]. In particular, Giuliani et al. [82] found that maintaining a low ratio of CD16+ to CD56+ cells appears to be important for pregnancy success, likely due to the pro-inflammatory nature of CD16+ cells.
Our preliminary data suggest that infection with P. gingivalis induces loss of CD56+ dNK cells and an increase in CD16+ dNK cells in the first trimester. The ratio of CD16+ cells to CD56+ cells increases to 0.63 with infection versus 0.32 in control animals, and to a lesser extent so does the ratio of NKp46+ cells to CD56+ cells with infection (0.24) versus control (0.20), indicating a shift towards an activated phenotype [85,86]. There is evidence that CD56bright cells have a crucial role in implantation, trophoblast migration, and spiral artery remodeling [64], and that these cells closely associate with dMΦ and decidual vessels in the first trimester [87,88].
Although our flow cytometric analysis of macaque decidual cells was not designed to comprehensively investigate T cell lineage and function (Figure 4), we did observe a higher percent and absolute count of CD3+ T cells in infected animals (data not shown). Within this subset, P. gingivalis infection also correlated with higher expression of PD-1 (46.8 ± 7% vs 4.35% control) and lower expression of IFN-γ (3.3 ± 1.5% vs 41% control), which may contribute to chronic infection by suppressing adaptive T cell responses.
In addition to lymphocytes, we examined markers of macrophages in the decidua to assess whether P. gingivalis infection alters the phenotype or function of these cells. As already mentioned, dMΦ also appear to have a role in spiral artery remodeling and trophoblast migration, typically bear markers associated with tissue remodeling and phagocytosis of debris, and have recently been divided into two subsets: one more regulatory, and the other more pro-inflammatory [67]. P. gingivalis infection resulted in a notable decrease in CD206+ dMΦs (Figure 5). CD206 is a marker of alternative activation and a mediator of phagocytosis of pathogens and debris, with a role in tissue remodeling [89]. A greater proportion of control dMΦs expressed CD206 in our pilot study compared with infected dMΦs (40% vs 3 ± 0.6%). A loss of alternatively activated macrophages in early pregnancy could have detrimental consequences on spiral artery remodeling [88]. Interestingly, although P. gingivalis infection is associated with chronic inflammation, dMΦs from infected animals appeared to secrete less TNF-α than dMΦs from controls. We observed only small decreases in FcγR1 receptor, CD64 and glycoprotein, CD1c with P. gingivalis infection, and no significant difference in HLA-DR (MHC class II) or proliferation marker Ki-67 expression between infected and control dMΦs. Although we cannot rule out the possibility that some of these differences are due to the variation inherent to a genetically diverse nonhuman primate model rather than a disease state, these results provide a possible mechanism for P. gingivalis-mediated effects on spiral artery remodeling. Further characterization will be necessary to determine the biological significance of our findings.
Figure 5.
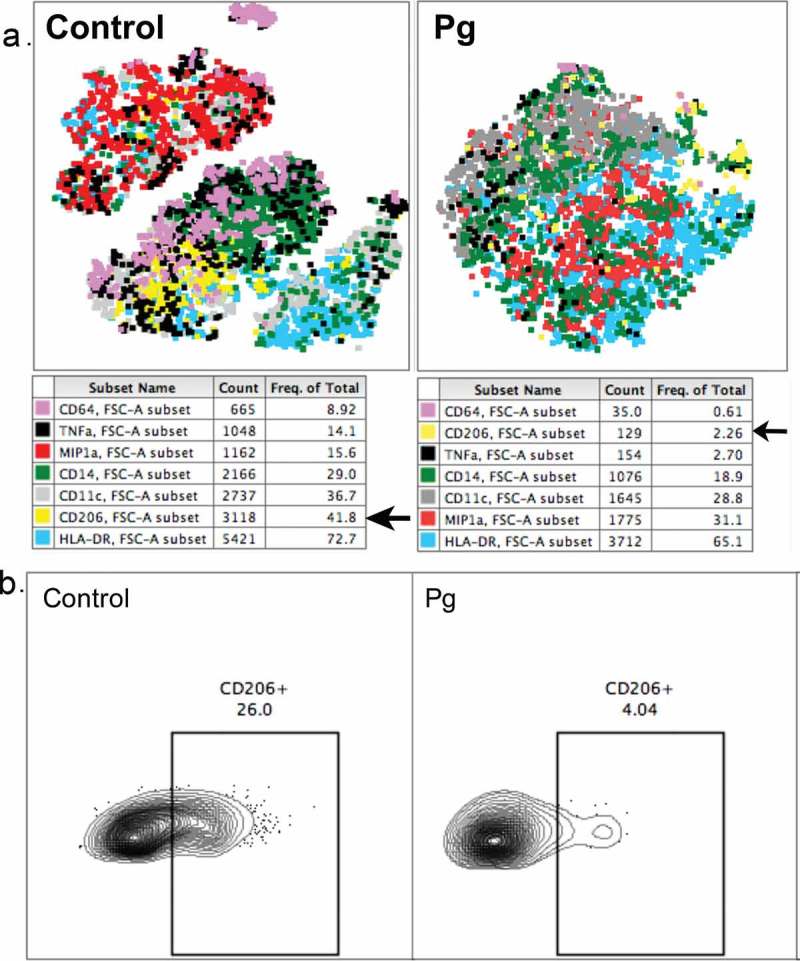
Unbiased, multidimensional tSNE clustering analysis of first-trimester decidual macrophages from uninfected and P. gingivalis infected macaques (A) shows a greater proportion of MIP1a+ cells and a lower proportion of CD206+ cells with P. gingivalis infection compared with controls. Each dot represents an individual cell, colored by marker expression as shown in the corresponding table below each graph. (B) Manual gating with FlowJo v10.3 confirms a decrease in CD206+ macrophages. Value indicates percentage of CD45+ cells.
Pg = infected with P. gingivalis; Control = uninfected, gestational age-matched control.
EVTs may also be a target for P. gingivalis-mediated DDP. HTR8/SvNeo cells are human EVTs [90,91] that were originally isolated from first trimester placental tissue and immortalized with the pSV3neo plasmid [92]. HTR8/SvNeo cells retain the EVT phenotype (i.e. the capacity to invade and secrete degradative enzymes) of the parent cell [92] and have been used extensively to elucidate P. gingivalis effects on trophoblasts in vitro. P. gingivalis can invade HTR8/SvNeo cells and inhibit their proliferation through induction of G1 arrest and apoptosis [93,94]. Further characterization of this phenotype shows that invasion of HTR8/SvNeo by P. gingivalis induces IFN-γ expression [94] and activates the DNA damage response in these cells that is facilitated by gingipain degradation of the p53 antagonist, MDM2 [95]. While EVT death could explain decreased density of these cells in the placental bed, paracrine-mediated inhibition of EVT invasion may also be a factor. Indeed, Hirohata et al. recently demonstrated that soluble factors secreted by P. gingivalis inhibit migration of HTR8/SvNeo through Matrigel [96]. Factors responsible for these effects may be components of outer membrane vesicles such as gingipains and fimbriae proteins [97]. This is yet to be determined either in vitro or in vivo.
P. gingivalis strain-specific effects on APO
It is already well established that P. gingivalis strains exhibit different degrees of pathogenicity based on their ability to invade host cells, disseminate to various organ/tissue sites, and induce disease [77,98–100]. This phenomenon also appears to occur in APOs, as indicated by experimental infection in pregnant rats [48]. To date, the two P. gingivalis strains that have been used extensively in pregnancy outcome studies are W83 and A7436. Although in silico comparison of the whole genome of these strains shows that they are closely related [99], W83 and A7436 differ in the degree and/or type of APO they produce.
P. gingivalis strain A7436 has been examined in various species including hamsters, mice, rats, and rabbits [48,56,57,101–103]. The most consistent pathologic feature observed with A7436 infection in pregnant animals is FGR and/or fetal resorption in mice and hamsters [56,57,101,102]. In addition, A7436-induced pathology is primarily restricted to the maternal side of the intrauterine compartment and consists of uterine vasculitis, endometritis, and DDP [48] (Figure 3). Inflammation of fetal membranes (i.e. extensive leukocyte infiltrates into the chorioamnion and/or umbilical cord) is not a common feature of A7436 infection [48]. Interestingly, extremely high doses (109 CFU) of A7436 given intravenously to pregnant rats induces moderate decidual necrosis without any evidence of inflammation in the labyrinth, chorioamnion, or amniotic cavity [48]. Taken together, this suggests that A7436, by itself, is unlikely to trigger the overt inflammatory responses that initiate spontaneous preterm labor [15]. Instead, the pathologic features observed with A7436 are more in line with implantation failure or poor placentation, which often occur with recurrent miscarriage, fetal growth restriction, or preeclampsia [14,75,104,105].
Unlike A7436, W83 is more likely to induce spontaneous preterm delivery, at least in mice [106,107]. In this model, periodontal disease is initiated before breeding by inoculating W83 into molar pulp chambers, which produces increased maternal serum levels of TNF-α, IL-17, IL-6, and IL-1β and spontaneous preterm delivery [106]. Intrauterine lesions associated with this outcome consist of necrosis of the decidua, chorionic plate (fetal side of the placenta), and fetal membranes with moderate infiltration of neutrophils and macrophages into the tissue [106]. Subsequent studies with this animal model showed that fetal membranes expressed higher levels of TNF-α and IL-1β than uninfected controls [107], which would be consistent with spontaneous preterm birth initiated by chorioamnionitis [15]. Although less frequent, FGR is also observed in rodents experimentally infected with W83 [106,108], suggesting that some APOs may be common among most P. gingivalis strains.
Conclusion
Given the effect of P. gingivalis strain diversity on APO, determining simply whether or not an individual is colonized is insufficient for establishing who is at risk of having a complicated pregnancy. Furthermore, studies that specifically address the role of P. gingivalis virulence mechanisms at the maternal-fetal interface are needed in order to define how this microbe contributes to a wide array of APOs. Regardless of microbial strain diversity, the ability of P. gingivalis to disrupt first trimester processes, namely the physiologic remodeling of the uterine spiral arteries, suggests that timing of oral therapies during pregnancy is important and should begin as early as possible including pregnancy or, if possible, prior to conception. Although it has been established that P. gingivalis can induce dysbiosis of the oral mucosal microbiota, the impact of this effect on systemic illness, particularly APOs, requires further investigation. The notion that P. gingivalis invasion of the maternal-fetal interface could also promote placental dysbiosis linked to APOs is intriguing, but is yet to be tested. Additional studies are warranted to test this hypothesis and to elucidate the microbial mechanisms through which placental dysbiosis occurs.
Acknowledgements
We wish to thank Dr. Aleksandar Stanic-Kostic for his assistance with implementation of high-dimensional flow cytometry and dimension reduction analytical methods. Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development of the National Institutes of Health (NIH) under Award Number R15HD081439, and the Wisconsin National Primate Research Center (WNPRC) Pilot Research Project Grant 2P51-OD011106-56. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or WNPRC.
Biographies
Dr. Leticia Reyes is an Assistant Professor in Pathobiological Sciences with a research interest in infection-induced changes in placental development and subsequent effects on pregnancy outcome.
Dr. Priscilla Phillips is an Assistant Professor of Microbiology and Immunology with research interests in molecular mechanisms that underlie the pathogenesis of P. gingivalis-mediated oral and systemic disease.
Ms. Bryce Wolfe is a PhD candidate in Cellular and Molecular Pathology with a research interest in the pathogenesis of disease and immune responses at the maternal-fetal interface.
Dr. Thaddeus G. Golos is Professor and Chair of Comparative Biosciences and studies the maternal immune response and impact of infection at the maternal-fetal interface.
Ms. Molly Walkenhorst is a MS candidate and first year osteopathic medical student with interests in investigating the human microbiome and its contribution to systemic health.
Dr.Ann Progulske-Foxis Program Director and Distinguished Professor in Oral Biology with a research program focused on the molecular mechanisms of P. gingivalis pathogenesis in adult periodontitis and cardiovascular disease.
Dr. Mary Brown is Professor of Infectious Diseases and Immunology with a focus on intrauterine infections and pregnancy outcome.
Funding Statement
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development [R15HD081439]; Wisconsin National Primate Research Center [2P51-OD011106-56].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1]. Darveau RP, Hajishengallis G, Curtis MA.. Porphyromonas gingivalis as a potential community activist for disease. J Dent Res. 2012;91:816–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Sanz M, Kornman K; Working group 3 of joint EFPAAPw . Periodontitis and adverse pregnancy outcomes: consensus report of the Joint EFP/AAP Workshop on Periodontitis and Systemic Diseases. J Clin Periodontol. 2013;40 Suppl 14:S164–S169. [DOI] [PubMed] [Google Scholar]
- [3]. Barak S, Oettinger-Barak O, Machtei EE, et al. Evidence of periopathogenic microorganisms in placentas of women with preeclampsia. J Periodontol. 2007;78:670–676. [DOI] [PubMed] [Google Scholar]
- [4]. Swati P, Thomas B, Vahab SA, et al. Simultaneous detection of periodontal pathogens in subgingival plaque and placenta of women with hypertension in pregnancy. Arch Gynecol Obstet. 2012;285:613–619. [DOI] [PubMed] [Google Scholar]
- [5]. Chaparro A, Blanlot C, Ramírez V, et al. Porphyromonas gingivalis, Treponema denticola and toll-like receptor 2 are associated with hypertensive disorders in placental tissue: a case-control study. J Periodontal Res. 2013;48:802–809. [DOI] [PubMed] [Google Scholar]
- [6]. Vanterpool SF, Been JV, Houben ML, et al. Porphyromonas gingivalis within placental villous mesenchyme and umbilical cord stroma is associated with adverse pregnancy outcome. PLoS One. 2016;11:e0146157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]. Katz J, Chegini N, Shiverick KT, et al. Localization of P. gingivalis in preterm delivery placenta. J Dent Res. 2009;88:575–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. Ercan E, Eratalay K, Deren O, et al. Evaluation of periodontal pathogens in amniotic fluid and the role of periodontal disease in pre-term birth and low birth weight. Acta Odontol Scand. 2013;71:553–559. [DOI] [PubMed] [Google Scholar]
- [9]. Gonzales-Marin C, Spratt DA, Millar MR, et al. Levels of periodontal pathogens in neonatal gastric aspirates and possible maternal sites of origin. Mol Oral Microbiol. 2011;26:277–290. [DOI] [PubMed] [Google Scholar]
- [10]. Hasegawa-Nakamura K, Tateishi F, Nakamura T, et al. The possible mechanism of preterm birth associated with periodontopathic Porphyromonas gingivalis . J Periodontal Res. 2011;46:497–504. [DOI] [PubMed] [Google Scholar]
- [11]. Parthiban PS, Mahendra J, Logaranjani A, et al. Association between specific periodontal pathogens, Toll-like receptor-4, and nuclear factor-κB expression in placental tissues of pre-eclamptic women with periodontitis. J Investig Clin Dent. 2017;e12265 DOI: 10.1111/jicd.12265. [DOI] [PubMed] [Google Scholar]
- [12]. Srinivas SK, Parry S. Periodontal disease and pregnancy outcomes: time to move on? J Womens Health (Larchmt). 2012;21:121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Blanc V, O’Valle F, Pozo E, et al. Oral bacteria in placental tissues: increased molecular detection in pregnant periodontitis patients. Oral Dis. 2015;21:905–912. [DOI] [PubMed] [Google Scholar]
- [14]. Tang L, He G, Liu X, et al. Progress in the understanding of the etiology and predictability of fetal growth restriction. Reproduction. 2017;153:R227–R240. [DOI] [PubMed] [Google Scholar]
- [15]. Redline RW. Inflammatory response in acute chorioamnionitis. Semin Fetal Neonatal Med. 2012;17:20–25. [DOI] [PubMed] [Google Scholar]
- [16]. Gomez-Lopez N, Romero R, Xu Y, et al. A role for the inflammasome in spontaneous preterm labor with acute histologic chorioamnionitis. Reprod Sci. 2017. DOI: 10.1177/1933719116687656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Wu M, Chen SW, Su WL, et al. Sex hormones enhance gingival inflammation without affecting IL-1β and TNF-α in periodontally healthy women during pregnancy. Mediators Inflamm. 2016;2016:1–6. article ID 4897890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Güncü GN, Tözüm TF, Cağlayan F. Effects of endogenous sex hormones on the periodontium--review of literature. Aust Dent J. 2005;50:138–145. [DOI] [PubMed] [Google Scholar]
- [19]. Silva de Araujo Figueiredo C, Gonçalves Carvalho Rosalem C, Costa Cantanhede AL, et al. Systemic alterations and their oral manifestations in pregnant women. J Obstet Gynaecol Res. 2017;43:16–22. [DOI] [PubMed] [Google Scholar]
- [20]. Adriaens LM, Alessandri R, Spörri S, et al. Does pregnancy have an impact on the subgingival microbiota? J Periodontol. 2009;80:72–81. [DOI] [PubMed] [Google Scholar]
- [21]. Markou E, Eleana B, Lazaros T, et al. The influence of sex steroid hormones on gingiva of women. Open Dent J. 2009;3:114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Soory M. Bacterial steroidogenesis by periodontal pathogens and the effect of bacterial enzymes on steroid conversions by human gingival fibroblasts in culture. J Periodontal Res. 1995;30:124–131. [DOI] [PubMed] [Google Scholar]
- [23]. Cleys ER, Halleran JL, Enriquez VA, et al. Androgen receptor and histone lysine demethylases in ovine placenta. Plos One. 2015;10:e0117472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Sathishkumar K, Elkins R, Chinnathambi V, et al. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol. 2011;9:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Makieva S, Saunders PTK, Norman JE. Androgens in pregnancy: roles in parturition. Hum Reprod Update. 2014;20:542–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26]. Yokoyama M, Hinode D, Yoshioka M, et al. Relationship between Campylobacter rectus and periodontal status during pregnancy. Oral Microbiol Immunol. 2008;23:55–59. [DOI] [PubMed] [Google Scholar]
- [27]. Kornman KS, Loesche WJ. Effects of estradiol and progesterone on Bacteroides melaninogenicus and Bacteroides gingivalis . Infect Immun. 1982;35:256–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Garcia-Gomez E, Gonzalez-Pedrajo E, Camacho-Arroyo I. Role of sex steroid hormones in bacterial-host interactions. Biomed Res Int. 2013;2013:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Beklen A, Sorsa T, Konttinen YT. Toll-like receptors 2 and 5 in human gingival epithelial cells co-operate with T-cell cytokine interleukin-17. Oral Microbiol Immunol. 2009;24:38–42. [DOI] [PubMed] [Google Scholar]
- [30]. Moutsopoulos NM, Kling HM, Angelov N, et al. Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. J Autoimmun. 2012;39:294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Glowczyk I, Wong A, Potempa B, et al. Inactive Gingipains from P. gingivalis selectively skews T cells toward a Th17 phenotype in an IL-6 dependent manner. Front Cell Infect Microbiol. 2017;7:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Gao L, Zhao Y, Wang P, et al. Detection of Th17/Treg cells and related factors in gingival tissues and peripheral blood of rats with experimental periodontitis. Iran J Basic Med Sci. 2017;20:294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. De Aquino SG, Talbot J, Sônego F, et al. The aggravation of arthritis by periodontitis is dependent of IL-17 receptor A activation. J Clin Periodontol. 2017. DOI: 10.1111/jcpe.12743. [DOI] [PubMed] [Google Scholar]
- [34]. Figueiredo AS, Schumacher A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology. 2016;148:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Aagaard K, Ma J, Antony KM, et al. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6:237ra65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36]. Zheng J, Xiao X, Zhang Q, et al. The placental microbiome varies in association with low birth weight in full-term neonates. Nutrients. 2015;7:6924–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Antony KM, Ma J, Mitchell KB, et al. The preterm placental microbiome varies in association with excess maternal gestational weight gain. Am J Obstet Gynecol. 2015;212(653):e1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38]. Queiros da Mota V, Prodhom G, Yan P, et al. Correlation between placental bacterial culture results and histological chorioamnionitis: a prospective study on 376 placentas. J Clin Pathol. 2013;66:243–248. [DOI] [PubMed] [Google Scholar]
- [39]. Amarasekara R, Jayasekara RW, Senanayake H, et al. Microbiome of the placenta in pre-eclampsia supports the role of bacteria in the multifactorial cause of pre-eclampsia. J Obstet Gynaecol Res. 2015;41:662–669. [DOI] [PubMed] [Google Scholar]
- [40]. Doyle RM, Alber DG, Jones HE, et al. Term and preterm labour are associated with distinct microbial community structures in placental membranes which are independent of mode of delivery. Placenta. 2014;35:1099–1101. [DOI] [PubMed] [Google Scholar]
- [41]. Hajishengallis G, Liang S, Payne MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42]. Nakajima M, Arimatsu K, Kato T, et al. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS One. 2015;10:e0134234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]. Verma RK, Bhattacharyya I, Sevilla A, et al. Virulence of major periodontal pathogens and lack of humoral immune protection in a rat model of periodontal disease. Oral Dis. 2010;16:686–695. [DOI] [PubMed] [Google Scholar]
- [44]. Rele M, Giles M, Daley AJ. Invasive Haemophilus parainfluenzae maternal–infant infections: an Australasian perspective and case report. Aust N Z J Obstet Gynaecol. 2006;46:258–260. [DOI] [PubMed] [Google Scholar]
- [45]. Chen RV, Bradley JS. Haemophilus parainfluenzae sepsis in a very low birth weight premature infant: a case report and review of the literature. J Perinatol. 1999;19:315–317. [DOI] [PubMed] [Google Scholar]
- [46]. Mendz GL, Petersen R, Quinlivan JA, et al. Potential involvement of Campylobacter curvus and Haemophilus parainfluenzae in preterm birth. BMJ Case Rep. 2014;2014 DOI: 10.1136/bcr-2014-205282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47]. Cardines R, Daprai L, Giufrè M, et al. Genital carriage of the genus Haemophilus in pregnancy: species distribution and antibiotic susceptibility. J Med Microbiol. 2015;64:724–730. [DOI] [PubMed] [Google Scholar]
- [48]. Bélanger M, Reyes L, von Deneen K, et al. Colonization of maternal and fetal tissues by P orphyromona s gingivalis is strain-dependent in a rodent animal model. Am J Obstet Gynecol. 2008;199(1):86.e1–e7. [DOI] [PubMed] [Google Scholar]
- [49]. Zenobia C, Hajishengallis G. Porphyromonas gingivalis virulence factors involved in subversion of leukocytes and microbial dysbiosis. Virulence. 2015;6:236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50]. Zenobia C, Hasturk H, Nguyen D, et al. Porphyromonas gingivalis lipid A phosphatase activity is critical for colonization and increasing the commensal load in the rabbit ligature model. Infect Immun. 2014;82:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51]. Duriez M, Quillay H, Madec Y, et al. Human decidual macrophages and NK cells differentially express Toll-like receptors and display distinct cytokine profiles upon TLR stimulation. Front Microbiol. 2014;5:article 316:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52]. Gonzalez JM, Xu H, Ofori E, et al. Toll-like receptors in the uterus, cervix, and placenta: is pregnancy an immunosuppressed state? Am J Obstet Gynecol. 2007;197:296.e1–e6. [DOI] [PubMed] [Google Scholar]
- [53]. Liu H, Redline RW, Han YW. Fusobacterium nucleatum induces fetal death in mice via stimulation of TLR4-mediated placental inflammatory response. J Immunol. 2007;179:2501–2508. [DOI] [PubMed] [Google Scholar]
- [54]. Han YW, Redline RW, Li M, et al. Fusobacterium nucleatum induces premature and term stillbirths in pregnant mice: implication of oral bacteria in preterm birth. Infect Immun. 2004;72:2272–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55]. Al-Qutub MN, Braham PH, Karimi-Naser LM, et al. Hemin-dependent modulation of the lipid A structure of P orphyromonas gingivalis lipopolysaccharide. Infect Immun. 2006;74:4474–4485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56]. Arce RM, Barros SP, Wacker B, et al. Increased TLR4 expression in murine placentas after oral infection with periodontal pathogens. Placenta. 2009;30:156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57]. Lin D, Smith MA, Elter J, et al. Porphyromonas gingivalis infection in pregnant mice is associated with placental dissemination, an increase in the placental Th1/Th2 cytokine ratio, and fetal growth restriction. Infect Immun. 2003;71:5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58]. Roberts DJ, Post MD. The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol. 2008;61:1254–1260. [DOI] [PubMed] [Google Scholar]
- [59]. Harris LK. Review: trophoblast-vascular cell interactions in early pregnancy: how to remodel a vessel. Placenta. 2010;31 Suppl:S93–S98. [DOI] [PubMed] [Google Scholar]
- [60]. Evans J, Salamonsen LA, Winship A, et al. Fertile ground: human endometrial programming and lessons in health and disease. Nat Rev Endocrinol. 2016;12:654–667. [DOI] [PubMed] [Google Scholar]
- [61]. Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. 2013;31:387–411. [DOI] [PubMed] [Google Scholar]
- [62]. Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol. 2002;2:656–663. [DOI] [PubMed] [Google Scholar]
- [63]. Cristiani CM, Palella E, Sottile R, et al. Human NK cell subsets in pregnancy and disease: toward a new biological complexity. Front Immunol. 2016;7:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64]. Golic M, Haase N, Herse F, et al. Natural killer cell reduction and uteroplacental vasculopathy. Hypertension. 2016;68:964–973. [DOI] [PubMed] [Google Scholar]
- [65]. Robson A, Harris LK, Innes BA, et al. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J. 2012;26:4876–4885. [DOI] [PubMed] [Google Scholar]
- [66]. Lash GE, Pitman H, Morgan HL, et al. Decidual macrophages: key regulators of vascular remodeling in human pregnancy. J Leukoc Biol. 2016;100:315–325. [DOI] [PubMed] [Google Scholar]
- [67]. Brown MB, Von Chamier M, Allam AB, et al. M1/M2 macrophage polarity in normal and complicated pregnancy. Front Immunol. 2014;5:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68]. Co EC, Gormley M, Kapidzic M, et al. Maternal decidual macrophages inhibit NK cell killing of invasive cytotrophoblasts during human pregnancy. Biol Reprod. 2013;88:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69]. Renaud SJ, Graham CH. The role of macrophages in utero-placental interactions during normal and pathological pregnancy. Immunol Invest. 2008;37:535–564. [DOI] [PubMed] [Google Scholar]
- [70]. Whitley GS, Cartwright JE. Trophoblast-mediated spiral artery remodelling: a role for apoptosis. J Anat. 2009;215:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71]. Ferreira LM, Meissner TB, Tilburgs T, et al. HLA-G: at the interface of maternal-fetal tolerance. Trends Immunol. 2017;38:272–286. [DOI] [PubMed] [Google Scholar]
- [72]. Vigliani MB, Bakardjiev AI. Intracellular organisms as placental invaders. Fetal Matern Med Rev. 2014;25:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73]. Robbins JR, Skrzypczynska KM, Zeldovich VB, et al. Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes . PLoS Pathog. 2010;6:e1000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74]. Khong Y, Brosens I. Defective deep placentation. Best Pract Res Clin Obstet Gynaecol. 2011;25:301–311. [DOI] [PubMed] [Google Scholar]
- [75]. Brosens I, Pijnenborg R, Vercruysse L, et al. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol. 2011;204:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76]. Romero R, Kusanovic JP, Chaiworapongsa T, et al. Placental bed disorders in preterm labor, preterm PROM, spontaneous abortion and abruptio placentae. Best Pract Res Clin Obstet Gynaecol. 2011;25:313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77]. Rodrigues PH, Reyes L, Chadda AS, et al. Porphyromonas gingivalis strain specific interactions with human coronary artery endothelial cells: a comparative study. PLoS One. 2012;7:e52606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78]. Golos TG, Bondarenko GI, Breburda EE, et al. Immune and trophoblast cells at the rhesus monkey maternal-fetal interface. Methods Mol Med. 2006;122:93–108. [DOI] [PubMed] [Google Scholar]
- [79]. Chen H, Lau MC, Wong MT, et al. Cytofkit: a bioconductor package for an integrated mass cytometry data analysis pipeline. PLoS Comput Biol. 2016;12:e1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80]. Krijthe JH. Rtsne: T-distributed stochastic neighbor embedding using a barnes-hut implementation. 2015. Available from: https://github.com/jkrijthe/Rtsne. [Google Scholar]
- [81]. Stamenov G. Endometrial NK Cell Subpopulations CD16− CD56Bright and CD16− CD56Dim in women with recurrent implantation failure. Biotechnol Biotechnol Equipment. 2013;27:4123–4126. [Google Scholar]
- [82]. Giuliani E, Parkin KL, Lessey BA, et al. Characterization of uterine NK cells in women with infertility or recurrent pregnancy loss and associated endometriosis. Am J Reprod Immunol. 2014;72:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83]. Lachapelle MH, Miron P, Hemmings R, et al. Endometrial T, B, and NK cells in patients with recurrent spontaneous abortion. Altered profile and pregnancy outcome. J Immunol. 1996;156:4027–4034. [PubMed] [Google Scholar]
- [84]. Fukui A, Fujii S, Yamaguchi E, et al. Natural killer cell subpopulations and cytotoxicity for infertile patients undergoing in vitro fertilization. Am J Reprod Immunol. 1999;41:413–422. [DOI] [PubMed] [Google Scholar]
- [85]. Zhang Y, Zhao A, Wang X, et al. Expressions of natural cytotoxicity receptors and NKG2D on decidual natural killer cells in patients having spontaneous abortions. Fertil Steril. 2008;90:1931–1937. [DOI] [PubMed] [Google Scholar]
- [86]. El Costa H, Tabiasco J, Berrebi A, et al. Effector functions of human decidual NK cells in healthy early pregnancy are dependent on the specific engagement of natural cytotoxicity receptors. J Reprod Immunol. 2009;82:142–147. [DOI] [PubMed] [Google Scholar]
- [87]. Presicce P, Senthamaraikannan P, Alvarez M, et al. Neutrophil recruitment and activation in decidua with intra-amniotic IL-1beta in the preterm rhesus macaque. Biol Reprod. 2014;92:Article 56, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88]. Faas MM, De Vos P. Uterine NK cells and macrophages in pregnancy. Placenta. 2017;56:44–52. [DOI] [PubMed] [Google Scholar]
- [89]. Azad AK, Rajaram MV, Schlesinger LS. Exploitation of the macrophage mannose receptor (CD206) in infectious disease diagnostics and therapeutics. J Cytol Mol Biol. 2014;10: 1 pii: 1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90]. Irving JA, Lysiak JJ, Graham CH, et al. Characteristics of trophoblast cells migrating from first trimester chorionic villus explants and propagated in culture. Placenta. 1995;16:413–433. [DOI] [PubMed] [Google Scholar]
- [91]. Irving JA, Lala PK. Functional role of cell surface integrins on human trophoblast cell migration: regulation by TGF-beta, IGF-II, and IGFBP-1. Exp Cell Res. 1995;217:419–427. [DOI] [PubMed] [Google Scholar]
- [92]. Graham CH, Hawley TS, Hawley RG, et al. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp Cell Res. 1993;206:204–211. [DOI] [PubMed] [Google Scholar]
- [93]. Inaba H, Kuboniwa M, Bainbridge B, et al. Porphyromonas gingivalis invades human trophoblasts and inhibits proliferation by inducing G1 arrest and apoptosis. Cell Microbiol. 2009;11:1517–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94]. Ren H, Li Y, Jiang H, et al. Porphyromonas gingivalis induces IL-8 and IFN-gamma secretion and apoptosis in human extravillous trophoblast derived HTR8/SVneo cells via activation of ERK1/2 and p38 signaling pathways. Placenta. 2016;45:8–15. [DOI] [PubMed] [Google Scholar]
- [95]. Inaba H, Kuboniwa M, Sugita H, et al. Identification of signaling pathways mediating cell cycle arrest and apoptosis induced by Porphyromonas gingivalis in human trophoblasts. Infect Immun. 2012;80:2847–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96]. Hirohata N, Komine-Aizawa S, Tamura M, et al. P. gingivalis suppresses trophoblast invasion by soluble factors. J Periodontol. 2017;1–18. [DOI] [PubMed] [Google Scholar]
- [97]. Mantri CK, Chen C-H, Dong X, et al. Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis . Microbiologyopen. 2015;4:53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98]. Laine ML, Van Winkelhoff AJ. Virulence of six capsular serotypes of Porphyromonas gingivalis in a mouse model. Oral Microbiol Immunol. 1998;13:322–325. [DOI] [PubMed] [Google Scholar]
- [99]. Chen T, Siddiqui H, Olsen I. In silico comparison of 19 Porphyromonas gingivalis strains in genomics, phylogenetics, phylogenomics and functional genomics. Front Cell Infect Microbiol. 2017. DOI: 10.3389/fcimb.2017.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100]. Baek KJ, Ji S, Kim YC, et al. Association of the invasion ability of Porphyromonas gingivalis with the severity of periodontitis. Virulence. 2015;6:274–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101]. Collins JG, Windley HW 3rd, Arnold RR, et al. Effects of a Porphyromonas gingivalis infection on inflammatory mediator response and pregnancy outcome in hamsters. Infect Immun. 1994;62:4356–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102]. Lin D, Smith MA, Champagne C, et al. Porphyromonas gingivalis infection during pregnancy increases maternal tumor necrosis factor alpha, suppresses maternal interleukin-10, and enhances fetal growth restriction and resorption in mice. Infect Immun. 2003;71:5156–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103]. Boggess KA, Lieff S, Murtha AP, et al. Maternal periodontal disease is associated with an increased risk for preeclampsia. Obstet Gynecol. 2003;101:227–231. [DOI] [PubMed] [Google Scholar]
- [104]. Chen X, Man GCW, Liu Y, et al. Physiological and pathological angiogenesis in endometrium at the time of embryo implantation. Am J Reprod Immunol. 2017;78:e12693. [DOI] [PubMed] [Google Scholar]
- [105]. Pijnenborg R, Vercruysse L, Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. 2006;27:939–958. [DOI] [PubMed] [Google Scholar]
- [106]. Ao M, Miyauchi M, Furusho H, et al. Dental infection of Porphyromonas gingivalis induces preterm birth in mice. PLoS One. 2015;10:e0137249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107]. Miyoshi H, Konishi H, Teraoka Y, et al. Enhanced expression of contractile-associated proteins and ion channels in preterm delivery model mice with chronic odontogenic Porphyromonas gingivalis infection. Reprod Sci. 2016;23:838–846. [DOI] [PubMed] [Google Scholar]
- [108]. Michelin M, Teixeira S, Ando-Suguimoto E, et al. Porphyromonas gingivalis infection at different gestation periods on fetus development and cytokines profile. Oral Dis. 2012;18:648–654. [DOI] [PubMed] [Google Scholar]