

Abstract
Primary biliary cholangitis (PBC) is considered a model autoimmune disease due to its signature anti‐mitochondrial antibody (AMA) autoantibody, female predominance, and relatively specific portal infiltration and cholestasis. The identification and cloning of the major mitochondrial autoantigens recognized by AMA have served as an immunologic platform to identify the earliest events involved in loss of tolerance. Despite the relatively high concordance rate in identical twins, genome‐wide association studies have not proven clinically useful and have led to suggestions of epigenetic events. To understand the natural history and etiology of PBC, several murine models have been developed, including spontaneous models, models induced by chemical xenobiotic immunization, and by “designer” mice with altered interferon metabolism. Herein, we describe five such models, including 1) NOD.c3c4 mice, 2) dominant negative form of transforming growth factor receptor type II mice, 3) interleukin‐2R α−/− mice, 4) adenylate‐uridylate‐rich element Del−/− mice, and 5) 2‐octynoic acid‐conjugated bovine serum albumin immunized mice. Individually there is no perfect murine model, but collectively the models point to loss of tolerance to PDC‐E2, the major mitochondrial autoantigen, as the earliest event that occurs before clinical disease is manifest. Although there is no direct association of AMA titer and PBC disease progression, it is noteworthy that the triad of PBC monocytes, biliary apotopes, and AMA leads to an intense proinflammatory cytokine burst. Further, the recurrence of PBC after liver transplantation indicates that, due to major histocompatibility complex restriction, disease activity must include not only adaptive immunity but also innate immune mechanisms. We postulate that successful treatment of PBC may require a personalized approach with therapies designed for different stages of disease. (Hepatology Communications 2017;1:275–287)
Abbreviations
- 2‐OA
2‐octynoic acid
- 2‐OADC
2‐oxo acid dehydrogenase
- AMA
antimitochondrial antibodies
- APC
antigen‐presenting cell
- ARE
adenylate‐uridylate‐rich element
- BECs
biliary epithelial cells
- dnTGFβRII
dominant negative form of transforming growth factor β receptor type II
- HLA
human leukocyte antigen
- IFN
interferon
- IL
interleukin
- LA
lipoic acid
- NOD
nonobese diabetic
- PBC
primary biliary cholangitis
- PDC‐E2
E2 subunit of pyruvate dehydrogenase
- TGF
transforming growth factor
- Th1
T helper 1
- UDCA
ursodeoxycholic acid
Introduction
Primary biliary cholangitis (PBC) is a chronic cholestatic liver disease characterized by immune‐mediated destruction of small and medium‐sized intrahepatic bile ducts.1, 2 PBC predominantly affects women in their fifth and sixth decades of life, with a female to male ratio of 10:1. The serologic diagnostic hallmark of PBC is detection of anti‐mitochondrial antibodies (AMAs) targeting the 2‐oxo acid dehydrogenase complex (2‐OADC) enzymes located in the inner lipoyl domain of the mitochondria.3, 4 Typical histologic findings of PBC include dense infiltration of mononuclear cells in the vicinity of small or medium‐sized intrahepatic bile ducts, known as chronic nonsuppurative destructive cholangitis.5, 6 Numerous previous studies have suggested that immunologic activity against small biliary epithelial cells (BECs) leads to clinical disease.7, 8, 9, 10 In PBC, as with other autoimmune diseases, both genetic and environmental factors contribute to the development of pathology11, 12, 13, 14; however, the precise etiology of this disease remains unclear.15, 16
While the introduction of ursodeoxycholic acid (UDCA) for the treatment of PBC greatly improved the outcome,17 nearly 30% of patients treated with UDCA show an incomplete response and disease progression.18, 19, 20 Recently, obeticholic acid, a selective ligand of the farnesoid X receptor, was approved for patients who are refractory to UDCA.21 However, the efficacy of obeticholic acid is still suboptimal, and additional therapeutic approaches are urgently needed.7, 22, 23, 24, 25, 26
Our laboratory identified mitochondrial autoantigens recognized by AMAs as 2‐OADC in 1987.27 Since then, we have intensively investigated the etiology of PBC, a prototype organ‐specific autoimmune disease (Fig. 1). In this review, we assess our results regarding the etiology of PBC.
Figure 1.

Toward solving the etiological mystery.
Definition of AMA Epitopes
AMAs are the most disease‐specific autoantibodies in human immunopathology and are detected in 90%‐95% of patients with PBC.28, 29 A high titer of autoantibody in the sera of patients with PBC was observed by Mackay more than 60 years ago,30 and AMA was found to be an effective serologic tool for the diagnosis of PBC.31 However, the immunodominant epitopes of AMA were not determined until the identification of pyruvate dehydrogenase complex E2 subunit (PDC‐E2) as the mitochondrial autoantigen of PBC by complementary DNA cloning.27 2‐OADC, a family of mitochondrial enzymes located in the inner membrane of mitochondria, are targets of AMA and include PDC‐E2, branched chain 2‐oxo‐acid dehydrogenase complex E2, 2‐oxo‐glutarate dehydrogenase complex E2, and dihydrolipoamide dehydrogenase binding protein.32 All these E2 enzymes have a common structure consisting of an N‐terminal domain with a single or multiple attachment sites to lysine (173K in mammalian PDC‐E2) of lipoic acid (LA). The dominant epitope sites recognized by AMA are in contiguity with the LA attachment site(s) as the lipoyl domains of these target antigens.33, 34, 35 The amino acid residues critical to maintaining the structural integrity of PDC‐E2 lipoyl domain have been revealed by site‐directed mutagenesis.36 Furthermore, while AMA is typically not found in other liver diseases or autoimmune diseases, a positive AMA in otherwise healthy individuals indicates a substantial risk of future PBC development.37, 38, 39 These findings suggest that AMAs are not simply serologic markers for diagnosis but are important in the immunopathology of PBC.
Overlapping of T‐Cell Epitopes
The histologic signature of PBC includes a dense infiltration of mononuclear cells in the portal tracts near small or medium‐sized bile ducts. In the typical pathology of PBC, infiltrated lymphocytes are found adjacent to BECs in damaged bile ducts.15 Immunohistochemical examination of these lymphocytes reveals a predominance of CD4+ and CD8+ T cells with B and natural killer cells.40, 41 BECs and hepatocytes in the liver of patients with PBC also express large amounts of human leukocyte antigen (HLA) class I and II molecules.42, 43 Therefore, both CD4+ and CD8+ autoreactive T cells play a crucial role in the pathogenesis of PBC.
In the case of CD4+ T cells, Shimoda et al.44 established HLA DRB4 0101‐restricted PDC‐E2‐specific T‐cell clones from the peripheral blood of patients with PBC and mapped immunodominant T‐cell epitopes as PDC‐E2 peptide 163‐176 (GDLLAEIETDKATI), which overlapped with the B‐cell epitope of human PDC‐E2 (Table 1). Importantly, the frequency of PDC‐E2‐specific CD4+ T cells was 100‐fold to 150‐fold higher in the liver and hilar lymph nodes than in the peripheral blood.45 Our laboratory also characterized a major histocompatibility complex class I (HLA‐A2)‐restricted epitope for CD8+ T cells as PDC‐E2 peptide 159‐167 (KLSEGDLLA), which again mapped to the same region of the inner lipoyl domain as the autoantigen PDC‐E2 (Table 1).46 The frequency of CD8+ cytotoxic T lymphocytes specific for this epitope was 10‐fold higher in the liver than in the blood.40 Interestingly, cytotoxic T lymphocyte cell lines specific for PDC‐E2 can be efficiently generated from peripheral blood mononuclear cells of patients with PBC by using soluble PDC‐E2 complexed with anti‐PDC‐E2 autoantibodies, which results from cross‐presentation of the PDC‐E2 epitope by antigen‐presenting cells (APCs).46 The finding that autoantigen‐immune complexes can cross‐present the autoantigen with high efficiency reveals a unique role for anti‐PDC‐E2 antibodies in the pathogenesis of PBC.
Table 1.
OVERLAPPING EPITOPES OF HUMAN PDC‐E2 RECOGNIZED BY B CELLS, CD4+ T CELLS, AND CD8+ T CELLS
| 155 | 160 | 165 | 170 | 175 | 180 | 185 | |
|---|---|---|---|---|---|---|---|
| B cell | KVGEKLSEGDLLAEIETDK*ATIGFEVQEEGY | ||||||
| CD4+ T cell | KVGEKLSEGDLLAEIETDK*ATIGFEVQEEGY | ||||||
| CD8+ T cell | KVGEKLSEGDLLAEIETDK*ATIGFEVQEEGY | ||||||
Bold characters define an epitope on the PDC‐E2 recognized by each cell type.
173K*, attachment site of lipoic acid.
Why Biliary Epithelial Cells? The “ABC” of PBC
We determined that AMA and autoreactive helper and cytotoxic T cells contain a shared peptide sequence of the human PDC‐E2 (Table 1). However, PDC‐E2 is a ubiquitous protein located in nearly all nucleated cells in the human body. Why do autoreactive T cells specific for PDC‐E2 elicit cytotoxicity against only biliary epithelial cells? We should note that PBC recurs even after liver transplantation, indicating that the immunopathologic susceptibility of BECs in PBC is not major histocompatibility complex‐specific, but a general feature shared with autologous BECs. To answer this question, we demonstrated that only human intrahepatic BECs could maintain PDC‐E2 immunologically intact within apoptotic blebs (apotopes) during apoptosis but not control epithelial cells. This supports data in which AMA‐containing sera reacted with PDC‐E2 on apoptotic BECs without permeabilization.47 We then examined the ability of BECs to induce cytokine secretion from mature monocyte‐derived macrophages, with and without AMAs and observed intense inflammatory cytokine production in the presence of a unique triad consisting of BEC apotopes, macrophages from patients with PBC, and AMAs.8 Macrophages from healthy controls did not produce inflammatory cytokines even when cocultured with apoptotic bodies from human intrahepatic BECs and AMAs (Fig. 2). Thus, we propose that A (AMA, apotope, and APC), B (blebs from apoptotic BECs), and C (complex formation and cytokine secretion) constitute the crucial triad in the inflammatory cascade of PBC.
Figure 2.
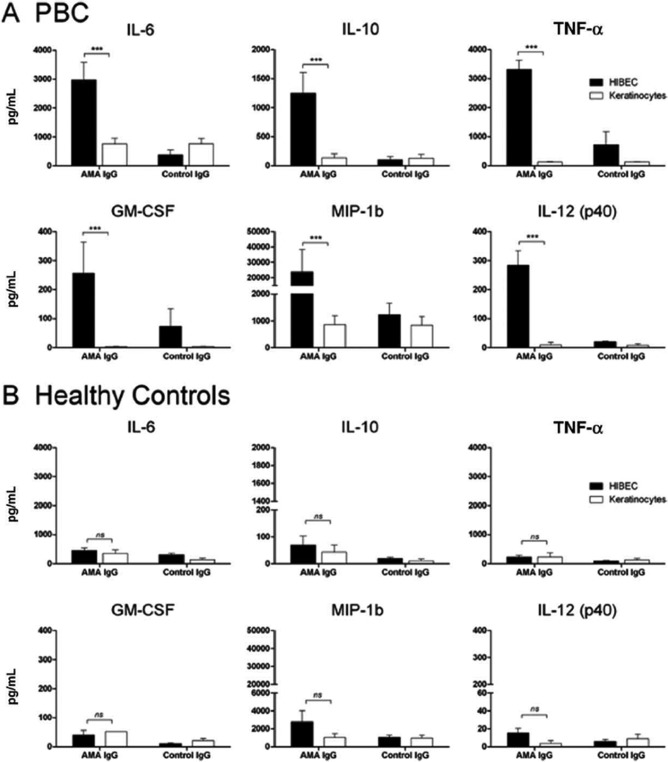
Macrophages from patients with PBC and from healthy controls. (A) Macrophages from patients with PBC cocultured with apoptotic bodies from HIBECs, and secreted proinflammatory cytokines in the presence of AMAs. (B) Macrophages from healthy controls did not produce inflammatory cytokines, even when cocultured with apoptotic bodies from HIBECs in the presence of AMAs. Abbreviations: GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; HIBECs, human intrahepatic BECs; IL‐6, interleukin 6; IL‐10, interleukin ‐10; IL12p40; interleukin 12 p40 subunit; MIP‐1b, MIP, macrophage inflammatory protein 1b; TNF‐α, tumor necrosis factor alpha. Error bar represents ± standard error of the mean. Level of significance is denoted as ***P < 0.001; two tailed Mann‐Whitney test with 95% CI. Reproduced with permission, Lleo et al., Hepatology 2010;52:987‐998.8
Genetic and Environmental Influences: Xenobiotic Modification of PDC‐E2
As with many other autoimmune diseases, genetic factors are known to play a decisive role in conferring susceptibility to PBC. The prevalence of PBC is higher in families with an affected member.48 High concordance rates among monozygotic twins have also been observed.49, 50 To identify the genetic background and immunologic pathways responsible for PBC development, multiple genome‐wide association studies have been conducted in several populations.51, 52, 53, 54, 55, 56 Although differences in several genes were reported, their clinical implications and relevance remain elusive. In fact, in PBC and all other autoimmune diseases, the results of genome‐wide association studies have been disappointing, and recent efforts have been directed to both deep sequencing and consideration of epigenetic events.57, 58, 59, 60, 61, 62, 63, 64
Despite the importance of genetics, environmental influences should not be underestimated in their ability to trigger or exacerbate PBC. We previously reported a large‐scale epidemiological study that evaluated risk factors and comorbidities in PBC, including an interview‐based study of 1,032 patients and controls matched for number, sex, age, race, and geographic location. In families with a first‐degree relative with PBC, history of urinary tract infections, past cigarette smoking, use of reproductive hormone replacement, and to a minor degree, frequent use of nail polish were associated with increased risk in PBC.65 Other studies suggested significant clustering of patients with PBC, particularly surrounding toxic sites.66, 67 These epidemiological data, along with the crucial role of AMA in the immunopathology of PBC, prompted us to search for environmental mimotopes in the form of xenobiotics.
We performed a detailed, quantitative, structure‐activity relationship analysis on xenobiotics. We found 107 potential xenobiotic mimics that coupled to the lysine residue of the immunodominant 15‐amino acid peptide of the PDC‐E2 inner lipoyl domain. PBC sera were more reactive with a number of xenobiotic‐modified PDC‐E2 peptides than with the native lipoylated peptide. Among them, 2‐octynamide, the conjugate derived from 2‐octynoic acid (2‐OA) present in cosmetics, lipsticks, and some chewing gums, was unique in both its quantitative structure‐activity relationship analysis and reactivity.68 We evaluated the chemical structure that leads to enhanced AMA recognition and found that 2‐nonyamide provided an optimal chemical structure of the xenobiotic‐modified epitope recognized by AMA‐positive PBC sera (Fig. 3).69 Indeed, significant molecular mimicry between lipoamide and 2‐nonynamide was demonstrated (Fig. 4). These findings illustrate that xenobiotic modification of PDC‐E2 with chemicals abundant in daily life plays a role in generating immunogenic neoantigens and breaking tolerance in PBC.
Figure 3.
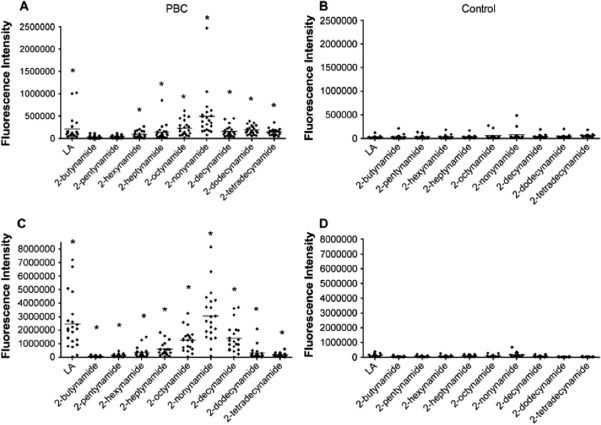
Reactivity of 2‐alkynamide‐modified PDC‐E2 peptide with (A,C) PBC sera and (B,D) healthy control sera were analyzed by microarray for IgG (A,B) and IgM (C,D) reactivity against PDC‐E2 peptide modified with 2‐alkynoic acids. Reproduced with permission, Rieger et al., J Autoimmun 2006;27:7‐16.69 Abbreviation: Ig, immunoglobulin. Level of significance is denoted as ***P < 0.05 (unpaired t‐test); horizontal bar indicates mean of pixel intensity.
Figure 4.
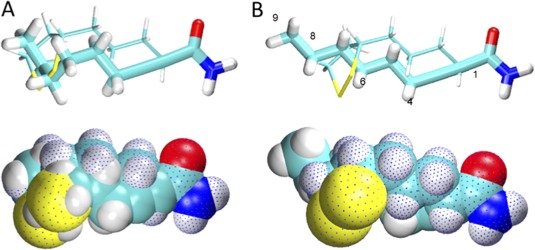
Molecular mimicry between lipoamide and 2‐nonynamide. Superimposed models of lipoamide (dotted) and 2‐nonynamide in space‐filled and bond representations with 2‐nonynamide in either (A) “corkscrew” or (B) straight chain conformation. Reproduced with permission, Rieger et al., J Autoimmun 2006; 27:7‐16.69
Efforts to Establish Mouse Models of PBC
In addition to in vitro studies, mouse models are important for understanding the etiology and natural history of PBC. Patients newly diagnosed with PBC have often past their initial phase. Animal models that reflect many important aspects of the disease are therefore needed to explore the initiating events and interactions between genetic and environmental factors. The animal model should have the same physiological mechanisms observed in human PBC, such as female predominance; chronic cholestasis; AMA production; histologic features, including lymphocyte infiltration into the liver; and bile duct involvement.
In particular, recognition of a strong gender (female) is essential to understand PBC. Sex hormones, X‐linked genes, and sex‐specific microbiota may contribute to the immune difference between male and female individuals.70, 71, 72, 73 However, the physiological mechanisms accounting for the strong female predominance in PBC remain unclear.74 PBC risk factors may function synergistically in accelerating the loss of tolerance. One theory proposes that haploinsufficiency for specific X‐linked genes leads to female susceptibility to PBC and that enhanced monosomy X in the peripheral lymphocytes of affected women induce PBC.75, 76 Data from another study suggest a potential association of PBC and the loss of the Y chromosome.77 Epigenetic analysis revealed a significant difference in the X chromosome DNA methylation profile of CD4+ T, CD8+ T, and CD14+ cells in patients with PBC, in particular with an aberrant demethylation on the CXCR3 promoter.78 However, how these genetic and environmental factors interact with the immune system to elicit autoimmunity in PBC remains enigmatic.
To date, we have established several murine models that develop autoimmune cholangitis resembling PBC in a spontaneous or xenobiotically induced manner (Table 2). These mice share some of the important clinical phenotypes of PBC.79 There are three mice models that spontaneously develop autoimmune cholangitis: nonobese diabetic (NOD).c3c4 mice, dominant negative form of transforming growth factor (TGF)‐β receptor type II (dnTGFβRII) mice, and interleukin (IL)‐2R α−/− mice. The NOD.c3c4 mice have multiple B6‐ and B10‐derived insulin‐dependent diabetes‐resistant alleles on chromosomes 3 and 4, respectively. These mice are protected from autoimmune diabetes but spontaneously develop lymphocytic peribiliary infiltrates and AMA positivity.80, 81 Notably, AMAs were detected in female mice, indicating female predominance as in human PBC. However, pathologic examination of the liver revealed biliary polycystic diseases in both the intrahepatic and extrahepatic biliary ducts and little evidence of chronic nonsuppurative destructive cholangitis. The dnTGFβRII mice also mimicked phenotypes of human PBC.82 These mice are transgenic for the expression of a dominantly negative form of TGF‐β receptor type II directed by the CD4 promoter. DnTGFβRII mice spontaneously produce AMAs directed to the same mitochondrial autoantigens as human PBC. Lymphocytic liver infiltration with periportal inflammation is analogous to the histologic profile of human PBC. The complexity of the IL‐12/IL‐23 cytokine milieu in autoimmunity in dnTGFβRII mice was examined by generating a series of cytokine knockouts: interferon (IFN)γ−/−, IL‐12p35−/−, IL‐12/IL‐23p40−/−, IL‐23p19−/−, and IL‐17A−/− dnTGFβRII mice. Collectively, our data indicated that the IL‐12/T helper type 1 (Th1) pathway is essential for biliary disease pathogenesis, and IFN‐γ production is significant for triggering Th1 cell responses in this model.83, 84, 85, 86
Table 2.
CHARACTERISTICS OF PBC MOUSE MODELS
| Spontaneous Model | Induced Model | ||||
|---|---|---|---|---|---|
| NOD.c3c4 | DnTGFβRII | IL‐2Rα−/− | ARE Del−/− | 2‐OA‐BSA Immunized | |
| Female dominance | Yes | No | No | Yes | No |
| Cholestasis | ‐ | + | ‐ | + | + |
| AMA seropositivity | 50‐60% | 100% | 100% | 100% | 100% |
| Portal inflammation | +++ | +++ | +++ | Yes | + |
| Granulomas | + | − | − | + | + |
| Other features | Biliary polycystic lesions | Moderate colitis | Severe anemia, inflammatory bowel diseases, and short life span | Peritonitis | |
Abbreviations: BSA, bovine serum albumin; −, not detected; +, present; +++, strongly present.
The third spontaneous mouse model is the IL‐2R α−/− mice, which lack the IL‐2R cytokine crucial for differentiation of regulatory T cells and their eventual reduction.87 These mice develop portal inflammation, biliary ductular damage, and a Th1 cytokine bias, resembling human PBC. In addition, AMAs are targeted to the inner lipoyl domain of PDC‐E2. However, female predominance was not observed.
We have also examined possible environmental triggers of autoimmune cholangitis in mice, particularly chemical xenobiotics. We immunized C57BL/6 mice with 2‐OA, which was suggested as a candidate xenobiotic present in the environment in our previous study,68 coupled to bovine serum albumin. We found that anti‐PDC‐E2 antibodies were present in the serum as early as 4 weeks after immunization (Fig. 5), indicating loss of tolerance to PDC‐E2 with xenobiotic immunization. In addition, these mice demonstrated portal infiltration of CD4+ and CD8+ T cells, granulomas, and elevated tumor necrosis factor‐α and IFN‐γ expression levels.88 Using several unique gene‐deleted mice immunized with 2‐OA‐bovine serum albumin,89 we also found that both IL‐12/Th1 and IL‐23/Th17 were involved in autoimmune cholangitis. The IL‐12/Th1 signaling pathway elicited the pathology, while deletion of IFN‐γ prevented autoimmune cholangitis (Fig. 6).
Figure 5.
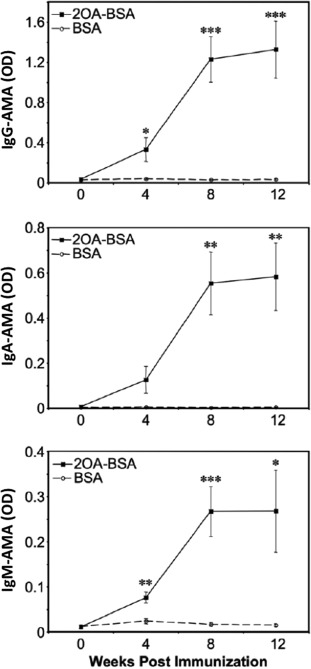
Detection and quantification of anti‐PDC‐E2 antibody in sera of 2‐OA‐BSA‐immunized mice by using enzyme‐linked immunosorbent assay at 2‐week intervals after immunization. A significant increase in OD was observed after immunization with xenobiotic modification compared to control. Reproduced with permission, Wakabayashi et al., Hepatology 48:531‐540.88 Abbreviation: BSA, bovine serum albumin; Ig, immunoglobulin; OD, optical density. Level of significance denoted as *P < 0.05, **P < 0.01, ***P < 0.001. Error bar represents ± standard deviation.
Figure 6.
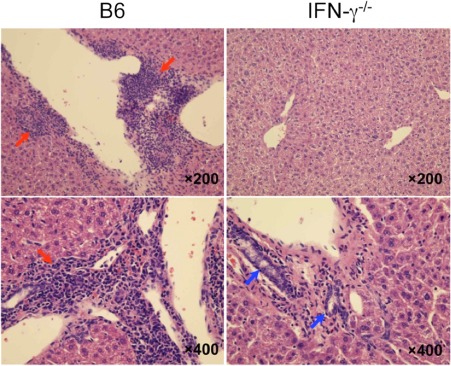
IFN‐γ knockout completely abolished autoimmune cholangitis. Portal inflammatory changes and interlobular bile duct damage (red arrows) were observed in wild‐type mice (B6 mice) and normal bile ducts in IFN‐γ−/− mice (blue arrows). Reproduced with permission Kawata et al., PLoS One 2013;8:e74225.89
Adenylate‐Uridylate‐Rich Element Del−/− Mice as a Novel PBC Model
We are currently focusing on IFN‐γ using a “designer” mouse with dysregulation of IFN‐γ, in which the adenylate‐uridylate‐rich element (ARE) of the IFN‐γ 3′‐untranslated region is deleted and IFN‐γ is constitutively produced.90 Through various assays, we found that IFN‐γ is crucial to the pathogenesis of autoimmune cholangitis in this model.89, 91 We should note that activation of naïve CD4 T cells from healthy women produces higher levels of IFN‐γ and lower levels of IL‐17 than in healthy men.92 Increased IFN‐γ levels have also been observed in patients with autoimmune diseases.70, 93
ARE Del−/− mice spontaneously developed many manifestations similar to human PBC, including nonsuppurative destructive cholangitis, AMA production, and elevated serum total bile acid levels.94 These features were also found predominantly in female mice. In male ARE Del−/− mice, portal inflammation was rarely observed and serum titers of AMA were elevated but not significantly compared to wild‐type mice (Fig. 7). Total bile acid levels were comparable. In addition, gene expression analysis revealed that up‐regulated genes in female ARE Del−/− mice specifically overlapped with the gene expression signature of BECs in human PBC. Therefore, female ARE Del−/− mice closely mimic human PBC.95
Figure 7.
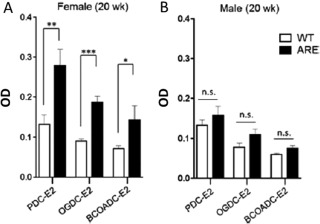
AMA levels in ARE Del−/− (A) female and (B) male mice at 20 weeks. Abbreviations: BCOADC‐E2, E2 component of branched chain 2‐oxo‐acid dehydrogenase complex; n.s., not significant; OGDC‐E2, E2 component of 2‐oxo‐acid dehydrogenase complex; WT, wild‐type. Error bar represents ± standard deviation. All data are representative of at least three independent experiments. Level of significance is denoted as *P < 0.05; **P < 0.01; ***P < 0.001 (unpaired student t‐test). Reproduced with permission, Bae et al., Hepatology 2016;64:1189‐1201.94
Female predominance occurs in ARE Del−/− mice likely because female hormones and genetics cause immune cells in female mice to favor production of additional IFN‐γ‐producing cells. In contrast, male mice may be protected by androgens, which favor up‐regulation of regulatory cells and down‐regulation of IFN‐γ‐producing cells. Female hormones activate T lymphocytes to express higher levels of IFN‐γ in female mice in this mouse model. Although numerous murine spontaneous and induced models have been reported as PBC mouse models,79, 85 no single model exhibits female dominance as observed in ARE Del−/− mice.
ARE Del−/− mice also provide clues regarding the immunopathology of PBC. IFN‐γ may play a pathogenic role in BECs during the initiation stage of PBC, and changes in expression levels of IFN‐γ are critical to the development of PBC in susceptible individuals. Furthermore, we demonstrated that transfer of CD4 T cells from ARE Del−/− mice to B5/Rag1−/− mice (an immune‐deficient strain producing no mature T or B cells) induced moderate portal and parenchymal inflammation, indicating that CD4+ T cells contribute to the induction of cholangitis.
Using ARE Del−/− mice, we learned the following: First, this is a female‐predominant model for PBC that allows for intensive investigation of the female predominance of the disease. We comprehensively analyzed sexual dimorphic physiological systems, including hormones, immune differences, and the microbiome. We can manipulate estrogen and estrogen receptors on cells to determine their effect on the pathology. We also examined the female microbiome and found that it was similar to the male microbiome; we can raise these mice in a germ‐free environment, followed by the introduction of a selected set of bacteria. Second, this model will help determine which gene pathways help or hinder the progress of PBC in both sexes. Third, this model allows us to study the interaction of multiple cellular messengers and their relationships with cellular immune transformation to the autoimmune state. Finally, this model has implications for clinical research. The heterogeneity and the natural history of PBC can be examined from an early asymptomatic stage to a late stage with full liver involvement, and new biomarkers at different stages may be identified. This mouse is an effective tool for assessing the effect of drugs currently used and those under development and for the design and screening of novel more‐effective compounds for curing PBC.
Where We Are and Future Directions
Since the cloning and identification of PDC‐E2 as the major mitochondrial autoantigen of PBC in 1987, our understanding of the immunologic mechanisms, pathologic process, genetics, etiology, and natural history of PBC has significantly increased.4, 7, 8, 15, 16, 65, 79, 91, 96, 97, 98, 99, 100 The vast amount of information gained from these studies points to the thesis that the loss of tolerance to the PDC‐E2 lipoyl domain is a linchpin in the development of the biliary pathology of PBC.
To help understand these events, Fig. 8 illustrates a hypothetical pathway from tolerance breakdown to biliary pathology in PBC. BECs express PDC‐E2 on apotopes in an immunologically intact form during apoptosis. Notably, the capability to do this is observed only in BECs and not in other epithelial cells, possibly explaining the tissue specificity of PBC. Intact lipoylated PDC‐E2, presumably after modification with xenobiotics, such as 2‐octynamide or 2‐nonyamide, from the environment are endocytosed by APCs and presented to CD4+ or CD8+ T cells. Immune complexes with PDC‐E2 and anti‐PDC‐E2 autoantibody cross‐present autoantigens in a more efficient manner. Finally, orchestrated immunologic responses against BECs with activated CD4+ and CD8+ T cells, AMA, and immunoglobulin A transcytosis result in subsequent pathology. We currently have ARE Del−/− model mice that mimic human PBC, including female dominance, and will use this model to elucidate the immunopathology of PBC. Expanding our knowledge regarding this pathology from a very early stage of disease will provide a foundation for “curing” PBC.
Figure 8.
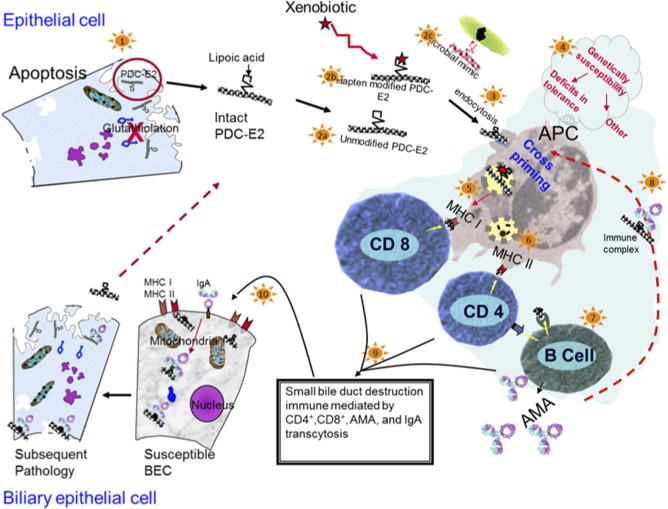
Tolerance breakdown against PDC‐E2 expressed on apotopes through xenobiotic modification, leading to activation of autoreactive T cells and cholangitis in PBC. Abbreviations: Ig, immunoglobulin; MHC, major histocompatibility complex.
Authors in bold designate shared co‐first authorship.
Potential conflict of interest: Nothing to report.
Supported in part by National Institutes of Health grants DK39588, DK090019, and DK067003.
REFERENCES
- 1. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. New Engl J Med 2005;353:1261‐1273. [DOI] [PubMed] [Google Scholar]
- 2. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ; American Association for Study of Liver Diseases . Primary biliary cirrhosis. Hepatology 2009;50:291‐308. [DOI] [PubMed] [Google Scholar]
- 3. Leung P, Coppel R, Ansari A, Munoz S, Gershwin M. Antimitochondrial antibodies in primary biliary cirrhosis. Semin Liver Dis 1997;17:61‐69. [DOI] [PubMed] [Google Scholar]
- 4. Leung PS, Choi J, Yang G, Woo E, Kenny TP, Gershwin ME. A contemporary perspective on the molecular characteristics of mitochondrial autoantigens and diagnosis in primary biliary cholangitis. Expert Rev Mol Diagn 2016;16:697‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DE, Lindor K, et al. Changing nomenclature for PBC: from ‘cirrhosis’ to ‘cholangitis’. Hepatology 2015;62:1620‐1622. [DOI] [PubMed] [Google Scholar]
- 6. Rubin E, Schaffner F, Popper H. Primary biliary cirrhosis. Chronic non‐suppurative destructive cholangitis. Am J Pathol 1965;46:387‐407. [PMC free article] [PubMed] [Google Scholar]
- 7. Hisamoto S, Shimoda S, Harada K, Iwasaka S, Onohara S, Chong Y, et al. Hydrophobic bile acids suppress expression of AE2 in biliary epithelial cells and induce bile duct inflammation in primary biliary cholangitis. J Autoimmun 2016;75:150‐160. [DOI] [PubMed] [Google Scholar]
- 8. Lleo A, Bowlus CL, Yang GX, Invernizzi P, Podda M, Van de Water J, et al. Biliary apotopes and anti‐mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 2010;52:987‐998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rong G, Zhong R, Lleo A, Leung PS, Bowlus CL, Yang GX, et al. Epithelial cell specificity and apotope recognition by serum autoantibodies in primary biliary cirrhosis. Hepatology 2011;54:196‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rong GH, Yang GX, Ando Y, Zhang W, He XS, Leung PS, et al. Human intrahepatic biliary epithelial cells engulf blebs from their apoptotic peers. Clin Exp Immunol 2013;172:95‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hardtke‐Wolenski M, Dywicki J, Fischer K, Hapke M, Sievers M, Schlue J, et al. The influence of genetic predisposition and autoimmune hepatitis inducing antigens in disease development. J Autoimmun 2017;78:39‐45. [DOI] [PubMed] [Google Scholar]
- 12. Hirschfield GM, Siminovitch KA. Genetics in PBC: what do the “risk genes” teach us? Clin Rev Allergy Immunol 2015;48:176‐181. [DOI] [PubMed] [Google Scholar]
- 13. Kerstein A, Schuler S, Cabral‐Marques O, Fazio J, Hasler R, Muller A, et al. Environmental factor and inflammation‐driven alteration of the total peripheral T‐cell compartment in granulomatosis with polyangiitis. J Autoimmun 2017;78:79‐91. [DOI] [PubMed] [Google Scholar]
- 14. Nielsen PR, Kragstrup TW, Deleuran BW, Benros ME. Infections as risk factor for autoimmune diseases ‐ a nationwide study. J Autoimmun 2016;74:176‐181. [DOI] [PubMed] [Google Scholar]
- 15. Gershwin ME, Mackay IR. The causes of primary biliary cirrhosis: convenient and inconvenient truths. Hepatology 2008;47:737‐745. [DOI] [PubMed] [Google Scholar]
- 16. Zhang H, Carbone M, Lleo A, Invernizzi P. Geoepidemiology, genetic and environmental risk factors for PBC. Dig Dis 2015;33 Suppl 2:94‐101. [DOI] [PubMed] [Google Scholar]
- 17. Poupon RE, Poupon R, Balkau B. Ursodiol for the long‐term treatment of primary biliary cirrhosis. The UDCA‐PBC Study Group. N Engl J Med 1994;330:1342‐1347. [DOI] [PubMed] [Google Scholar]
- 18. Ali AH, Lindor KD. Obeticholic acid for the treatment of primary biliary cholangitis. Expert Opin Pharmacother 2016;17:1809‐1815. [DOI] [PubMed] [Google Scholar]
- 19. de Vries E, Beuers U. Management of cholestatic disease in 2017. Liver Int 2017;37 Suppl 1:123‐129. [DOI] [PubMed] [Google Scholar]
- 20. Hirschfield GM, Gershwin ME, Strauss R, Mayo MJ, Levy C, Zou B, et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: a proof‐of‐concept study. Hepatology 2016;64:189‐199. [DOI] [PubMed] [Google Scholar]
- 21. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 22. He XS, Gershwin ME, Ansari AA. Checkpoint‐based immunotherapy for autoimmune diseases ‐ opportunities and challenges. J Autoimmun 2017;79:1‐3. [DOI] [PubMed] [Google Scholar]
- 23. Mousa HS, Carbone M, Malinverno F, Ronca V, Gershwin ME, Invernizzi P. Novel therapeutics for primary biliary cholangitis: toward a disease‐stage‐based approach. Autoimmun Rev 2016;15:870‐876. [DOI] [PubMed] [Google Scholar]
- 24. Choi J, Selmi C, Leung PS, Kenny TP, Roskams T, Gershwin ME. Chemokine and chemokine receptors in autoimmunity: the case of primary biliary cholangitis. Expert Rev Clin Immunol 2016;12:661‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang JB, Wang YH, Yang W, Lu FT, Ma HD, Zhao ZB, et al. Successful treatment of murine autoimmune cholangitis by parabiosis: implications for hematopoietic therapy. J Autoimmun 2016;66:108‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Syu BJ, Loh CE, Hsueh YH, Gershwin ME, Chuang YH. Dual roles of IFN‐gamma and IL‐4 in the natural history of murine autoimmune cholangitis: IL‐30 and implications for precision medicine. Sci Rep 2016;6:34884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gershwin M, Mackay I, Sturgess A, Coppel R. Identification and specificity of a cDNA encoding the 70 kd mitochondrial antigen recognized in primary biliary cirrhosis. J Immunol 1987;138:3525‐3531. [PubMed] [Google Scholar]
- 28. Van de Water J, Gershwin M, Leung P. The autoepitope of the 74‐kD mitochondrial autoantigen of primary biliary cirrhosis corresponds to the functional site of dihydrolipoamide acetyltransferase. J Exp Med 1988;167:1791‐1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oertelt S, Rieger R, Selmi C, Invernizzi P, Ansari AA, Coppel RL, et al. A sensitive bead assay for antimitochondrial antibodies: chipping away at AMA‐negative primary biliary cirrhosis. Hepatology 2007;45:659‐665. [DOI] [PubMed] [Google Scholar]
- 30. Mackay IR. Primary biliary cirrhosis showing a high titer of autoantibody; report of a case. N Engl J Med 1958;258:185‐188. [DOI] [PubMed] [Google Scholar]
- 31. Klatskin G, Kantor FS. Mitochondrial antibody in primary biliary cirrhosis and other diseases. Ann Intern Med 1972;77:533‐541. [DOI] [PubMed] [Google Scholar]
- 32. Dubel L, Tanaka A, Leung P, Van de Water J, Coppel R, Roche T, et al. Autoepitope mapping and reactivity of autoantibodies to the dihydrolipoamide dehydrogenase‐binding protein (E3BP) and the glycine cleavage proteins in primary biliary cirrhosis. Hepatology 1999;29:1013‐1018. [DOI] [PubMed] [Google Scholar]
- 33. Leung P, Chuang D, Wynn R, Cha S, Danner D, Ansari A, et al. Autoantibodies to BCOADC‐E2 in patients with primary biliary cirrhosis recognize a conformational epitope. Hepatology 1995;22:505‐513. [PubMed] [Google Scholar]
- 34. Moteki S, Leung P, Dickson E, Van Thiel D, Galperin C, Buch T, et al. Epitope mapping and reactivity of autoantibodies to the E2 component of 2‐oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2‐oxoglutarate dehydrogenase complex. Hepatology 1996;23:436‐444. [DOI] [PubMed] [Google Scholar]
- 35. Surh C, Coppel R, Gershwin M. Structural requirement for autoreactivity on human pyruvate dehydrogenase‐E2, the major autoantigen of primary biliary cirrhosis. Implication for a conformational autoepitope. J Immunol 1990;144:3367‐3374. [PubMed] [Google Scholar]
- 36. Wang J, Budamagunta MS, Voss JC, Kurth MJ, Lam KS, Lu L, et al. Antimitochondrial antibody recognition and structural integrity of the inner lipoyl domain of the E2 subunit of pyruvate dehydrogenase complex. J Immunol 2013;191:2126‐2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mattalia A, Quaranta S, Leung P, Bauducci M, Van de Water J, Calvo P, et al. Characterization of antimitochondrial antibodies in health adults. Hepatology 1998;27:656‐661. [DOI] [PubMed] [Google Scholar]
- 38. Metcalf J, Mitchison H, Palmer J, Jones D, Bassendine M, James O. Natural history of early primary biliary cirrhosis. Lancet 1996;348:1399‐1402. [DOI] [PubMed] [Google Scholar]
- 39. Yang F, Wang Q, Wang Z, Miao Q, Xiao X, Tang R, et al. The natural history and prognosis of primary biliary cirrhosis with clinical features of autoimmune hepatitis. Clin Rev Allergy Immunol 2016;50:114‐123. [DOI] [PubMed] [Google Scholar]
- 40. Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van de Water J, et al. Quantitative and functional analysis of PDC‐E2‐specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest 2002;109:1231‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shimoda S, Harada K, Niiro H, Shirabe K, Taketomi A, Maehara Y, et al. Interaction between Toll‐like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology 2011;53:1270‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bjorkland A, Festin R, Mendel‐Hartvig I, Nyberg A, Loof L, Totterman TH. Blood and liver‐infiltrating lymphocytes in primary biliary cirrhosis: increase in activated T and natural killer cells and recruitment of primed memory T cells. Hepatology 1991;13:1106‐1111. [DOI] [PubMed] [Google Scholar]
- 43. Krams SM, Van de Water J, Coppel RL, Esquivel C, Roberts J, Ansari A, et al. Analysis of hepatic T lymphocyte and immunoglobulin deposits in patients with primary biliary cirrhosis. Hepatology 1990;12:306‐313. [DOI] [PubMed] [Google Scholar]
- 44. Shimoda S, Nakamura M, Ishibashi H. HLA DRB4 0101‐restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune disease. J Exp Med 1995;181:1835‐1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimoda S, Van de Water J, Ansari A, Nakamura M, Ishibashi H, Coppel R, et al. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J Clin Invest 1998;102:1831‐1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kita H, Lian ZX, Van de Water J, He XS, Matsumura S, Kaplan M, et al. Identification of HLA‐A2‐restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross‐presented by dendritic cells. J Exp Med 2002;195:113‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lleo A, Selmi C, Invernizzi P, Podda M, Coppel RL, Mackay IR, et al. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009;49:871‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bach N, Schaffner F. Familial primary biliary cirrhosis. J Hepatol 1994;20:698‐701. [DOI] [PubMed] [Google Scholar]
- 49. Selmi C, Mayo MJ, Bach N, Ishibashi H, Invernizzi P, Gish RG, et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: genetics, epigenetics, and environment. Gastroenterology 2004;127:485‐492. [DOI] [PubMed] [Google Scholar]
- 50. Webb GJ, Siminovitch KA, Hirschfield GM. The immunogenetics of primary biliary cirrhosis: a comprehensive review. J Autoimmun 2015;64:42‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cordell HJ, Han Y, Mells GF, Li Y, Hirschfield GM, Greene CS, et al. International genome‐wide meta‐analysis identifies new primary biliary cirrhosis risk loci and targetable pathogenic pathways. Nat Commun 2015;6:8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hirschfield GM, Liu X, Han Y, Gorlov IP, Lu Y, Xu C, et al. Variants at IRF5‐TNPO3, 17q12‐21 and MMEL1 are associated with primary biliary cirrhosis. Nat Genet 2010;42:655‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kar SP, Seldin MF, Chen W, Lu E, Hirschfield GM, Invernizzi P, et al. Pathway‐based analysis of primary biliary cirrhosis genome‐wide association studies. Genes Immun 2013;14:179‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu X, Invernizzi P, Lu Y, Kosoy R, Lu Y, Bianchi I, et al. Genome‐wide meta‐analyses identify three loci associated with primary biliary cirrhosis. Nat Genet 2010;42:658‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mells GF, Floyd JA, Morley KI, Cordell HJ, Franklin CS, Shin SY, et al. Genome‐wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat Genet 2011;43:329‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, et al. Genome‐wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet 2012;91:721‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Meroni PL, Penatti AE. Epigenetics and systemic lupus erythematosus: unmet needs. Clin Rev Allergy Immunol 2016;50:367‐376. [DOI] [PubMed] [Google Scholar]
- 58. Pollock RA, Abji F, Gladman DD. Epigenetics of psoriatic disease: a systematic review and critical appraisal. J Autoimmun 2017;78:29‐38. [DOI] [PubMed] [Google Scholar]
- 59. Xie YQ, Ma HD, Lian ZX. Epigenetics and primary biliary cirrhosis: a comprehensive review and implications for autoimmunity. Clin Rev Allergy Immunol 2016;50:390‐403. [DOI] [PubMed] [Google Scholar]
- 60. Shu Y, Hu Q, Long H, Chang C, Lu Q, Xiao R. Epigenetic variability of CD4+CD25+ Tregs contributes to the pathogenesis of autoimmune diseases. Clin Rev Allergy Immunol 2017;52:260‐272. [DOI] [PubMed] [Google Scholar]
- 61. Ricano‐Ponce I, Zhernakova DV, Deelen P, Luo O, Li X, Isaacs A, et al. Refined mapping of autoimmune disease associated genetic variants with gene expression suggests an important role for non‐coding RNAs. J Autoimmun 2016;68:62‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao LP, Alshiekh S, Zhao M, Carlsson A, Larsson HE, Forsander G, et al. Next‐generation sequencing reveals that HLA‐DRB3, ‐DRB4, and ‐DRB5 may be associated with islet autoantibodies and risk for childhood type 1 diabetes. Diabetes 2016;65:710‐718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Long H, Yin H, Wang L, Gershwin ME, Lu Q. The critical role of epigenetics in systemic lupus erythematosus and autoimmunity. J Autoimmun 2016;74:118‐138. [DOI] [PubMed] [Google Scholar]
- 64. Marzorati S, Lleo A, Carbone M, Gershwin ME, Invernizzi P. The epigenetics of PBC: the link between genetic susceptibility and environment. Clin Res Hepatol Gastroenterol 2016;40:650‐659. [DOI] [PubMed] [Google Scholar]
- 65. Gershwin ME, Selmi C, Worman HJ, Gold EB, Watnik M, Utts J, et al. Risk factors and comorbidities in primary biliary cirrhosis: a controlled interview‐based study of 1032 patients. Hepatology 2005;42:1194‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ala A, Stanca CM, Bu‐Ghanim M, Ahmado I, Branch AD, Schiano TD, et al. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology 2006;43:525‐531. [DOI] [PubMed] [Google Scholar]
- 67. Jones D, Watt F, Metcalf J, Bassendine M, James O. Familial primary biliary cirrhosis reassesed: a geographically‐based population study. J Hepatol 1999;30:402‐407. [DOI] [PubMed] [Google Scholar]
- 68. Amano K, Leung P, Rieger R, Quan C, Wang X, Marik J, et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: identification of antibodies against a common environmental, cosmetic, and food additive, 2‐octynoic acid. J Immunol 2005;174:5874‐5883. [DOI] [PubMed] [Google Scholar]
- 69. Rieger R, Leung PS, Jeddeloh MR, Kurth MJ, Nantz MH, Lam KS, et al. Identification of 2‐nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J Autoimmun 2006;27:7‐16. [DOI] [PubMed] [Google Scholar]
- 70. Rubtsova K, Marrack P, Rubtsov AV. TLR7, IFNgamma, and T‐bet: their roles in the development of ABCs in female‐biased autoimmunity. Cell Immunol 2015;294:80‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fish EN. The X‐files in immunity: sex‐based differences predispose immune responses. Nat Rev Immunol 2008;8:737‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Markle JG, Frank DN, Mortin‐Toth S, Robertson CE, Feazel LM, Rolle‐Kampczyk U, et al. Sex differences in the gut microbiome drive hormone‐dependent regulation of autoimmunity. Science 2013;339:1084‐1088. [DOI] [PubMed] [Google Scholar]
- 73. Rosser EC, Mauri C. A clinical update on the significance of the gut microbiota in systemic autoimmunity. J Autoimmun 2016;74:85‐93. [DOI] [PubMed] [Google Scholar]
- 74. Sun Y, Haapanen K, Li B, Zhang W, Van de Water J, Gershwin ME. Women and primary biliary cirrhosis. Clin Rev Allergy Immunol 2015;48:285‐300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Invernizzi P, Miozzo M, Battezzati PM, Bianchi I, Grati FR, Simoni G, et al. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004;363:533‐535. [DOI] [PubMed] [Google Scholar]
- 76. Selmi C, Invernizzi P, Miozzo M, Podda M, Gershwin ME. Primary biliary cirrhosis: does X mark the spot? Autoimmun Rev 2004;3:493‐499. [DOI] [PubMed] [Google Scholar]
- 77. Lleo A, Oertelt‐Prigione S, Bianchi I, Caliari L, Finelli P, Miozzo M, et al. Y chromosome loss in male patients with primary biliary cirrhosis. J Autoimmun 2013;41:87‐91. [DOI] [PubMed] [Google Scholar]
- 78. Lleo A, Zhang W, Zhao M, Tan Y, Bernuzzi F, Zhu B, et al. DNA methylation profiling of the X chromosome reveals an aberrant demethylation on CXCR3 promoter in primary biliary cirrhosis. Clin Epigenetics 2015;7:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Katsumi T, Tomita K, Leung PS, Yang GX, Gershwin ME, Ueno Y. Animal models of primary biliary cirrhosis. Clin Rev Allergy Immunol 2015;48:142‐153. [DOI] [PubMed] [Google Scholar]
- 80. Irie J, Wu Y, Wicker LS, Rainbow D, Nalesnik MA, Hirsch R, et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J Exp Med 2006;203:1209‐1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Koarada S, Wu Y, Fertig N, Sass DA, Nalesnik M, Todd JA, et al. Genetic control of autoimmunity: protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J Immunol 2004;173:2315‐2323. [DOI] [PubMed] [Google Scholar]
- 82. Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, et al. Anti‐mitochondrial antibodies and primary biliary cirrhosis in TGF‐beta receptor II dominant‐negative mice. J Immunol 2006;177:1655‐1660. [DOI] [PubMed] [Google Scholar]
- 83. Ando Y, Yang GX, Tsuda M, Kawata K, Zhang W, Nakajima T, et al. The immunobiology of colitis and cholangitis in interleukin‐23p19 and interleukin‐17A deleted dominant negative form of transforming growth factor beta receptor type II mice. Hepatology 2012;56:1418‐1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tsuda M, Zhang W, Yang GX, Tsuneyama K, Ando Y, Kawata K, et al. Deletion of interleukin (IL)‐12p35 induces liver fibrosis in dominant‐negative TGFbeta receptor type II mice. Hepatology 2013;57:806‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang J, Yang GX, Tsuneyama K, Gershwin ME, Ridgway WM, Leung PS. Animal models of primary biliary cirrhosis. Semin Liver Dis 2014;34:285‐296. [DOI] [PubMed] [Google Scholar]
- 86. Yoshida K, Yang GX, Zhang W, Tsuda M, Tsuneyama K, Moritoki Y, et al. Deletion of interleukin‐12p40 suppresses autoimmune cholangitis in dominant negative transforming growth factor beta receptor type II mice. Hepatology 2009;50:1494‐1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wakabayashi K, Lian ZX, Moritoki Y, Lan RY, Tsuneyama K, Chuang YH, et al. IL‐2 receptor alpha(‐/‐) mice and the development of primary biliary cirrhosis. Hepatology 2006;44:1240‐1249. [DOI] [PubMed] [Google Scholar]
- 88. Wakabayashi K, Lian ZX, Leung PS, Moritoki Y, Tsuneyama K, Kurth MJ, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology 2008;48:531‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kawata K, Tsuda M, Yang GX, Zhang W, Tanaka H, Tsuneyama K, et al. Identification of potential cytokine pathways for therapeutic intervention in murine primary biliary cirrhosis. PLoS One 2013;8:e74225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hodge DL, Berthet C, Coppola V, Kastenmuller W, Buschman MD, Schaughency PM, et al. IFN‐gamma AU‐rich element removal promotes chronic IFN‐gamma expression and autoimmunity in mice. J Autoimmun 2014;53:33‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yang CY, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, et al. IL‐12/Th1 and IL‐23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology 2014;59:1944‐1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang MA, Rego D, Moshkova M, Kebir H, Chruscinski A, Nguyen H, et al. Peroxisome proliferator‐activated receptor (PPAR)alpha and ‐gamma regulate IFNgamma and IL‐17A production by human T cells in a sex‐specific way. Proc Natl Acad Sci U S A 2012;109:9505‐9510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Pelfrey CM, Cotleur AC, Lee JC, Rudick RA. Sex differences in cytokine responses to myelin peptides in multiple sclerosis. J Neuroimmunol 2002;130:211‐223. [DOI] [PubMed] [Google Scholar]
- 94. Bae HR, Leung PS, Tsuneyama K, Valencia JC, Hodge DL, Kim S, et al. Chronic expression of interferon‐gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology 2016;64:1189‐1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Vergani D, Mieli‐Vergani G. Mouse model of primary biliary cholangitis with a striking female predominance: a new powerful research tool. Hepatology 2016;64:1024‐1027. [DOI] [PubMed] [Google Scholar]
- 96. Shuai Z, Wang J, Badamagunta M, Choi J, Yang G, Zhang W, et al. The fingerprint of antimitochondrial antibodies and the etiology of primary biliary cholangitis. Hepatology 2017;65:1670‐1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sun Y, Zhang W, Evans JF, Floreani A, Zou Z, Nishio Y, et al. Autotaxin, pruritus and primary biliary cholangitis (PBC). Autoimmun Rev 2016;15:795‐800. [DOI] [PubMed] [Google Scholar]
- 98. Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2017; doi:10.1136/gutjnl.2016.313332. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 99. Wang J, Yang G, Dubrovsky AM, Choi J, Leung PS. Xenobiotics and loss of tolerance in primary biliary cholangitis. World J Gastroenterol 2016;22:338‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Webb GJ, Hirschfield GM. Primary biliary cholangitis in 2016: high‐definition PBC: biology, models and therapeutic advances. Nat Rev Gastroenterol Hepatol 2017;14:76‐78. [DOI] [PubMed] [Google Scholar]