

Abstract
Background and Objectives
Given moderate heritability and significant heterogeneity among addiction phenotypes, successful genome-wide association studies (GWAS) are expected to need very large samples. As sample sizes grow, so can genetic diversity leading to challenges in analyzing these data. Methods for empirically assigning individuals to genetically informed ancestry groups are needed.
Methods
We describe a strategy for empirically assigning ancestry groups in ethnically diverse GWAS data including extensions of principal component analysis (PCA) and population matching through minimum Mahalanobis distance. We apply these methods to data from Spit for Science (S4S): the University Student Survey, a study following college students longitudinally that includes genetic and environmental data on substance use and mental health (n=7603).
Results
The genetic-based population assignments for S4S were 48.7% European, 22.5% African, 10.4% Americas, 9.2% East Asian, and 9.2% South Asian descent. Self-reported census categories “More than one race” and “Unknown”as well as “Hawaiian/Pacific Islander” and “American Indian/Native Alaskan” were empirically assigned representing a +9% sample retention over conventional methods. Although there was high concordance between self-reported race and empirical population-match (+0.924), there was reduction in variance for most ancestry PCs for genetic-based population assignments.
Conclusions
We were able to create more genetically homogenous groups and reduce sample and marker loss through cross-ancestry meta-analysis, potentially increasing power to detect etiologically relevant variation. Our approach provides a framework for empirically assigning genetic ancestry groups which can be applied to other ethnically-diverse genetic studies.
Scientific Significance
Given the important public health impact and demonstrable gains in statistical power from studying diverse populations, empirically sound practices for genetic studies are needed.
Keywords: genetics and addiction, GWAS, cross-population meta-analysis, psychiatric genetics, principal component analysis, Mahalanobis distance, genetic ancestry
Background and Objectives
Genome-wide association studies (GWAS) have successfully discovered thousands of variants robustly associated with a variety of human traits including psychiatric outcomes long known to be heritable.1 Although addiction related GWAS typically have used thousands of individuals in their samples, there is compelling evidence across a variety of traits that even larger sample sizes will be required to discover robustly associated genetic variants.2,3 This leads to the phenomenon that as larger samples are collected and ascertained, maintaining homogeneity across multiple domains including phenotype, culture, or genetic ancestry becomes increasingly difficult.
An important motive for understanding the effects of ancestry on GWAS is preventing genomic inflation due to population stratification. Population stratification occurs when both disease prevalence and allelic frequency differences exist in the subpopulation sampled. Such stratification may lead to false positive associations of genetic signals.4 Since up to millions of markers are tested across the genome, many will show differences in allele frequency between populations and the overall distribution of test statistics will be inflated, leading to an increase in false positives, hence the term “genomic inflation.”
There are several approaches by which ancestry is considered in GWAS. One is self identified ancestry (SIA)/ethnicity/race. There is some utility in using this information as a starting point for addressing population stratification. When subsetting a sample based on SIA, the number of markers violating Hardy Weinberg Equilibrium (HWE) and measures of genomic inflation are greatly reduced. However, SIA alone is usually insufficient even in apparently homogenous samples.5
Another approach to address ancestry in genetic studies is the use of ancestry informative markers (AIMS) which are a fixed small set of markers (1500–5000) shown to have high differentiation between ancestral populations. AIMS are useful when no GWAS data are available such as in candidate gene or target genotyping for replication studies. There is not perfect agreement between SIA and AIMs as SIA may provide more information on social and environmental exposures while AIMS may provide a better estimate of ancestry.6 However, if GWAS data are available, limiting the ancestry analyses to a small fixed set of markers is not ideal.5,6
Admixture analysis assumes a fixed number of ancestral populations and estimates a percent ancestry of each ancestral population for each sample. The resulting quantitative values are substantially more informative than SIA, can be used to assess sample homogeneity, and divide the sample into sub-populations if there are large differences. There is a variety of software designed to perform these analyses including ADMIXTURE,5 FRAPPE,7 sNMF.8 However, creating more homogenous subgroups using admixture results can be challenging if there are no clear clusterings of individuals. Subdividing by global ancestry may not be sufficient to address population stratification when local population structures exist.
The most common approach to assessing ancestry and population structure is the use of ancestry principal component analysis (PCA), which has been applied to adjust for global and local population structures such as Eastern-Western European differences9 and regional differences in China.10 The principal components (PCs) can be used to exclude outliers from otherwise homogenous groups and, importantly, used as covariates in association analyses to reduce effects of population stratification. However, the statistical justification for excluding outliers is often not clear (visual inspection), simplistic, or non-multivariate. Samples that show evidence for admixture or have missing self reported race/ethnicity/ancestry are often excluded which unnecessarily reduces sample size. Critically, the choice of PCs included in GWAS as covariates is commonly not done empirically. That is, not appropriately assessing if they are associated with the trait of interest or with some other technical artefact such as batch effect. The practice of blindly including PCs in GWAS (typically 10–20) can negatively impact results. First, if too many PCs are included association models may become overfit, which can reduce the power to detect etiologically relevant variation (inflated type-II error rates). Conversely, if too few PCs are included, which is particularly salient for admixed and diverse samples, population stratification can remain potentially leading to false positive associations (increased type-I error rates).
The standard practice in GWAS is not only to test directly genotyped markers but also to test unobserved markers where the genotypes are estimated using imputation. In addition to many quality control (QC) steps requiring homogenous samples, ancestry can influence imputation and some methods recommend within group imputation (MaCH, BEAGLE)11,12 while other methods, such as IMPUTE2, recommend using diverse reference panels.13 There are also methods specifically intended to be used in admixed populations such as MaCHadmix.14 Therefore the choice to perform imputation within or across groups will depend on the imputation method’s recommended practices. Regardless of method, imputation quality can vary across groups due to a variety of factors including reference panel diversity and single nucleotide polymorphism (SNP) array density, content, and design. Meta-analysis of data from diverse populations offers the opportunity to increase the power to detect loci through increased sample size and improve the resolution of fine-mapping of causal variants.15 It is expected that there will be some differences in causal loci between diverse populations and how well GWAS findings translate from one population to another depends on heterogeneity in allelic effects between distantly related populations.
Until recently, most GWAS were conducted in relatively homogeneous samples.16 For many samples collected in the US, the analyses were limited to individuals of European and African ancestry and excluded groups with too few individuals for statistical analysis. However, as samples become increasingly large and diverse, there is a need to establish practices for cosmopolitan samples. Here we describe procedures for empirically assigning individuals to genetically informed ancestry groups for cross-population meta-analysis with the goal of maximizing power for discovering etiologically relevant genetic variants while minimizing false positive associations due to population stratification. Our approach is applied to data from Spit for Science (S4S): the University Student Survey, an ongoing study following college students longitudinally that consists of both genetic and environmental data collected on substance use and mental health. Our framework for empirically assigning genetic ancestry groups can be applied to other cross-population studies, including those of admixed ancestry such as African- and Asian-Americans.
Methods
Sample collection
Spit for Science is a longitudinal study of college students enrolled in a large, urban university in the mid-Atlantic, as described previously.17,18 Briefly, incoming students age 18 or older were eligible to complete online phenotypic assessments that covered a range of behavioral and health-related phenotypes, as well as demographic information including self-identified ethnicity. Follow-up assessments were completed in subsequent spring semesters. Study data were collected and managed using REDCap electronic data capture tools19 hosted at Virginia Commonwealth University. REDCap (Research Electronic Data Capture) is a secure, web-based application designed to support data capture for research studies, providing 1) an intuitive interface for validated data entry; 2) audit trails for tracking data manipulation and export procedures; 3) automated export procedures for seamless data downloads to common statistical packages; and 4) procedures for importing data from external sources. All students that initiated the survey were first led through a consent process that further explained the study and their participation. Individuals who completed the online survey were also eligible to provide a deoxyribonucleic acid (DNA) sample. The current analyses are based on students who matriculated in the Fall of 2011 (n = 2714), 2012 (n = 2486), and 2013 (n = 2403), for a total of n = 7603. Of these, 98% provided a DNA sample.
Genotyping and imputation
Genotyping was performed at Rutgers University Cell and DNA Repository using the Affymetrix BioBank array (653k) which contains both common GWAS framework variants (296k) for imputation and functional variants (357k) including rare high impact exome variants (272k), indels (18k), eQTLs (16k) and miscellaneous markers (51k). QC excluded Off Target Variants found by SNPolisher, high missingness SNPs (5%) and samples (2%), and post sample filtering high missingness SNPs (2%) similar to the Psychiatric Genomics Consortium (PGC).20 There were 6534 samples passing DNA and initial genotyping QC. An additional 209 samples were removed due to excessive relatedness (n = 194) and/or Pi_Hat > 0.1 (n = 180), leaving 6325 samples and 560138 variants for imputation. Imputation was conducted using SHAPEIT2,21 IMPUTE2,22 and the 1000 genomes phase 3 (1KGP) reference panel (n = 2504) which includes 81,706,022 variants.23,24 The post imputation and pre-GWAS filtering included excluding rare (MAF <0.005) and low quality (INFO<0.5) variants, SNPTEST v2.5.225 was used to calculate Hardy-Weinberg Equilibrium (HWE) which yielded 17,461,305 variants. Group (super-population or census race) GWAS filtering was performed using within grouping specific HWE (p-value>10−6) and sample size based MAFs. Instead of using a fixed MAF threshold for each group, the minimum observed minor allele count (MAC) of 40 was used as this minimum is robust for most association analyses performed in GWAS.26
Ancestry principal components
Ancestry PCs can be estimated from the sample itself or from an external reference such as the 1KGP and projected onto the GWAS sample. The use of an external reference panel has advantages such as not needing to exclude relatives or poorly performing samples, and some of the loadings can be interpreted (i.e., what ancestral population it reflects) based on reference panel populations. Here, 1KGP phase 3 variants (2504 samples, 26 populations), found in common with the post QC filtered S4S genotypes were merged together. Regions with high LD were excluded27,28 and the common set of variants was then pruned (r2 < 0.1) using PLINK 1.929,30 (–indep-pairwise 1500 150 0.1) to yield 109,259 semi-independent variants for ancestry analyses. EIGENSOFT and SmartPCA27,31 were used to perform PCA using only the 1KGP phase 3 reference panel to determine SNP weights for each eigenvector. This solution was then projected onto the S4S data to generate 10 PCs.
Genetic based population assignment
The 1KGP reference panel was used for S4S population assignments. The 1KGP consists of 26 populations including Han Chinese in Bejing China (CHB), Japanese in Tokyo Japan (JPT), Southern Han Chinese (CHS), Chinese Dai in Xishuangbanna China (CDX), Kinh in Ho Chi Minh City Vietnam (KHV), Utah residents from the USA with Northern and Western Ancestry (CEU), Toscani in Italia (TSI), Finnish in Finland (FIN), British in England and Scotland (GBR), Iberian Population in Spain (IBS), Yoruba in Ibadan Nigeria (YRI), Luhya in Webuye Kenya (LWK), Gambian in Western Divisions in the Gambia (GWD), Mende in Sierra Leone (MSL), Esan in Nigeria (ESN), Americans of African Ancestry in Southwest USA (ASW), African Caribbeans in Barbados (ACB), Mexican Ancestry from Los Angeles USA (MXL), Puerto Ricans from Puerto Rico (PUR), Colombians from Medellin Colombia (CLM), Peruvians from Lima Peru (PEL), Gujarati Indian from Houston, Texas USA (GIH), Punjabi from Lahore Pakistan (PJL), Bengali from Bangladesh (BEB), Sri Lankan Tamil from the UK (STU), and Indian Telugu from the UK (ITU). The 26 1KGP populations are divided into five super populations: African (AFR), admixed from the Americas (AMR), East Asian (EAS), South Asian (SAS), and European (EUR).
Using all 10 ancestry PCs, we began by calculating the median and variance for each 1KGP population and then calculating the Mahalanobis distance(30) for each 1KGP sample for all 26 populations (Figure 1a). We chose to apply Mahalanobis distance, a common approach for detecting outliers, to assign the best population match in multivariate space as it accounts for both mean distance and group variances. Reference population outliers (> 4 SD from population median, n = 61) were then removed (Figure 1b) and the procedure was repeated for all 1KGP samples. Every S4S sample was then assigned to the 1KGP population with the minimum Mahalanobis distance. The S4S samples were then collapsed into their respective super-population assignment (Figure 1c).
Figure 1a.
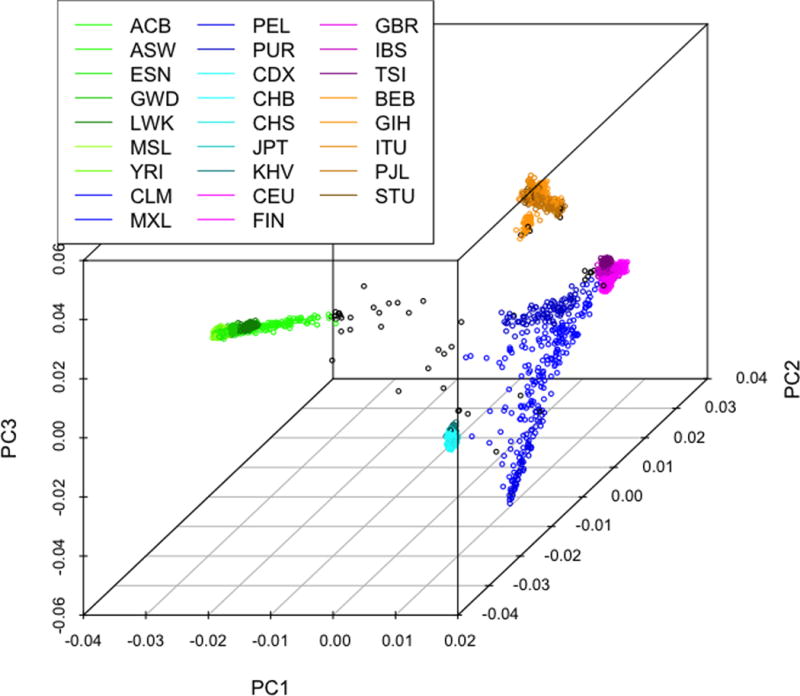
1KGP population assignment. The first three principal components (PC) plotted for each of the 26 1KGP populations with Mahalanobis distance outliers marked in black.
Figure 1b.
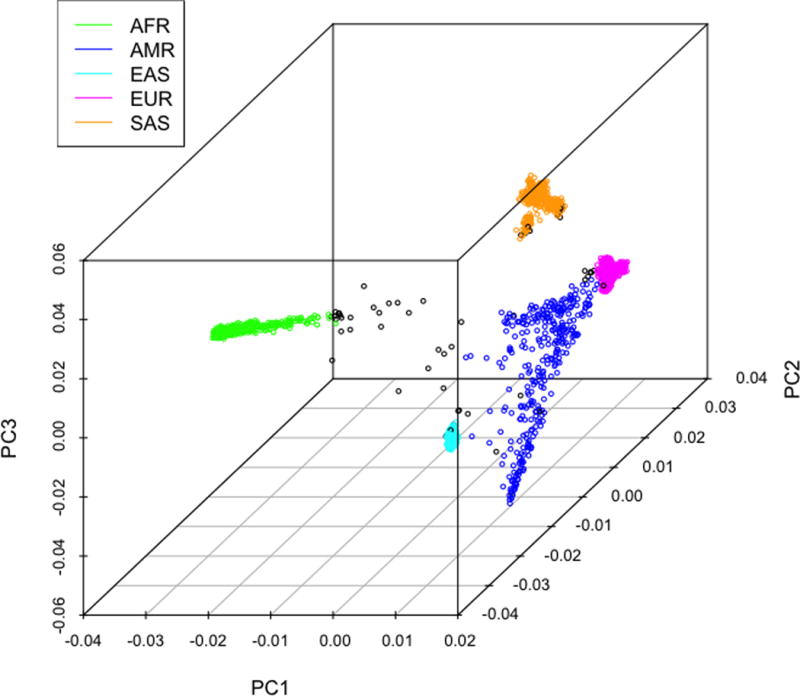
1KGP super-population assignment. The first three PCs plotted for each of the 5 1KGP super-populations with Mahalanobis distance outliers marked in black.
Figure 1c.
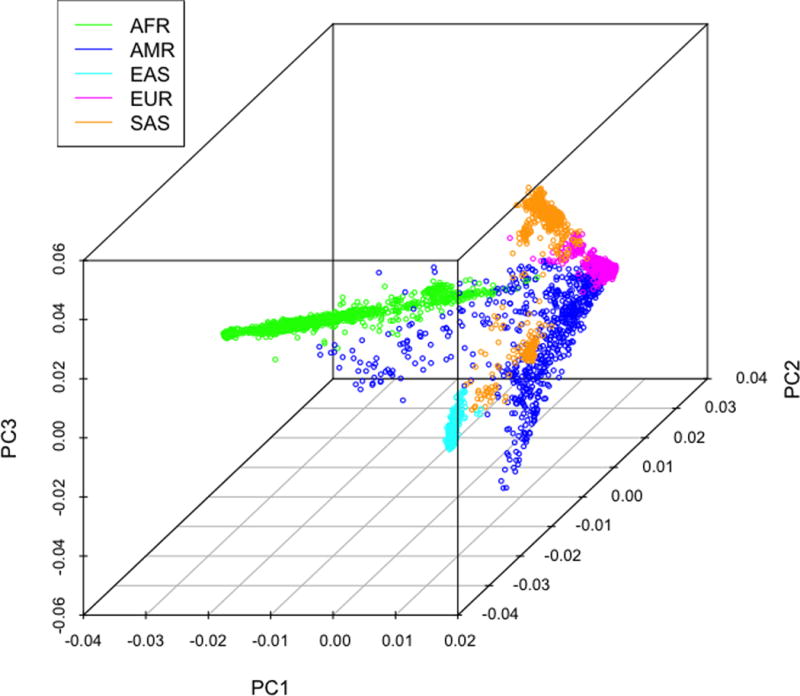
S4S super-population assignment. The first three PCs plotted for each of the 5 genetic based super-populations determined using Mahalanobis distance.
Results
Study population
Responses for self-identified ethnicity were as follows: American Indian/Alaska Native (n = 35); Asian (n = 1223); Black/African American (n = 1464); Hispanic/Latino (n = 450); More than one race (n = 467); Native Hawaiian/Other Pacific Islander (n = 50); Unknown (n = 30); and White (n = 3763). Participants could also elect not to answer (n = 108). As noted previously, the sample of participants corresponds closely to the overall demographics of the university student population.18
Genetic based population assignment
To assess population matching, the assigned versus known population and super-population for each 1KGP sample was examined. The average 1KGP cross-population mismatch rate was 0.09 (ranging from 0.03 for AFR to 0.185 for SAS). However, there were no cross super-population mismatches. For S4S, the concordance rate between super-population assignment based on minimum Mahalanobis distance and self-reported census race was greater than 92.4% with 4.6% of the samples within the major census categories changing super-populations. As expected, the census category Asian split into EAS and SAS super-populations. Furthermore, samples in “More than one race” and “Unknown” groups were empirically assigned. Additional details are shown in Table 1 and plots of the first three PCs for the empirically matched S4S super-population assignments are shown in Figure 1c. Although there is high concordance between self-reported census race and empirical genetic population match, there is reduction in variance for most ancestry PCs for the genetic based super population assignments. Specifically, of the 10 PCs constructed variances were smaller for all 10 PCs in the EUR, 8 in the AFR, 8 in EAS, 7 in SAS but only 2 in the AMR group. Variance reduction was statistically significant via the sign-test for the EUR (p = 0.0008), AFR (p = 0.029), and EAS super population groups (p = 0.029). Although 7 of 10 PC variances were smaller in the SAS group, the reduction did not reach statistical significance (p = 0.103). Plots of the first three PCs for each self-reported census race and super-population assignments showing the reduced variance can be seen in Figure 2.
Table 1.
S4S self-reported census race and concordance with genetic ancestry match by Mahalanobis distance
| S4S Census Race/Ethnicity | n | EUR | AFR | EAS | SAS | AMR | Conc. | Sample Prop. |
|---|---|---|---|---|---|---|---|---|
| White | 3098 | 2932 | 11 | 3 | 37 | 115 | 0.946 | 0.490 |
| Black/African American | 1256 | 6 | 1215 | 2 | 3 | 30 | 0.967 | 0.198 |
| Asian | 998 | 11 | 2 | 553 | 418 | 14 | 0.973 | 0.158 |
| Hispanic/Latino | 381 | 17 | 11 | 0 | 1 | 352 | 0.924 | 0.060 |
| More than one race | 390 | 38 | 147 | 2 | 81 | 122 | 0.062 | |
| Unknown/Missing | 134 | 61 | 23 | 5 | 28 | 17 | 0.021 | |
| Hawaiian/Pacific Islander | 38 | 4 | 0 | 18 | 14 | 2 | 0.006 | |
| American Indian /Native Alaskan | 30 | 10 | 11 | 0 | 2 | 7 | 0.005 | |
| Total | 6325 | 3079 | 1420 | 583 | 584 | 659 |
Note: S4S = data from Spit for Science the University student survey, 1KGP = 1000 Genomes Project reference panel, EUR = matched to 1KGP European populations, AFR = matched to 1KGP African populations, EAS = matched to 1KGP East Asian populations, SAS = matched to 1KGP South Asian populations, AMR = matched to 1KGP Americas populations, Conc. = concordance rate between self-reported census race/ethnicity and genetic ancestry match, Prop. = proportion of sample.
Figure 2.
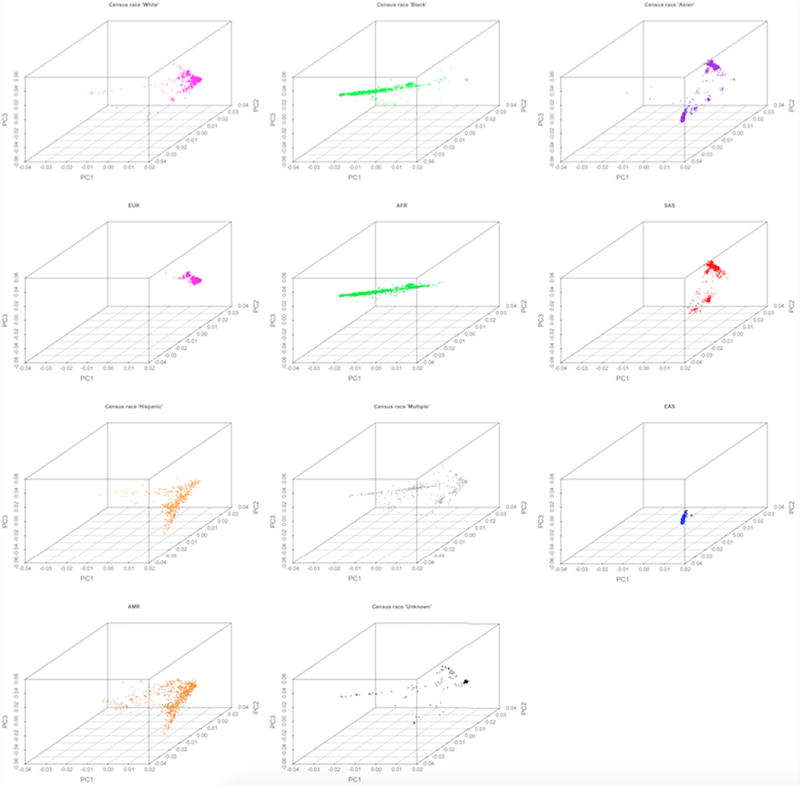
The first three principal components (PC) plotted for self-reported census race and genetic super-population assignments. Comparison of self-reported census race and genetic super-population assignment plots indicate a reduction of variance in PCs for genetic based super-population assignments.
Sample and marker retention
Conventional GWAS strategies typically remove those subjects that are missing self-reported census race, those endorsing more than one census race, or those census categories with small sample sizes. By applying our population matching methodology we were able to create more genetically homogenous groups while retaining those groups that are commonly removed from analyses. As shown in Table 1, were able to retain greater than 9% of the sample, which would otherwise have been excluded.
There are established QC metrics that are used to remove problematic variants prior to running GWAS analyses, including markers in violation of HWE, low minor allele frequency (MAF), and poor imputation quality. These QC procedures need to be carefully considered when applied to diverse samples as there can be significant differences on these metrics by ancestry. Therefore standard QC procedures were applied to each genetically assigned super-population separately. Post GWAS filtering was performed using ancestry specific HWE (p >10−6) and sample size based MAFs. As a result, there was greater marker retention by applying QC to empirically assigned ancestry groups and then meta-analyzed across groups. When analyzing all samples together, 2.7×106 markers were removed due to excessive violation of HWE (p<10−6), representing 16% of markers passing minimum INFO and MAF thresholds. As shown in Table 2, for individual groups, few markers (0.1%) were filtered due to HWE except for census Asian (0.8%) which had a higher frequency of markers failing HWE than other census categories. By performing meta-analysis across assigned ancestry groups, as compared to census categories or all samples together, we were able to retain 220,689 – 1,930,671 SNPs, which would have otherwise been removed from analysis (Table 2).
Table 2.
Post imputation marker filtering by MAF and HWE for self-reported census race and genetic population assignment for an example GWAS of unrelated individuals using 17,461,305 imputed variants.
| Group | n | min. MAF | MAF fail | HWE fail | MAF/HWE fail | |
|---|---|---|---|---|---|---|
| ALL | 5880 | 0.0034 | 77,826 | 2,716,350 | 2,794,175 | |
|
| ||||||
| WHT | 3002 | 0.0067 | 6,965,188 | 10,004 | 6,974,728 | |
| EUR | 2972 | 0.0067 | 7,036,314 | 9,308 | 7,045,613 | |
|
| ||||||
| BLK | 1202 | 0.0166 | 3,333,134 | 4,169 | 3,337,283 | |
| AFR | 1329 | 0.0150 | 2,973,215 | 4,579 | 2,977,773 | |
|
| ||||||
| ASN | 921 | 0.0217 | 9,182,668 | 143,288 | 9,325,808 | |
| EAS | 552 | 0.0362 | 10,561,171 | 2,829 | 10,563,961 | |
| SAS | 447 | 0.0447 | 10,093,522 | 1,774 | 10,095,181 | |
|
| ||||||
| HSP | 357 | 0.0560 | 10,450,319 | 925 | 10,451,231 | |
| AMR | 580 | 0.0345 | 9,402,185 | 2,229 | 9,404,397 | |
|
| ||||||
| Analysis | nmax | nmin | nmedian | Total SNPs | ||
|
| ||||||
| ALL | 5880 | – | – | 14,667,130 | ||
|
| ||||||
| Census Race | 5482 | 1,202 | 4,204 | 16,377,112 | ||
|
| ||||||
| Pop. Match | 5880 | 1,027 | 4,104 | 16,597,801 | ||
Note: SNP = single nucleotide polymorphism, n = sample size, min. MAF = minimum minor allele frequency where at least 40 minor alleles are observed in the sample, MAF pass = number of SNPs passed MAF, HWE = number of SNPs passed Hardy Weinberg Equilibrium threshold, Total fail = number of SNPs failing MAF or HWE threshold, ALL = all data available for genetic analysis, WHT = self-identified census race (SIA) white, EUR = matched to 1KGP European population, BLK = SIA black/African American, AFR = matched to 1KGP African population, ASN = SIA Asian, EAS = matched to 1KGP East Asian population, SAS = matched to 1KGP South Asian population, HSP = SIA Hispanic/Latino, AMR = matched to 1KGP Americas population, Census Race = total sample available for analysis using SIA, Pop. Match = total sample available for analysis using genetic population assignment, Analysis = ALL is all subjects analyzed together, Census Race or Pop. Match is each group analyzed separately and then meta-analyzed, nmax = maximum number of individuals in analysis, nmin and nmedian are the minimum and median number of individuals available per meta-analysis, Total SNPs = total SNPs available for genome-wide association analysis.
Discussion and Conclusions
As GWAS studies grow larger and more inclusive, guidance on how best to perform GWAS in highly diverse samples is needed. Here we outline a strategy for empirically assigning samples to more homogenous ancestry groups based on reference populations. This approach minimizes overall sample and marker loss and reduces within group genetic variance with the potential to increase discovery and replication power without increased inflation due to population stratification.
We demonstrate the utility of a multivariate approach to assigning samples to more homogeneous genetic groups by using minimum Mahalanobis distance based on 1KGP reference populations. By doing so, we were able to balance maximal genetic similarity within group assignment with minimal sample loss due to unknown and self-reported mixed ancestry. We were able to include 9% of the sample that may have been excluded otherwise. As more cohorts are combined through large scale collaborations, maximizing sample retention, particularly for understudied ancestry groups, while reducing population stratification remains an important endeavour.
Another practice supported herein is to perform pre-GWAS QC on homogenous ancestry groups rather than on the entire cohort. In addition to known QC metrics sensitive to population ancestry, such as heterozygosity rate and IBD, we show by applying QC on ancestry groups rather than on the entire sample there is a reduction in marker loss due to MAF and HWE thresholds. By removing markers that would be retained in subpopulation analyses potential etiologically relevant associations may be missed.
Although we see modest gains in QC metrics by applying our practices, there are limitations to consider. For example, even though the concordance rate between super-population assignment and self-reported census race was high (+92.4%), we cannot rule out the possibility that a portion of the 4.6% of samples that changed super-population category were due to sample mix-ups, which could impact results. However, it should be noted that our method can correct some response biases due to misrepresentation by respondents whether intentionally or unintentionally (e.g.; ethnic identity, truthfulness, technical problems). For example, 37 S4S respondents self-identified as census category white were empirically matched to SAS, representing 6.3% of the S4S SAS super population. Although a seemingly large proportion of cross-super population mismatch, this rate is consistent with previous reports of census race category endorsements of South Asians in the US (e.g.; Indian, Pakistani, Bangladeshi).32
There are limitations to applying Mahalanobis distance matching methods. First, Mahalanobis distance is generally employed to identify multivariate outliers. Here, we have used it to find the closest group for each sample in multivariate space. However, it is possible the sample may still be an outlier from the closest group, especially if the individual is from a population not well represented on the reference panel. Indeed, the 1KGP reference panel is missing human populations including those from Oceania and the Middle East. An important scientific venture will be the inclusion and representation of all human populations, which will see the current method improved with respect to both resolution and applicability.
An additional limitation of this method is the requirement of a large starting sample is needed since dividing the sample into genetically homogenous subpopulations leads to smaller groups. Indeed, significant structure remained in the SAS (n = 584) and AMR super-populations (n = 659) but further dividing the populations would yield problematically small sample sizes. Therefore, the need to create more homogenous subpopulations must be balanced against losing power from small subsamples.
Despite these limitations, the population-matching paradigm presented herein has several important extensions beyond standard GWAS such as reducing problems of stratification in conventional genetic methods including gene and pathway based tests, functional enrichment, aggregate genetic risk profiles, and rare variant analyses. Furthermore, the development of methods that facilitate inclusion of more diverse, and often understudied, populations in genetic studies will have significant downstream public health impacts, as made necessary by long-standing disparities in addiction research.
Acknowledgments
Spit for Science: The University Student Survey is funded by R37 AA011408 (to KSK) from the National Institute on Alcohol Abuse and Alcoholism, with support for DMD through K02 AA018755. Additional support for the project was obtained through National Institutes of Health P20 AA107828, P50 AA022537, Virginia Commonwealth University, and UL1 RR031990 from the National Center for Research Resources and National Institutes of Health Roadmap for Medical Research. BTW is supported by R37 AA011408 and NIH P50 grant AA022537. REP is supported by NIH T32 grant MH020030. ACE is supported by K01 grant AA021399. We would like to thank the students for making this study a success, as well as the many VCU faculty, students, and staff who contributed to the design and implementation of the project.
Footnotes
Conflict of Interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this paper.
References
- 1.Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:1001. doi: 10.1093/nar/gkt1229. Database issue. Accessed March 14, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hart AB, Kranzler HR. Alcohol dependence genetics: Lessons learned from genome-wide association studies (GWAS) and post-GWAS analyses. Alcohol Clin Exp Res. 2015;39(8):1312–1327. doi: 10.1111/acer.12792. Accessed March 14, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Price AL, Spencer CCA, Donnelly P. Progress and promise in understanding the genetic basis of common diseases. Proc Biol Sci. 2015;282:1821. doi: 10.1098/rspb.2015.1684. Accessed March 14, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36(5):512–517. doi: 10.1038/ng1337. Accessed March 11, 2016. [DOI] [PubMed] [Google Scholar]
- 5.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. Accessed March 11, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barnholtz-Sloan JS, McEvoy B, Shriver MD, Rebbeck TR. Ancestry estimation and correction for population stratification in molecular epidemiologic association studies. Cancer Epidemiol Biomarkers Prev. 2008;17(3):471–477. doi: 10.1158/1055-9965.EPI-07-0491. Accessed March 13, 2016. [DOI] [PubMed] [Google Scholar]
- 7.Vinkhuyzen AA, Wray NR, Yang J, Goddard ME, Visscher PM. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu Rev Genet. 2013;47:75–95. doi: 10.1146/annurev-genet-111212-133258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frichot E, Mathieu F, Trouillon T, Bouchard G, François O. Fast and efficient estimation of individual ancestry coefficients. Genetics. 2014;196(4):973–983. doi: 10.1534/genetics.113.160572. Accessed March 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ripke S, Wray NR, Lewis CM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18(4):497–511. doi: 10.1038/mp.2012.21. Accessed August 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Converge Consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523(7562):588–591. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. American journal of human genetics. 2007;81(5):1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34(8):816–834. doi: 10.1002/gepi.20533. Accessed April 12, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3: Genes, Genomes, Genetics. 2011;1(6):457–470. doi: 10.1534/g3.111.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu EY, Li M, Wang W, Li Y. MaCH-admix: Genotype imputation for admixed populations. Genet Epidemiol. 2013;37(1):25–37. doi: 10.1002/gepi.21690. Accessed March 14, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morris AP. Transethnic meta-analysis of genomewide association studies. Genet Epidemiol. 2011;35(8):809–822. doi: 10.1002/gepi.20630. Accessed April 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M. Genome-wide association studies in diverse populations. Nat Rev Genet. 2010;11(5):356–366. doi: 10.1038/nrg2760. Accessed April 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Webb BT, Edwards AC, Wolen AR, et al. Molecular genetic influences on normative and problematic alcohol use in a population-based sample of college students. Front Genet. 2017;8:30. doi: 10.3389/fgene.2017.00030. Accessed Apr 9, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dick DM, Nasim A, Edwards AC, et al. Spit for science: Launching a longitudinal study of genetic and environmental influences on substance use and emotional health at a large US university. Front Genet. 2014;5:47. doi: 10.3389/fgene.2014.00047. Accessed March 11, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi: 10.1016/j.jbi.2008.08.010. Accessed April 21, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10(1):5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 22.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5(6):e1000529. doi: 10.1371/journal.pgen.1000529. Accessed July 18, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sudmant PH, Rausch T, Gardner EJ, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526(7571):75–81. doi: 10.1038/nature15394. Accessed March 15, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. Accessed March 15, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini J, Howie B, Myers S, McVean G, Donnelly P, Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes; nature genetics. Nat Genet. 2007 doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 26.Bigdeli TB, Neale BM, Neale MC. Statistical properties of single-marker tests for rare variants. Twin Res Hum Genet. 2014;17(3):143–150. doi: 10.1017/thg.2014.17. Accessed April 13, 2016. [DOI] [PubMed] [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. Accessed August 13, 2016. [DOI] [PubMed] [Google Scholar]
- 28.Price AL, Weale ME, Patterson N, et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet. 2008;83(1):139. doi: 10.1016/j.ajhg.2008.06.005. Accessed April 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. Accessed August 13, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patterson N, Price A, Reich D. Population structure and eigenanalysis. PLOS Genetics. 2006;2(12):e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morning A. The racial self-identification of south asians in the united states. Journal of Ethnic and Migration Studies. 2001;27(1):61–79. doi: 10.1080/13691830125692. http://www.tandfonline.com/doi/abs/10.1080/13691830125692. [DOI] [Google Scholar]