

Abstract
Purpose of review
The only currently approved treatment for primary sclerosing cholangitis (PSC) is liver transplantation, with a median time to transplant of 12–18 years after diagnosis. There are a number of emerging drugs that have the potential to meet this critically unmet need that will be summarized and discussed herein.
Recent findings
Although the cause of PSC is unknown, there are a number of novel therapeutics under development. These drugs target presumed pathogenic mechanisms largely extrapolated from ex-vivo and in-vivo preclinical models, as well as translational observations.
Summary
Future therapeutic strategies for PSC may include a multitude of complex pathogenic mechanisms encompassing pathways of immunomodulation, the microbiome and inflammation-related fibrosis.
Keywords: bile acids, management, primary sclerosing cholangitis, therapies
Introduction
Classic primary sclerosing cholangitis (PSC) is a progressive, cholestatic liver disease of unknown cause, characterized by an inflammatory, fibro-obliterative process that may affect both the extrahepatic and intrahepatic bile ducts. The epidemiology, pathophysiology and general management of adult and pediatric PSC was recently reviewed [1■,2■]. The only available curative therapy is liver transplantation; up to 12.7% of patients will die on the waitlist, whereas 15–25% of transplanted patients will develop recurrent disease [3■,4■]. The current review will consider the key publications relevant to the subject of emerging pharmacologic therapies in PSC.
Although the cause of PSC is unknown, there are several prevailing hypotheses that have driven the search for novel therapeutics. The current therapeutic pipelines for PSC are based on four categories: bile acid pool modification (‘toxic-bile’ hypothesis) (summarized in Fig. 1 [5] and Table 1); intestinal microbiome modification (‘leaky gut’ hypothesis); immunomodulation of receptors that potentiate recruitment of intestinally derived lymphocytes to the hepatobiliary milieu (‘aberrant gut lymphocyte homing’ hypothesis) (summarized in Fig. 2) and lastly, modification of pathways involved in liver fibrogenesis.
Figure 1.
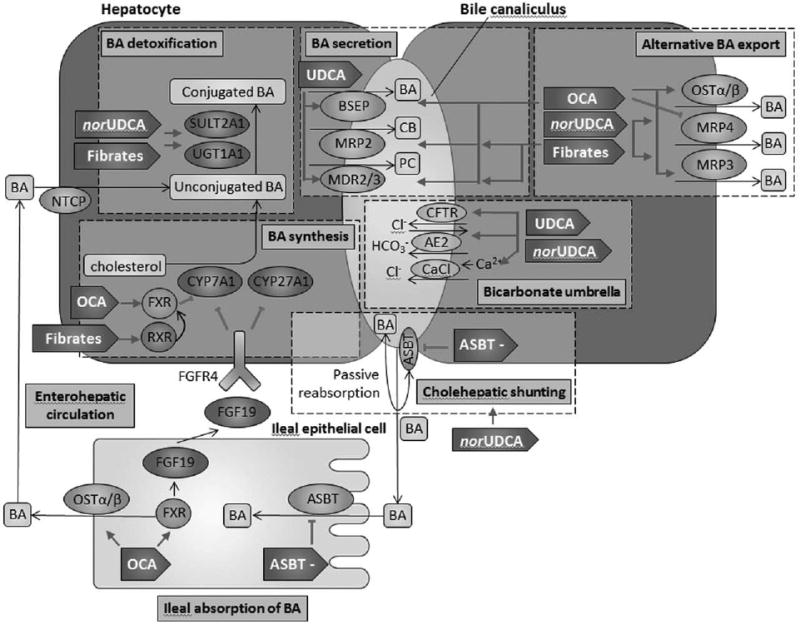
Current and emerging therapies for primary sclerosing cholangitis that modulate bile acid synthesis, detoxification, secretion and reabsorption. AE2, Cl−/HCO3− anion exchanger 2; ASBT, apical sodium-bile acid transporter; ASBT–, apical sodium-bile acid transporter inhibitor; BA, bile acids; BSEP, bile salt export pump; CaCl, Ca2+-dependent chloride channel; CB, conjugated bilirubin; CFTR, cystic fibrosis transmembrane conductance regulator; FGF19, fibroblast growth factor 19; FGFR4, fibroblast growth factor receptor 4; FXR, farsenoid X receptor; MDR2/3, multidrug resistance 2/3; MRP2, multidrug resistance-associated protein 2; MRP3, multidrug resistance-associated protein 3; MRP4, multidrug resistance-associated protein 4; NTCP, sodium/taurocholate cotransporting polypeptide; norUDCA, 24-norUrsodeoxycholic acid; OCA, obetichoic acid; OSTα/β, organic solute transporter α/β; PC, phosphatidylcholine; PSC, primary sclerosing cholangitis; RXR, retinoid X receptor; SULT2A1, sulfotransferase family 2A member 1; UDCA, ursodeoxycholic acid; UGT1A1, uridine diphosphate-glucoronosyltransferase family 1 member A1. Modified from [5].
Table 1. Potential therapeutics for primary sclerosing cholangitis that modulate bile acids.
| Process | Targets | Drug | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| UDCA | norUDCA | OCA | Fibrates | ASBT– | ||
| BA synthesis | FXR |
|
||||
| RXR |
|
|||||
| CYP7A1 |
|
|
||||
| CYP27A1 |
|
|
||||
|
| ||||||
| BA detoxification | SULT2A1 |
|
|
|
||
| UGT1A1 |
|
|
|
|||
|
| ||||||
| BA secretion | BSEP |
|
|
|||
|
| ||||||
| MRP2 |
|
|
||||
| MDR2/3 |
|
|
||||
|
| ||||||
| Alternative BA export | OSTα/β |
|
||||
| MRP4 |
|
|
||||
| MRP3 |
|
|
||||
|
| ||||||
| Bicarbonate umbrella | CFTR |
|
|
|||
| AE2 |
|
|
||||
| CaCl |
|
|
||||
|
| ||||||
| Cholehepatic shunting | ASBT |
|
||||
|
| ||||||
| Enterohepatic circulation | ASBT |
|
||||
| FXR |
|
|||||
| OSTα/β (ileal) |
|
|||||
| FGF19 |
|
|||||
Checkboxes indicate where each drug acts on individual targets. ASBT, apical sodium-bile acid transporter; ASBT–, apical sodium-bile acid transporter inhibitor; BA, bile acids; BSEP, bile salt export pump; CaCl, Ca2+-dependent chloride channel; CFTR, cystic fibrosis transmembrane conductance regulator; FXR, farsenoid X receptor; MDR2/3, multidrug resistance 2/3; MRP2, multidrug resistance-associated protein 2; MRP3, multidrug resistance-associated protein 3; MRP4, multidrug resistance-associated protein 4; norUDCA, 24-norursodeoxycholic acid; OCA, obetichoic acid; OSTα/β, organic solute transporter α/β; PSC, primary sclerosing cholangitis; RXR, retinoid X receptor; SULT2A1, sulfotransferase family 2A member 1; UDCA, ursodeoxycholic acid; UGT1A1, uridine diphosphate-glucoronosyltransferase family 1 member A1.
Figure 2.
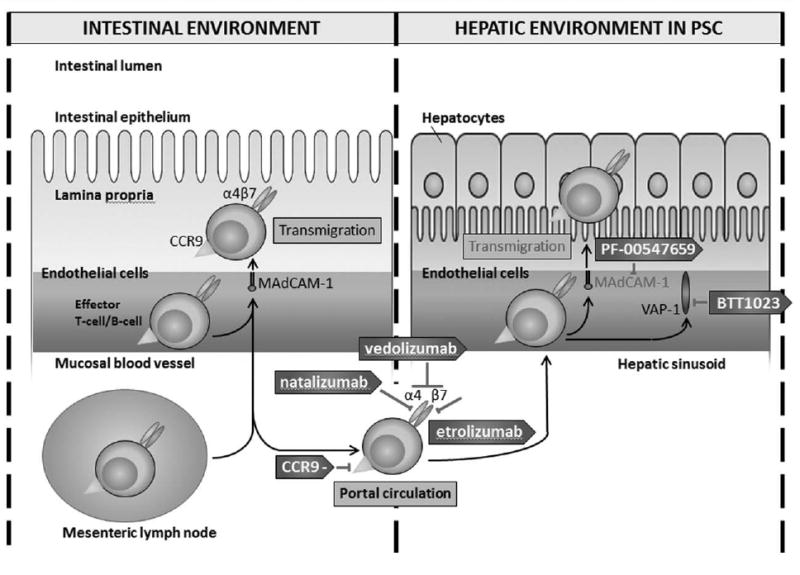
Therapies that are proposed to inhibit the transmigration of gut-derived lymphocytes into the hepatic environment in primary sclerosing cholangitis. In primary sclerosing cholangitis, mucosal vascular address in cell adhesion molecule 1 is aberrantly expressed. This allows aberrant binding and transmigration of gut-derived lymphocytes via α4β7 integrin. Binding and transmigration is further enhanced by vascular adhesion protein-1, which is constitutively present on hepatic endothelial cells and allows lymphocyte recruitment. CCR9–, chemokine receptor 9 inhibitor; MAdCAM-1, mucosal vascular addressin cell adhesion molecule 1; PSC, primary sclerosing cholangitis; VAP-1, vascular adhesion protein-1.
The challenge of primary sclerosing cholangitis trials
The discovery of effective therapies for PSC has been hampered by lack of knowledge regarding its pathogenesis. This impacts the rate of therapeutic development and limits the identification of key surrogate endpoints and the creation of animal models that can recapitulate the complex nature of PSC [6■■,7]. Robust surrogate endpoints for PSC are urgently needed as clinical endpoints such as transplant-free survival or decompensation require lengthy follow-up and large patient cohorts that are virtually impossible to achieve in clinical trials. In lieu of hard clinical endpoints, alkaline phosphatase (ALP) has often been used as a surrogate measure of outcome, but as discussed below, this is controversial [8]. Better biomarkers for cholestasis and/or biliary fibrosis are clearly needed for this disease. Also, we need the capability to identify patients with rapidly progressive disease and preferentially enroll these patients into clinical trials.
Lessons from ursodeoxycholic acid studies
Although originally, administration of exogenous bile acid therapy was postulated to replace toxic endogenous bile with a more nontoxic form [i.e. ursodeoxycholic acid (UDCA)], it was later discovered that bile acids are not simply passive bystanders, but are signaling molecules that participate in maintaining their own homeostasis [9]. In animal models, UDCA has been demonstrated to have effects on the secretion of bile acids through Ca2+, protein kinase C, mitogen-activated protein kinase- and integrin-dependent mechanisms, leading to an increase in bile salt export pump (BSEP) and multidrug resistance-associated protein 2 (MRP2) [10–14]. UDCA also stimulates HCO3− secretion, potentiating the ‘biliary HCO3− umbrella’, a constitutive mechanism that protects cholangiocytes from the injurious effects of hydrophobic bile [15,16]. It has negligible effects on the synthesis of bile acids [17].
Despite numerous studies on the utility of the synthetic bile acid UDCA in PSC, its use remains highly controversial [18,19]. Clearly, high-dose UDCA (28–30 mg/kg/day) worsens outcomes with greater progression to liver transplantation and the development of varices despite biochemical improvement in serum ALP, highlighting the uncertainty of ALP as a surrogate marker [20]. Furthermore, despite studies demonstrating that lower doses of UDCA (13–25 mg/kg/day) are associated with significant improvement in serum ALP – these studies failed to demonstrate improvements in transplant-free survival [21,22]. Significantly, stratification of patients into those with or without improvement in ALP (i.e. normalization or a drop in ALP to <1.5 times the upper limit of normal) demonstrate that patients have improved survival regardless of whether this biochemical change occurs spontaneously or following treatment with UDCA [23–24,25■,26]. Due to the uncertainty around the true effect of UDCA, and the lack of a biomarker that may identify those who are more likely to have a biochemical response to UDCA, the use of UDCA in PSC remains controversial [18,19,27,28]. In children, the association between ALP and outcomes is even less clear as ALP levels vary with both age and sex due to fluctuations in the bone-derived isoenzyme [29]. The experience with UDCA highlights the difficulty in designing PSC trials and the vulnerability of relying on a surrogate biomarker such as ALP.
Emerging therapies and trials
The following sections will outline the novel therapies currently under investigation for PSC, starting with those that modulate bile acids, followed by those that modulate the microbiome, immunomodulatory processes and finally fibrogenesis.
norUrsodeoxycholic acid
24-norUrsodeoxycholic acid (norUDCA, Dr Falk Pharma GmbH, Freiburg, Baden-Württemberg, Germany) is a derivative of UDCA that lacks a methylene group, providing resistance to conjugation with taurine or glycine [30]. As a weak acid, unconjugated norUDCA can be passively absorbed by cholangiocytes and secreted by hepatocytes, unlike UDCA, which in its unconjugated form can only be absorbed through the apical sodium bile acid transporter (ASBT) [31]. This allows norUDCA to efficiently avoid full enterohepatic circulation, instead undergoing a process called ‘cholehepatic shunting’, in which bile acids are reabsorbed by cholangiocytes and returned to hepatocytes rather than undergoing active transport across the terminal ileum [32,33]. This leads to significantly greater choleresis than with UDCA [32]. In addition, nor-UDCA causes choleresis of more hydrophobic bile, with a significant increase in biliary glutathioine and bicarbonate secretion in mouse models [34]. Unlike UDCA, norUDCA can also increase the expression of basolateral efflux pumps (i.e. Mrp4 and Mrp3) and upregulate phase I and II detoxification enzymes (Sult2a1, Ugt1a1, Cyp2b10 and Cyp3a11), the latter leading to a significant decrease in serum bile acids [34].
In a 12-week phase II dose finding study, 159 patients were randomized to placebo or a daily dose of norUDCA of 500, 1000 or 1500mg (NCT01755507) [35]. Intention-to-treat analysis demonstrated a significant reduction (12.3–26.0%) in serum ALP, compared with the placebo group. Similar changes in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were noted. Thus, norUDCA may be a promising treatment for PSC due to its apparent lack of toxicity and greater choleretic effects when compared with UDCA; a phase III study is necessary to examine the effectiveness of this drug in PSC.
Obeticholic acid
In 2016, obeticholic acid (OCA, Intercept Pharmaceuticals, New York City, New York, United States) was approved for use in primary biliary cholangitis, a cholestatic liver disease of presumed autoimmune origin that afflicts the interlobular and septal bile ducts; it is currently undergoing investigation in PSC (NCT02177136) [36]. OCA, a ligand for the farnesoid X receptor (FXR), has over 100 times greater potency for FXR than its endogenous analog, chenodeoxycholic acid [37]. OCA is postulated to reduce toxic bile production and induce secretion through FXR-mediated pathways. FXR is expressed in the liver and the small intestine (highest levels in the terminal ileum) [5]. Activation of FXR inhibits CYP7A1 (the rate limiting step of bile acid production) both directly, through translational activation of the short heterodimer protein (SHP), as well as indirectly, through the release of fibroblast growth factor (FGF)-19, which binds to FGF-4 on hepatocytes and leads to additional CYP7A1 inhibition [5,38,39]. OCA also decreases exposure to toxic bile by upregulating BSEP, MDR2/3, MRP2 and OSTα/β, which leads to increased canalicular bile secretion [40–42]. And finally, OCA also induces the expression of SULT2A1 and UGT1A1 [43,44■]. These processes combined should limit hepatocyte accumulation of toxic bile acids, thereby reducing liver injury and potentially ameliorating the pathogenesis of PSC.
Fibrates
Fibrates are peroxisome proliferator-activated receptor (PPAR) agonists that were originally designed for treatment of hyperlipidemia and have recently been reviewed as potential therapies in cholestatic liver disease [45]. Each fibrate has a different affinity for individual PPAR receptors, of which there are three isoforms (α, β/δ and γ), with bezafibrate having equal activity with all three isoforms and fenofibrate having 10-fold higher selectivity for PPAR-α than PPAR-γ [46]. PPARs act as ligand-activated transcription factors, and once activated, they form a heterodimer with the retinoid X receptor and bind distinct DNA-response elements [45]. Similar to OCA, fibrates can downregulate bile acid synthesis (by suppressing CYP7A1 and CYP27A1) and stimulate cannalicular secretion (by increasing expression of MDR2/3, MRP2) [45]. Fibrates may also act similarly to norUDCA by upregulating bile acid detoxification enzymes such as UGT1A1 and SULT2A1 [45].
Two pilot studies evaluating the efficacy of fibrates in PSC have been published [47,48]. Together, these include 21 patients treated over 6–12 months. Patients demonstrated significant decreases in ALP, with adverse events that included myalgias, nausea and worsening of psoriasis. Larger, placebo-controlled studies are warranted.
Apical sodium bile acid transporter inhibitors
Bile acids undergo enterohepatic circulation, with reabsorption of more than 95% of bile acids occurring at the terminal ileum via ASBT in the brush border membrane [49]. Two animal studies involving Mdr2−/− mice have demonstrated that ASBT inhibitors reduce serum bile acid levels and profibrogenic gene expression, while improving hepatic biochemical profiles (i.e. ALP and ALT) [50■,51■]. A phase 2 study of an ASBT inhibitor (SHP625/Lum001; Lumena Pharmaceuticals, San Diego, California, United States) in PSC demonstrated significant reductions in bile acids from baseline, but no significant change in ALP and other liver biochemistries (NCT02061540) [52■].
Antimicrobial therapy and modulation of the microbiome: the ‘leaky gut’ hypothesis
Failure of the intestinal barrier is posited to lead to bacterial translocation into the classically sterile portal and biliary system via the gut-liver axis, thereby exposing biliary epithelium to foreign microbial antigens [53]. Supportive evidence for this hypothesis includes studies demonstrating the presence of bacteria in the bile of explanted livers, as well as the presence of Helicobacter pylori in hilar hepatic ducts and antibodies against Chlamydia species lipopolysaccharide in the sera of PSC patients [54–57].
Exposure to bacterial products triggers pattern recognition receptors such as Toll-like receptors, causing cholangiocytes to assume an activated phenotype, leading to the release of cytokines and chemokines like TNF-α, IL-6 and IL-8, as well as growth factors and other signaling molecules [58]. This proinflammatory state may precipitate cholestasis and periductular fibrosis [58].
In turn, bacteria that comprise the intestinal microbiome may be influenced by bile composition. Recently, it has been demonstrated that increases in taurocholic acid secretion induced by a diet high in milk-fat may potentiate an abundance of Bilophila wadsworthia, a sulphate-reducing bacteria associated with ulcerative colitis [59]. This suggests that changes in bile acid composition may alter the intestinal microbiota and lead to dysbiosis, an aberrant microbial ecology that may cause disease through alterations in immune homeostasis [60].
In addition to their antimicrobial effects, antibiotics such as minocycline have been shown to have anti-inflammatory properties that may provide additional therapeutic actions in PSC. The clinical efficacy of vancomycin with metronidazole (NCT01085760) or without metronidazole (NCT01802073, NCT02605213 and NCT02137668), minocycline and rifaximin (Salix Pharmaceuticals, Raleigh, North Carolina, United States) have all been under recent or current investigation.
Vancomycin has been evaluated in three studies (total patients: 14 pediatric, 17 adult) in doses ranging from 50 mg/kg/day (pediatric, treatment for mean 54±43 months) to 150mg three to four times daily (adult, treatment for 12 weeks) [61,62]. The latter study also investigated metronidazole 250 or 500 mg three times daily. In children, vancomycin was associated with a reduction in ALT and gamma-glutamyl transferase (GGT). In adults, both doses were associated with decreased ALP. Counter-intuitively, only low-dose vancomyin and metronidazole led to decreased bilirubin and Mayo PSC risk scores.
In an open-label study evaluating rifaximin (550-g bid) in 16 patients over 12 weeks, there were no significant improvements in serum ALP, bilirubin, GGT or Mayo PSC risk score [63■]. Conversely, an open-label study of minocycline (100-mg bid) over 1 year demonstrated a significant improvement in serum ALP and Mayo PSC risk score, with no changes in bilirubin or albumin [64]. Minocycline's therapeutic effects may be in part because of its antiinflammatory and antiapoptotic properties [65]. It can also decrease inducible nitric oxide synthase, which is overexpressed in PSC and deleterious to cholangiocytes, and it also upregulates IL-10, a potent anti-inflammatory cytokine that limits auto-reactivity [66–69].
Fecal microbiota transplantation, probiotics and beyond
If modulation of the microbiome becomes a key component in the therapeutic arsenal for PSC, it is likely paramount that it is targeted toward the development of distinct intestinal microbial profiles, rather than a broad-spectrum destruction of the resident flora. This is exemplified by a recent study demonstrating that germ-free Mdr2−/− mice have worsening fibrosis, ductular reaction and ductopenia, with higher ALP, AST and bilirubin compared with conventionally caged mice [70■■]. Senescence, which has been implicated in the pathogenesis of PSC, was also worse in germ-free models and was abrogated by the addition of UDCA, a metabolite of commensal flora. This demonstrates that microbial metabolites may play a vital protective role in preventing injury to the biliary epithelium, suggesting that an alternative route to novel PSC therapeutics may involve targeted modification of metabolomics profiles which in healthy hosts are normally microbially derived.
Another major concern regarding antibiotic therapy is the evolution of antibiotic resistance, such as vancomycin-resistant enterococcus [71,72]. Ideally, future clinical trials will include studies to elucidate specific microbial metabolic pathways that could be targeted, thus eliminating the need for chronic antimicrobial treatment.
Future therapies may also include fecal microbiota transplantation (FMT) (NCT02424175) or the use of probiotics (NCT00161148). Fecal transplantation has been shown to be effective for diarrhea due to recurrent Clostridium difficile, but the long-term consequences of this approach are unclear [73]. Similarly, pooled data from two randomized controlled studies involving patients with ulcerative colitis suggest that FMT may be of benefit; however, randomized, placebo-controlled studies are needed to further validate its effectiveness in inflammatory bowel disease (IBD) [74■].
Immunomodulatory agents: the ‘aberrant gut lymphocyte’ hypothesis
The hypothesis postulates that intestinally derived lymphocytes aberrantly express receptors that allow for recruitment to the hepatobiliary system in which they incite a proinflammatory cascade [75]. Mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1), an endothelial cell adhesion molecule expressed on mesenteric lymph nodes and in vessels of the lamina propria of the intestinal milieu, may be integral to this aberrant signaling process [76]. MAdCAM-1 permits the recruitment of lymphocytes into the intestinal mucosa by binding to the α4β7 integrin and has been implicated in IBD pathogenesis [77]. MAdCAM-1 is not constitutively expressed in the liver, but it appears to be inappropriately expressed on liver endothelium in PSC [76]. This allows gut-derived lymphocytes traveling through the enterohepatic circulation to bind to cholangiocytes, then infiltrate and incite local inflammation [75]. The adhesion of lymphocytes is further potentiated by vascular adhesion protein-1 (VAP-1), which is constitutively expressed on hepatic endothelial cells and upregulated in individuals with IBD [76,78].
There have been a number of molecules developed for IBD that may have a therapeutic effect in PSC. One of the first was natalizumab (Biogen, Cambridge, Massachusetts, United States), which targets the α4 integrin and can associate with either β1, which can bind to vascular cell adhesion molecule 1, or β7, which can bind to MAdCAM-1 [79]. Unfortunately, natalizumab caused reactivation of the John Cunningham virus leading to progressive multifocal leukoencephalopathy (attributed to the inhibition of α4β1) and has since been discontinued [80,81]. Studies evaluating vercirnon, a CCR9 inhibitor (ChemoCentryx, Mountain View, California, United States), failed to consistently reach primary endpoints in Crohn's disease, and so further drug development has ceased [82]. Therapies currently under investigation for IBD, which may gain relevance in PSC, are etrolizumab (a monoclonal antibody against β7) (NCT01461317 and NCT02394028) (Roche Holding AG, Basel, Switzerland) and PF-00547659 (a selective inhibitor of MAdCAM-1; Shire Pharmaceuticals, St. Helier, Jersey) [83–85]. Vedolizumab (a mAb against the α4β7 integrin; Takeda Pharmaceutical Company Limited, Chuo-ku, Osaka, Japan) was approved by the Federal Drug Administration for ulcerative colitis and Crohn's disease in 2014 and is currently under investigation for PSC (EudraCT2014-003942-28) [86–88]. BTT1023 (Biotie Therapies, Turku, Finland), an inhibitor against VAP-1, is also currently under investigation (NCT02239211).
Antifibrogenic therapies
The evolving interest in antifibrogenic therapies in noncholestatic liver disease has naturally given rise to investigations into their utility in cholestatic liver disease.
Chemokine receptor 2/chemokine receptor 5 inhibitors
Chemokine receptor 2 (CCR2) and chemokine receptor 5 (CCR5) are instrumental to liver fibrogenesis [89]. Located on monocytes/macrophages, Kupffer cells and hepatic stellate cells (HSC), they bind chemokine ligand type 2 and 5, leading to release of TNF-β, which then stimulates collagen production by activated HSCs [90■]. Cenicriviroc, a dual CCR2/CCR5 antagonist (Takeda Pharmaceutical Company Limited, Chuo-ku, Osaka, Japan and Tobira Therapeutics, San Francisco, California, United States), is currently under investigation for PSC (NCT02653625).
Simtuzumab
Simtuzumab (Gilead Sciences, Foster City, California, United States) is a mAb against LOXL2, an enzyme that promotes the formation of cross-linkages between collagen fibers as well as the activation and recruitment of fibroblasts [91]. It is increased in hepatitis B, hepatitis C, NASH and animal models of liver fibrosis [92,93■]. A randomized, placebo-controlled trial was recently completed, and published results are pending (NCT01672853), but given preliminary results, further drug development is on hold [94■].
Conclusion
There are a number of pharmacologic therapies under development or in clinical trials for PSC, which target the different proposed pathogenic pathways. The future of PSC treatment is highly promising, though it is important that future studies integrate biomarker discovery into their design, given the lack of clarity around the ideal surrogate endpoint in PSC, as well as tools to improve risk stratification.
Key Points.
PSC is an incurable, fibro-obliterative disease that affects the large bile ducts in the liver, the only treatment of which is currently liver transplant.
Novel therapeutics under development target four proposed pathways of PSC pathogenesis: bile acid pool modulation, intestinal microbiome modification, immunomodulation of receptors that potentiate hepatic infiltration of gut-derived lymphocytes, and inhibition of fibrogenesis.
The lack of a strong surrogate marker continues to hamper therapeutic development in PSC, as traditional clinical endpoints (i.e. decompensation, death and liver transplant) require infeasible follow-up times and cohort sizes.
It is critical that future therapeutic studies include discovery and validation pathways for novel biomarkers and tools to stratify disease severity.
Acknowledgments
None.
Financial support and sponsorship The authors have no conflicts of interest relevant to this article. This work was supported by NIH grants DK63947 (to G.J.G.), NIH/NIDDK DK57993 and the Mayo Center for Cell Signaling in Gastroenterology grant DK84567 (P30) (to N.F.L.), DK84960 (to K.N.L.), the Chris M. Carlos and Catharine N. Jockisch Carlos Foundation for PSC, and the Mayo Clinic.
Footnotes
Conflicts of interest: There are no conflicts of interest.
References and Recommended Reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
■ of special interest
■■ of outstanding interest
- 1■.Lazaridis KN, LaRusso NF. Primary sclerosing cholangitis. N Engl J Med. 2016;375:1161–1170. doi: 10.1056/NEJMra1506330. This is a review of the management, proposed pathogenesis and management of primary sclerosing cholangitis (PSC) in adults. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2■.Mieli-Vergani G, Vergani D. Sclerosing cholangitis in children and adolescents. Clin Liver Dis. 2016;20:99–111. doi: 10.1016/j.cld.2015.08.008. This is a review of the management, proposed pathogenesis and management of sclerosing cholangitis in the pediatric population, which includes a discussion regarding patients with overlapping features of autoimmune hepatitis. [DOI] [PubMed] [Google Scholar]
- 3■.Singal AK, Fang X, Kaif M, et al. Primary biliary cirrhosis has high wait-list mortality among patients listed for liver transplantation. Transpl Int. 2016 doi: 10.1111/tri.12877. [Epub ahead of print] This study highlights the mortality rate for patients with PSC on the transplant waitlist, which remains relatively high given that it is the only treatment option currently available. [DOI] [PubMed] [Google Scholar]
- 4■.Gordon FD, Goldberg DS, Goodrich NP, et al. Recurrent primary sclerosing cholangitis in the Adult-to-Adult Living Donor Liver Transplantation Cohort Study: comparison of risk factors between living and deceased donor recipients. Liver Transpl. 2016;22:1214–1222. doi: 10.1002/lt.24496. This study demonstrates that a significant number of patients with PSC will develop recurrent disease following liver transplantation, which is critically important given that many will require retransplantation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fickert P, Pollheimer MJ, Beuers U, et al. Characterization of animal models for primary sclerosing cholangitis (PSC) J Hepatol. 2014;60:1290–1303. doi: 10.1016/j.jhep.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6■■.Ponsioen CY, Chapman RW, Chazouilleres O, et al. Surrogate endpoints for clinical trials in primary sclerosing cholangitis: review and results from an International PSC Study Group consensus process. Hepatology. 2016;63:1357–1367. doi: 10.1002/hep.28256. This article highlights the critical importance of including the exploration and validation of surrogate markers in drug development and clinical trials. Through a Delphi consensus process, it was determined that histology, alkaline phosphatase (ALP) and transient elastography are the key surrogate markers available to date, but even these require further validation. [DOI] [PubMed] [Google Scholar]
- 7.Kim WR, Therneau TM, Wiesner RH, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc. 2000;75:688–694. doi: 10.4065/75.7.688. [DOI] [PubMed] [Google Scholar]
- 8.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 9.Bouscarel B, Fromm H, Nussbaum R. Ursodeoxycholate mobilizes intracellular Ca2+ and activates phosphorylase a in isolated hepatocytes. Am J Physiol. 1993;264:G243–G251. doi: 10.1152/ajpgi.1993.264.2.G243. [DOI] [PubMed] [Google Scholar]
- 10.Bouscarel B, Gettys TW, Fromm H, et al. Ursodeoxycholic acid inhibits glucagon-induced cAMP formation in hamster hepatocytes: a role for PKC. Am J Physiol. 1995;268:G300–G310. doi: 10.1152/ajpgi.1995.268.2.G300. [DOI] [PubMed] [Google Scholar]
- 11.Schliess F, Kurz AK, vom Dahl S, et al. Mitogen-activated protein kinases mediate the stimulation of bile acid secretion by tauroursodeoxycholate in rat liver. Gastroenterology. 1997;113:1306–1314. doi: 10.1053/gast.1997.v113.pm9322526. [DOI] [PubMed] [Google Scholar]
- 12.Haussinger D, Kurz AK, Wettstein M, et al. Involvement of integrins and Src in tauroursodeoxycholate-induced and swelling-induced choleresis. Gastroenterology. 2003;124:1476–1487. doi: 10.1016/s0016-5085(03)00274-9. [DOI] [PubMed] [Google Scholar]
- 13.Paumgartner G, Beuers U. Mechanisms of action and therapeutic efficacy of ursodeoxycholic acid in cholestatic liver disease. Clin Liver Dis. 2004;8:67–81. vi. doi: 10.1016/S1089-3261(03)00135-1. [DOI] [PubMed] [Google Scholar]
- 14.Jazrawi RP, de Caestecker JS, Goggin PM, et al. Kinetics of hepatic bile acid handling in cholestatic liver disease: effect of ursodeoxycholic acid. Gastroenterology. 1994;106:134–142. doi: 10.1016/s0016-5085(94)94899-2. [DOI] [PubMed] [Google Scholar]
- 15.Beuers U, Hohenester S, de Buy Wenniger LJ, et al. The biliary HCO(3)(−) umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. 2010;52:1489–1496. doi: 10.1002/hep.23810. [DOI] [PubMed] [Google Scholar]
- 16.Ellis E, Axelson M, Abrahamsson A, et al. Feedback regulation of bile acid synthesis in primary human hepatocytes: evidence that CDCA is the strongest inhibitor. Hepatology. 2003;38:930–938. doi: 10.1053/jhep.2003.50394. [DOI] [PubMed] [Google Scholar]
- 17.Chapman R, Fevery J, Kalloo A, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatology. 2010;51:660–678. doi: 10.1002/hep.23294. [DOI] [PubMed] [Google Scholar]
- 18.European Association for the Study of the Liver. EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51:237–267. doi: 10.1016/j.jhep.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 19.Lindor KD, Kowdley KV, Luketic VA, et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology. 2009;50:808–814. doi: 10.1002/hep.23082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindor KD. Ursodiol for primary sclerosing cholangitis. Mayo Primary Sclerosing Cholangitis-Ursodeoxycholic Acid Study Group. N Engl J Med. 1997;336:691–695. doi: 10.1056/NEJM199703063361003. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell SA, Bansi DS, Hunt N, et al. A preliminary trial of high-dose ursodeoxycholic acid in primary sclerosing cholangitis. Gastroenterology. 2001;121:900–907. doi: 10.1053/gast.2001.27965. [DOI] [PubMed] [Google Scholar]
- 22.Stanich PP, Bjornsson E, Gossard AA, et al. Alkaline phosphatase normalization is associated with better prognosis in primary sclerosing cholangitis. Dig Liver Dis. 2011;43:309–313. doi: 10.1016/j.dld.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindstrom L, Hultcrantz R, Boberg KM, et al. Association between reduced levels of alkaline phosphatase and survival times of patients with primary sclerosing cholangitis. Clin Gastroenterol Hepatol. 2013;11:841–846. doi: 10.1016/j.cgh.2012.12.032. [DOI] [PubMed] [Google Scholar]
- 24.Rupp C, Rossler A, Halibasic E, et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment Pharmacol Ther. 2014;40:1292–1301. doi: 10.1111/apt.12979. [DOI] [PubMed] [Google Scholar]
- 25■.Hilscher M, Enders FB, Carey EJ, et al. Alkaline phosphatase normalization is a biomarker of improved survival in primary sclerosing cholangitis. Ann Hepatol. 2016;15:246–253. doi: 10.5604/16652681.1193721. This retrospective cohort study demonstrated that normalization of alkaline phosphatase can occur in nearly 50% of patients with PSC within a year of diagnosis, and that this is an independent predictor of transplant-free and neoplasia-free survival. [DOI] [PubMed] [Google Scholar]
- 26.Lindor KD, Kowdley KV, Harrison ME, et al. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015;110:646–659. doi: 10.1038/ajg.2015.112. quiz 660. [DOI] [PubMed] [Google Scholar]
- 27.Tabibian JH, Lindor KD. Ursodeoxycholic acid in primary sclerosing cholangitis: if withdrawal is bad, then administration is good (right?) Hepatology. 2014;60:785–788. doi: 10.1002/hep.27180. [DOI] [PubMed] [Google Scholar]
- 28.Turan S, Topcu B, Gokce I, et al. Serum alkaline phosphatase levels in healthy children and evaluation of alkaline phosphatase z-scores in different types of rickets. J Clin Res Pediatr Endocrinol. 2011;3:7–11. doi: 10.4274/jcrpe.v3i1.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beuers U, Trauner M, Jansen P, et al. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol. 2015;62:S25–37. doi: 10.1016/j.jhep.2015.02.023. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann AF, Zakko SF, Lira M, et al. Novel biotransformation and physiological properties of norursodeoxycholic acid in humans. Hepatology. 2005;42:1391–1398. doi: 10.1002/hep.20943. [DOI] [PubMed] [Google Scholar]
- 31.Gurantz D, Schteingart CD, Hagey LR, et al. Hypercholeresis induced by unconjugated bile acid infusion correlates with recovery in bile of unconjugated bile acids. Hepatology. 1991;13:540–550. [PubMed] [Google Scholar]
- 32.Kip NS, Lazaridis KN, Masyuk AI, et al. Differential expression of cholangiocyte and ileal bile acid transporters following bile acid supplementation and depletion. World J Gastroenterol. 2004;10:1440–1446. doi: 10.3748/wjg.v10.i10.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fickert P, Wagner M, Marschall HU, et al. 24-norUrsodeoxycholic acid is superior to ursodeoxycholic acid in the treatment of sclerosing cholangitis in Mdr2 (Abcb4) knockout mice. Gastroenterology. 2006;130:465–481. doi: 10.1053/j.gastro.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Trauner M, Fickert P, Hirschfield G, et al. Norursodeoxycholic acid improves cholestasis in primary sclerosing cholangitis: results of a phase II dose finding study. J Hepatol. 2015;64:S208–S209. doi: 10.1016/j.jhep.2017.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Hirschfield GM, Mason A, Luketic V, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148:751–761. doi: 10.1053/j.gastro.2014.12.005. ; e8. [DOI] [PubMed] [Google Scholar]
- 36.Pellicciari R, Fiorucci S, Camaioni E, et al. 6Alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 37.Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 38.Thomas C, Pellicciari R, Pruzanski M, et al. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 39.Inagaki T, Choi M, Moschetta A, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Ananthanarayanan M, Balasubramanian N, Makishima M, et al. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- 41.Boyer JL, Trauner M, Mennone A, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1124–G1130. doi: 10.1152/ajpgi.00539.2005. [DOI] [PubMed] [Google Scholar]
- 42.Kast HR, Goodwin B, Tarr PT, et al. Regulation of multidrug resistance-associated protein 2 (ABCC2) by the nuclear receptors pregnane X receptor, farnesoid X-activated receptor, and constitutive androstane receptor. J Biol Chem. 2002;277:2908–2915. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- 43.Song CS, Echchgadda I, Baek BS, et al. Dehydroepiandrosterone sulfo-transferase gene induction by bile acid activated farnesoid X receptor. J Biol Chem. 2001;276:42549–42556. doi: 10.1074/jbc.M107557200. [DOI] [PubMed] [Google Scholar]
- 44■.Edwards J, Zhang Y, Jackson J, et al. Obeticholic acid does not affect the hepatic metabolism of hormonal birth control in human sandwich cultured hepatocytes. Hepatology. 2016;64:201A–202A. This abstract demonstrates that obetichoic acid increases transcription of UGT1A1. [Google Scholar]
- 45.Ghonem NS, Assis DN, Boyer JL. Fibrates cholestasis. Hepatology. 2015;62:635–643. doi: 10.1002/hep.27744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willson TM, Brown PJ, Sternbach DD, et al. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 47.Chazouilleres O, Corpechot C, Gaovar F, et al. Fenofibrate improves liver tests in primary sclerosing cholangitis with incomplete response to ursodeoxycholic acid. Hepatology. 2010;52:488A. [Google Scholar]
- 48.Dejman A, Clark V, Martin P, et al. Fenofibrate improves alkaline phosphatase in primary sclerosing cholangitis. Gastroenterology. 2013;144:S1028–S1029. [Google Scholar]
- 49.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50:2340–2357. doi: 10.1194/jlr.R900012-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50■.Baghdasaryan A, Fuchs CD, Osterreicher CH, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol. 2016;64:674–681. doi: 10.1016/j.jhep.2015.10.024. This study of Mdr2−/− mice demonstrated that pharmacologic inhibition of apical sodium bile acid transporter (ASBT) results in a reduction of bile acid secretion, ALP, aspartate aminotransferase, hepatic expression of proinflammatory and profibrogenic markers, leading to decreased histologic evidence of bile duct injury and fibrosis. [DOI] [PubMed] [Google Scholar]
- 51■.Miethke AG, Zhang W, Simmons J, et al. Pharmacological inhibition of apical sodium-dependent bile acid transporter changes bile composition and blocks progression of sclerosing cholangitis in multidrug resistance 2 knockout mice. Hepatology. 2016;63:512–523. doi: 10.1002/hep.27973. This study of Mdr2−/− mice recapitulated the same findings as the above study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52■.Shire Pharmaceuticals. Dublin: Shire Pharmaceuticals; 2016. Apr 29, [Accessed 15 December 2016]. Shire delivers strong Q1 2016 results with double-digit growth in revenue and Non GAAP earnings per ADS. press release. https://www.shire.com/-/media/shire/shireglobal/shirecom/pdffiles/newsroom/2016/q1–2016-er-2016–04–29.pdf. In a press release, Shire outlined the results of their open-label phase 2 investigating the use of an ASBT inhibitor in PSC, which demonstrated that patients had significant decreases in serum bile acids and pruritus scores, but not in ALP or other liver function tests. [Google Scholar]
- 53.Tabibian JH, O'Hara SP, Lindor KD. Primary sclerosing cholangitis and the microbiota: current knowledge and perspectives on etiopathogenesis and emerging therapies. Scand J Gastroenterol. 2014;49:901–908. doi: 10.3109/00365521.2014.913189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olsson R, Bjornsson E, Backman L, et al. Bile duct bacterial isolates in primary sclerosing cholangitis: a study of explanted livers. J Hepatol. 1998;28:426–432. doi: 10.1016/s0168-8278(98)80316-4. [DOI] [PubMed] [Google Scholar]
- 55.Krasinskas AM, Yao Y, Randhawa P, et al. Helicobacter pylori may play a contributory role in the pathogenesis of primary sclerosing cholangitis. Dig Dis Sci. 2007;52:2265–2270. doi: 10.1007/s10620-007-9803-7. [DOI] [PubMed] [Google Scholar]
- 56.Ponsioen CY, Defoer J, Ten Kate FJ, et al. A survey of infectious agents as risk factors for primary sclerosing cholangitis: are Chlamydia species involved? Eur J Gastroenterol Hepatol. 2002;14:641–648. doi: 10.1097/00042737-200206000-00009. [DOI] [PubMed] [Google Scholar]
- 57.Bjornsson ES, Kilander AF, Olsson RG. Bile duct bacterial isolates in primary sclerosing cholangitis and certain other forms of cholestasis – a study of bile cultures from ERCP. Hepatogastroenterology. 2000;47:1504–1508. [PubMed] [Google Scholar]
- 58.O'Hara SP, Tabibian JH, Splinter PL, et al. The dynamic biliary epithelia: molecules, pathways, and disease. J Hepatol. 2013;58:575–582. doi: 10.1016/j.jhep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rowan FE, Docherty NG, Coffey JC, et al. Sulphate-reducing bacteria and hydrogen sulphide in the aetiology of ulcerative colitis. Br J Surg. 2009;96:151–158. doi: 10.1002/bjs.6454. [DOI] [PubMed] [Google Scholar]
- 60.Round JL, Mazmanian SK. The gut microbiota shapes intestinal immune responses during health and disease. Nat Rev Immunol. 2009;9:313–323. doi: 10.1038/nri2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davies YK, Cox KM, Abdullah BA, et al. Long-term treatment of primary sclerosing cholangitis in children with oral vancomycin: an immunomodulating antibiotic. J Pediatr Gastroenterol Nutr. 2008;47:61–67. doi: 10.1097/MPG.0b013e31816fee95. [DOI] [PubMed] [Google Scholar]
- 62.Tabibian JH, Weeding E, Jorgensen RA, et al. Randomised clinical trial: vancomycin or metronidazole in patients with primary sclerosing cholangitis – a pilot study. Aliment Pharmacol Ther. 2013;37:604–612. doi: 10.1111/apt.12232. [DOI] [PubMed] [Google Scholar]
- 63■.Tabibian JH, Gossard A, El-Youssef M, et al. Prospective clinical trial of rifaximin therapy for patients with primary sclerosing cholangitis. Am J Ther. 2017;24:e56–e63. doi: 10.1097/MJT.0000000000000102. In this 12-week, open-label pilot study of rifaximin in patients with PSC, there was no significant change in ALP or other liver function tests, suggesting that rifaximin may not be an effective treatment in PSC, possibly due to its indiscriminate effects on the intestinal microbiota. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Silveira MG, Torok NJ, Gossard AA, et al. Minocycline in the treatment of patients with primary sclerosing cholangitis: results of a pilot study. Am J Gastroenterol. 2009;104:83–88. doi: 10.1038/ajg.2008.14. [DOI] [PubMed] [Google Scholar]
- 65.Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169:337–352. doi: 10.1111/bph.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Amin AR, Attur MG, Thakker GD, et al. A novel mechanism of action of tetracyclines: effects on nitric oxide synthases. Proc Natl Acad Sci U S A. 1996;93:14014–14019. doi: 10.1073/pnas.93.24.14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jaiswal M, LaRusso NF, Burgart LJ, et al. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 68.Ledeboer A, Sloane EM, Milligan ED, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 69.Henriksen EK, Melum E, Karlsen TH. Update on primary sclerosing cholangitis genetics. Curr Opin Gastroenterol. 2014;30:310–319. doi: 10.1097/MOG.0000000000000052. [DOI] [PubMed] [Google Scholar]
- 70■■.Tabibian JH, O'Hara SP, Trussoni CE, et al. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology. 2016;63:185–196. doi: 10.1002/hep.27927. This murine study utilizing the Mdr2−/− model demonstrated that the commensal intestinal microbiota play a crucial role in preventing biliary injury, and that microbial metabolites may be implicated in this process. Novel pathogenic pathways and corresponding biomarkers and therapeutics may be derived from further investigations into the unique role of the intestinal microbiota in biliary homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O'Driscoll T, Crank CW. Vancomycin-resistant enterococcal infections: epidemiology, clinical manifestations, and optimal management. Infect Drug Resist. 2015;8:217–230. doi: 10.2147/IDR.S54125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jones RN. The impact of antimicrobial resistance: changing epidemiology of community-acquired respiratory-tract infections. Am J Health Syst Pharm. 1999;56:S4–11. doi: 10.1093/ajhp/56.suppl_3.S4. [DOI] [PubMed] [Google Scholar]
- 73.Koenigsknecht MJ, Young VB. Faecal microbiota transplantation for the treatment of recurrent Clostridium difficile infection: current promise and future needs. Curr Opin Gastroenterol. 2013;29:628–632. doi: 10.1097/MOG.0b013e328365d326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74■.Moayyedi P. Fecal transplantation: any real hope for inflammatory bowel disease? Curr Opin Gastroenterol. 2016;32:282–286. doi: 10.1097/MOG.0000000000000285. This systematic review summarizes the evidence available for the use of fecal microbiota transplantation in inflammatory bowel disease, which, to date, is limited to two clinical trial in ulcerative colitis and four case series in Crohn's disease, highlighting the need for additional controlled randomized clinical trials. [DOI] [PubMed] [Google Scholar]
- 75.Adams DH, Eksteen B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat Rev Immunol. 2006;6:244–251. doi: 10.1038/nri1784. [DOI] [PubMed] [Google Scholar]
- 76.Grant AJ, Lalor PF, Hubscher SG, et al. MAdCAM-1 expressed in chronic inflammatory liver disease supports mucosal lymphocyte adhesion to hepatic endothelium (MAdCAM-1 in chronic inflammatory liver disease) Hepatology. 2001;33:1065–1072. doi: 10.1053/jhep.2001.24231. [DOI] [PubMed] [Google Scholar]
- 77.Briskin M, Winsor-Hines D, Shyjan A, et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997;151:97–110. [PMC free article] [PubMed] [Google Scholar]
- 78.Salmi M, Kalimo K, Jalkanen S. Induction and function of vascular adhesion protein-1 at sites of inflammation. J Exp Med. 1993;178:2255–2260. doi: 10.1084/jem.178.6.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ghosh S, Goldin E, Gordon FH, et al. Natalizumab for active Crohn's disease. N Engl J Med. 2003;348:24–32. doi: 10.1056/NEJMoa020732. [DOI] [PubMed] [Google Scholar]
- 80.Adelman B, Sandrock A, Panzara MA. Natalizumab and progressive multifocal leukoencephalopathy. N Engl J Med. 2005;353:432–433. doi: 10.1056/NEJMc055235. [DOI] [PubMed] [Google Scholar]
- 81.Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N Engl J Med. 2005;353:362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 82.Wendt E, Keshav S. CCR9 antagonism: potential in the treatment of Inflammatory Bowel Disease. Clin Exp Gastroenterol. 2015;8:119–130. doi: 10.2147/CEG.S48305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rosenfeld G, Parker CE, MacDonald JK, et al. Etrolizumab for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2015:CD011661. doi: 10.1002/14651858.CD011661.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.D'Haens G, Lee S, Tarabar D, et al. OP022 anti-MAdCAM-1 antibody (PF-00547659) for active refractory Crohn's disease: Results of the OPERA study. European Crohn's and Colitis Organization Conference, Journal of Crohn's and Colitis; Barcelona, Spain. 2015. [Google Scholar]
- 85.Vermeire S, Sandborn W, Danese S, et al. OP021 TURANDOT: a randomized, multicenter double-blind, placebo-controlled study of the safety and efficacy of anti-MAdCAM antibody PF-00547659 (PF) in patients with moderate to severe ulcerative colitis (UC). European Crohn's and Colitis Organisation Conference, Journal of Crohn's and Colitis; Barcelona, Spain. 2015. [Google Scholar]
- 86.Federal Drug Administration (FDA) FDA approves Entyvio to treat ulcerative colitis and Crohn's disease. 2014 May 20; news release. [Google Scholar]
- 87.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699–710. doi: 10.1056/NEJMoa1215734. [DOI] [PubMed] [Google Scholar]
- 88.Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn's disease. N Engl J Med. 2013;369:711–721. doi: 10.1056/NEJMoa1215739. [DOI] [PubMed] [Google Scholar]
- 89.Seki E, de Minicis S, Inokuchi S, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50:185–197. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90■.Lefebvre E, Moyle G, Reshef R, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One. 2016;11:e0158156. doi: 10.1371/journal.pone.0158156. This study demonstrated that cenicriviroc (a dual chemokine receptor 2/chemokine receptor 5 antagonist), when used in three rodent models of fibrosis (thioglycollate-induced peritonitis, diet-induced nonalcoholic steatohepatitis and renal fibrosis), significantly reduced macrophage recruitment, collagen expression and deposition and thus may be useful in preventing disease progression in PSC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 92.Vadasz Z, Kessler O, Akiri G, et al. Abnormal deposition of collagen around hepatocytes in Wilson's disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J Hepatol. 2005;43:499–507. doi: 10.1016/j.jhep.2005.02.052. [DOI] [PubMed] [Google Scholar]
- 93■.Ikenaga N, Peng ZW, Vaid KA, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017 doi: 10.1136/gutjnl-2016-312473. Epub ahead of print. This murine study demonstrated that lysyl oxidase-like 2 (LOXL2) is overexpressed in fibrosis, with particularly high levels localized to fibrotic septa, with pharmacologic inhibition of LOXL2 leading to decreased collagen deposition and histologic evidence of fibrosis, as well as diminished ductular reaction, indicating that it may limit both hepatic and biliary fibrosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94■.Third Quarter 2016 Gilead Sciences Earnings Conference Call. [Accessed 7 January 2017];Gilead Sciences. 2016 Nov 1; http://investors.gilead.com/phoenix.zhtml?-P=irol-eventDetails&c=69964&eventID=5237083. The results of the randomized, placebo-controlled clinical trial evaluating the safety and efficacy of simtuzumab are yet to be published; however, during a conference call to investors, it was stated that further drug development is on hold.