

Abstract
Chitosan was selectively monophosphorylated via reaction with phosphorus oxychloride (POCl3) to enhance water solubility while avoiding polyphosphate formation. The use of POCl3 resulted in negligible product degradation (i.e., breakdown of O-glycosidic bonds) even after a 3 d reaction period (<5% weight loss). X-ray photoelectron spectroscopy (XPS) characterization of the POCl3-phosphorylated chitosan (P-chitosan) revealed a phosphorus to nitrogen (P/N) atomic ratio of 0.30. Phosphorus-31 nuclear magnetic resonance (31P NMR) spectroscopy verified the monophosphorylation of chitosan’s primary and secondary alcohols, and primary amines. The calcium chelation efficiency for the phosphorylated product approached 0.05 mg Ca2+ per mg of P-chitosan as measured by inductively coupled plasma-optical emission spectrometry (ICP-OES), indicating improved chelation over native chitosan. This selective monophosphorylation approach proved useful for modifying other biopolymers, including cellulose and alginate.
Keywords: Biopolymer, phosphorylated chitosan, phosphorus oxychloride, 31P NMR, calcium chelation, remineralization
Abstract Graphic
Introduction
The medical field is increasingly turning to renewable biopolymers, such as cellulose,1–2 collagen,3–4 alginate,5–7 and chitosan,8–10 for tissue engineering and drug delivery applications due to their favorable in vivo properties.11 For tissue reconstruction, such polymers tend to elicit a reduced immune response, lower implant rejection rates, and decreased toxicity as a result of biomimicry (i.e., the ability to mimic the natural tissue environment).12–14 The glycosidic linkages of biopolymers also promote facile biodegradation via enzymatic hydrolysis.13–15
Chitosan, a polysaccharide derived from crustacean shells, consists of repeating units of N-acetylglucosamine and D-glucosamine. The primary alcohols and amines of these units are readily accessible and enable a diverse range of chemical modifications.8 As such, researchers have engineered chitosan-based materials for wound dressing,16 metal and dye chelation,16 drug delivery,17 sensing,18 fuel cell,19 and antibacterial20–21 applications. For example, Ishiara22 reported the modification of chitosan with azides to enhance the mechanical properties of the biopolymer for wound healing applications. Sashiwa et al. added α-galactosyl pendants to chitosan to inhibit binding of human antibodies and evade the host immune response.23 The authors demonstrated the benefits of the α-galactosyl pendants in a pig liver xenotransplant model.
Chitosan’s poor solubility in physiological media represents a critical shortcoming that limits therapeutic utility. In general, chitosan of >20 kDa molecular weight is soluble only in acidic aqueous solutions.9 Though certain chemical modifications may alter functional aspects of chitosan for a given application, often the modification does not influence the solubility since many of these entities are electronically neutral (e.g., sugars) at pH 7.4. A satisfactory alternative to chemical modifications is to work with lower molecular weight oligosaccharides (~10 kDa) that are water-soluble.21 However, a large molecular weight scaffold is preferred for many biomedical applications (e.g., wound dressings and polymeric membranes).
Phosphorylation is a popular chemical modification strategy for improving the water solubility of chitosan and maintaining its molecular weight.22,24 At pH >6.5, phosphorylated chitosan (P-chitosan) consists of deprotonated phosphate groups that enables high water solubility for biological and industrial applications. For example, P-chitosan solutions have been used to enhance Ca2+ remineralization of dentine resulting from the high surface adsorption/interaction that P-chitosan promotes with enamel.25–27 Of note, P-chitosan would lose effectiveness in the absence of water solubility and likely size (i.e., molecular weight).
Many researchers have phosphorylated chitosan predominantly by reaction with solutions of phosphoric acid, triethyl phosphate, and phosphorus pentoxide (P2O5).28–30 Unfortunately, chitosan is insoluble in many solvents, thus the ensuing heterogeneous reaction conditions result in poor phosphorylation and irreproducibility.29–30 To address this, Nishi et al. 28 reported an alternative phosphorylation reaction that employs methanesulfonic acid to solubilize the chitosan. Insoluble P2O5 was employed as the phosphorylating reagent, a chemical compound that results in the undesirable formation of polyphosphoric acids/ions.31 These polyphosphate byproducts are either free or bound to the chitosan through inter- and intramolecular bridging, making their removal difficult, and complicating adequate characterization of the resulting P-chitosan’s structure. In general, previous reports on chitosan phosphorylation have not adequately characterized the resulting product. Indeed, 31P NMR data is rarely provided resulting in questionable or even outright neglected phosphorylation conversion efficiencies.
The shortcomings of P2O5-based phosphorylation warrant further exploration into alternative phosphorylating reagents. Reactants that facilitate monophosphorylation (i.e., polyphosphate formation does not occur) are essential to both prevent intra- and intermolecular polyphosphate bridging and enable more accurate characterization. To this end, we explored the use of phosphorus oxychloride (POCl3) to generate P-chitosan. Phosphorus oxychloride is a liquid phosphorylating reagent utilized in the synthesis of phosphate esters and cross-linked starch and cellulose because of its high solubility in organic solvents.32–36 Furthermore, negligible self-reaction should prevent the formation of polyphosphates, making it a promising alternative to P2O5. Herein, we report the selective monophosphorylation of chitosan using POCl3 in methanesulfonic acid to produce a water-soluble product of high molecular weight.
Experimental
Materials
Low molecular weight chitosan (degree of acetylation: 27%), phosphorus pentoxide (P2O5), phosphorus oxychloride (POCl3), low viscosity alginic acid sodium salt, methanesulfonic acid (MSA), cellulose, deuterium oxide (D2O), D-glucosamine, D-glucosamine 1-phosphate, D-glucosamine 6-phosphate, and N-acetylglucosamine were purchased from Sigma (St. Louis, MO). Tetrahydrofuran (THF), sodium hydroxide (NaOH), nitric acid (HNO3), orthophosphoric acid (H3PO4), diethyl ether, and calcium chloride (CaCl2) dihydrate were purchased from Fisher Scientific (Fair Lawn, NJ). Water was purified to a resistivity of 18.2 MΩ·cm and a total organic content ≤6 ppb using a Millipore Milli-Q UV Gradient A10 System (Bedford, MA)
Synthesis of phosphorylated chitosan
Chitosan (200 mg) was added to a round-bottomed flask and dissolved in 10 mL methanesulfonic acid. After complete dissolution, 2.060 mL POCl3 were injected into this stirring solution. The reaction was allowed to proceed at room temperature for up to 72 h. To end the reaction, 1.40 mL of water were added to the flask and stirred for another 15 min. The solution was then transferred to centrifuge tubes, precipitated with diethyl ether, and centrifuged (4000 × g for 3 min) to pellet the P-chitosan. The P-chitosan was washed twice with THF, once with ethanol, dried under vacuum overnight, and stored at −20 °C until use.
1H and 31P nuclear magnetic resonance (NMR) spectroscopy
All NMR spectra were collected at 23 °C in 5 mm NMR tubes using a Bruker AVIII 600 MHz spectrometer equipped with a Quattro Nucleus Probe (QNP) C-P-N cryoprobe. Samples were prepared at ~2 mg/mL in D2O (1H NMR) or a 1:3 volumetric ratio of D2O to 50 mM NaOH (31P NMR). Proton spectra were acquired using a conventional 1-D pulse sequence with 16 scans. Phosphorus spectra were collected using a standard proton decoupled pulse sequence with 800 scans. Data was processed using Bruker’s TopSpin software. Relevant 1H NMR data of P-chitosan (600 MHz, D2O, δ): 1.9 (C7: CHNHCOCH3), 2.67 (SO3CH3), 3.1 (C2: CHCHNH3+), 3.4–3.9 (C3, C4, C5, C6: OHCH, OCHCH(OH)CH(NH2), OHCH2CH, OHCH2CH.
X-ray photoelectron spectroscopy (XPS)
Samples for XPS analysis were prepared by casting 20 μL of 1 mg/mL P-chitosan in water onto gold-sputtered glass slides and evaporating the water under vacuum overnight. The sample-coated gold substrates were analyzed using a Kratos Axis Ultra DLD X-ray photoelectron spectrometer equipped with a monochromatic Al Kα X-ray excitation source (base pressure = 6 × 10−9 torr). Emitted photoelectrons were measured at a 90° take-off angle. A pass energy of 20 eV was utilized for all high-resolution scans. A standard Shirley baseline correction algorithm was employed after data collection with relative sensitivity factors from the Kratos Vision software to calculate atomic concentrations.
Molecular weight determination
The average molecular weight and polydispersity of the polymers were determined using gel permeation chromatography (GPC). Low molecular weight chitosan (1 mg/mL in 2 vol % acetic acid) and P-chitosan (1 mg/mL in water) solutions were passed through a 0.22 μm diameter syringe filter and injected (100 μL) onto a Waters 2695 GPC column (flow rate = 0.925 mL/min) equipped with a Waters 2414 refractometric detector. Three Ultrahydrogel 1000, 7.8×300 mm columns were connected in series with an Ultrahydrogel guard column. Molecular weight calibration curves were created using polyethylene oxide (PEO) standards in a range from 25–881 kDa.
Calcium chelation
P-chitosan (10.0 mg) was dissolved in a 10 mM CaCl2 solution (2.5 mL) at 37 °C. The mixture was stirred in a 37 °C incubator for 2 min. The solution was then transferred to a centrifuge tube, diluted with 10.0 mL THF, and centrifuged at 4000 × g for 2 min. After decanting the liquid into a glass vial, the solvent was evaporated under vacuum. The remaining calcium chloride was digested in a known volume of 2 vol % HNO3 prior to calcium content analysis by inductively coupled plasma-optical emission spectrometry (ICP). The ICP was first calibrated using calcium standards (0.1–25 ppm) prepared in 2 vol % HNO3. The calcium emission intensity at 317.93 nm was used for quantification.
Results and Discussion
The use of anhydrous liquid organic acids during the synthesis of P-chitosan is critical to prevent undesirable hydrolysis of the phosphorylating reagent. While other organic acids are available, methanesulfonic acid (MSA) is one of the few liquid organic acids that is sufficiently acidic enough to fully solubilize chitosan. Phosphorylating reagents such as P2O5 are insoluble in organic acids like MSA, but this issue can be resolved by employing POCl3.
Chitosan’s reactivity with POCl3 was initially examined using 1H NMR spectroscopy (Fig. 1). The protonation of chitosan’s primary amine was followed throughout the reaction and washing steps by monitoring the associated alpha-carbon (C2) proton peak. When protonated, the peak appears at 3.1 ppm. Washing P-chitosan with 50 mM NaOH, which deprotonates the amine, shifts the peak at 3.1 ppm upfield to 2.6 ppm (Fig. S1). In addition to the protonation state of the amine, 1H NMR may be used to confirm the presence of ionically-bound MSA. Following extensive washing of P-chitosan to remove free acid, the peak near 2.7 ppm from the methyl hydrogens of MSA revealed that the methanesulfonate anion serves as a counterion to chitosan’s protonated amine. Subsequential washings with 2 M HCl replaces the methanesulfonate with a chloride counterion (Fig. S2). Unfortunately, actual phosphorylation is not directly observed using 1H NMR.
Fig. 1.

1H NMR spectrum of phosphorylated chitosan (molecular structure shown above).
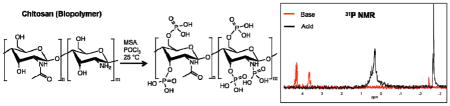
31P NMR spectroscopy may be employed to directly examine P-chitosan phosphorylation. Sample preparation under basic conditions was essential to differentiate 31P NMR phosphate peaks from one another. When samples are prepared solely in neutral D2O (Fig. 2a), a broad peak appears near 0.5 ppm, similar in location to that of phosphoric acid. Under basic conditions (13 mM NaOH in D2O) the appended phosphate groups are deprotonated, resulting in adequate peak separation due to a downfield shift (Fig. 2b). The separated peaks at 4.30 and 3.65 ppm are assignable to the phosphates attached to the primary and secondary alcohols of chitosan, respectively. Spectra acquired from solutions of D-glucosamine 1-phosphate and D-glucosamine 6-phosphate corroborate these peak transitions and assignments (Fig. S3 and S4).
Fig. 2.

31P NMR spectra of phosphorylated chitosan (molecular structure shown above) prepared in (a) D2O and (b) 1:3 volumetric ratio of D2O to 50 mM NaOH. Numeric labels (1, 2, 3) indicate the associated phosphate group.
The 31P NMR peak near −2.5 ppm (Fig. 2) is assigned to the monophosphorylation of chitosan’s primary amine. While Wang et al. implied phosphorylation of the amine, clear evidence (31P NMR characterization) was not provided as to support primary amine phosphorylation.30 We sought to confirm amine phosphorylation by reacting other biopolymers (i.e., alginate, cellulose) lacking primary amines with POCl3 as well. 31P NMR analysis of both phosphorylated alginate and cellulose revealed no peaks near −2.5 ppm (Fig. S5 and S6), confirming the identity of the −2.5 ppm peak as an amine-bound phosphate (both spectra contained peaks at ~4 ppm indicating phosphorylation of their alcohol groups). In contrast to prior work,30 we believe that only the primary amines and not the acetylated amines (i.e., amides) are participating in the reaction. The electron-withdrawing properties of the carbonyl group, in conjunction with resonance, likely reduce the nucleophilicity of the nitrogen atom, preventing the amide from attacking POCl3. To verify this hypothesis, we synthesized both phosphorylated D-glucosamine containing no amide groups and N-acetylglucosamine with amide groups under identical conditions that were used to prepare P-chitosan. As predicted, the 31P NMR spectrum of phosphorylated D-glucosamine revealed a peak at −2.5 ppm, while that of N-acetylglucosamine lacked the −2.5 ppm peak (Fig. S7 and S8). These results confirm that POCl3 phosphorylation only occurs at the alcohols and primary amines of chitosan.
The POCl3 reaction was also compared to the P2O5 reaction following a published protocol.28 While both phosphorylated amine and alcohol peaks were clearly apparent in the 31P NMR spectra (Fig. 3) after chitosan phosphorylation with P2O5, several additional peaks appeared at more negative chemical shifts that were indicative of polyphosphate formation. In fact, the NMR data reveals the presence of both pyrophosphates37 (−6 to −11 ppm) and tripolyphosphates37 (−21 to −25 ppm). In contrast to the P2O5 reaction, the POCl3 reaction generates no polyphosphates, allowing for more reliable characterization and inherent P-chitosan homogeneity.
Fig. 3.

31P NMR spectra of phosphorylated chitosan synthesized by the (a) POCl3 method and (b) P2O5 method. Peaks located between −6 and −11 are assigned as pyrophosphates, while those located between −20 and −25 are tripolyphosphates.
Controlling phosphorylation efficiency
The extent of phosphorylation as a function of POCl3 and the chitosan concentration was characterized using X-ray photoelectron spectroscopy (XPS). The nitrogen atomic percent (N 1s peak) was used as a normalization factor across syntheses as changes in nitrogen content are unlikely to occur during phosphorylation. The molar ratio of POCl3 relative to chitosan alcohol groups was varied from 1 to 10, while the overall reaction time (48 h) and chitosan concentration (20 mg/mL) were kept constant. At molar ratios of POCl3 <10, phosphorylation was not observed at any appreciable level (i.e., below the XPS limit of detection). An increase in the P/N ratio to 0.232 was measured after reaction with a 10-molar excess of POCl3. The large excess of POCl3 required is attributed to the slower and less favorable reaction kinetics under strongly acidic conditions.
Tuning the phosphorylation efficiency through simple changes in the reaction conditions (e.g., chitosan concentration) is highly desirable for chemical versatility. As shown in Table 1, the P/N ratio correlated well with the concentration of chitosan from 5 to 80 mg/mL. We also monitored the Cl/N atomic ratio (zero for all concentrations) to ensure post-synthetic removal of unreacted POCl3 and appended chlorine groups. Likewise, the P/N ratio was measured as a function of reaction time for a 20 mg/mL chitosan solution and 10-molar excess of POCl3 (Fig. 4). The phosphorus content increased steadily over the course of 3 d, allowing for precise tunability (up to 72 h) of the phosphate content. An ability to decrease the reaction time while maintaining high P/N ratios was also observed by heating the reaction. For example, increasing the temperature of the 3 h reaction to 37 °C resulted in atomic ratios (P/N = 0.162) near those obtained at 25 °C for 24 h (P/N = 0.142).
Table 1.
Chitosan concentration effect on phosphorylation P/N atomic ratio.a
| Chitosan concentration (mg/mL) | P/N atomic ratio |
|---|---|
| 5 | n/a |
| 20 | 0.232 ± 0.029 |
| 40 | 0.279 ± 0.038 |
| 80 | 0.436 ± 0.066 |
Error bars represent the standard deviation from n≥3 separate preparations using a 48 h reaction time. The chloride-to-nitrogen atomic ratio was zero for all concentrations.
Fig. 4.

Atomic P/N ratio as a function of reaction time for reactions with 20 mg/mL chitosan (10 molar ratio of POCl3).
Molecular weight
Degradation of P-chitosan is a concern given that the phosphorylation reaction occurs in a strong organic acid. The O-glycosidic bonds that link monomers of chitosan together are prone to cleavage in acidic environments.38–39 The change in P-chitosan molecular weight versus reaction time was investigated using gel permeation chromatography (GPC). Of note, the molecular weight of unmodified chitosan was ~85,000 g/mol (experimentally determined), a value within the range of typical low molecular weight chitosan materials (20,000–130,000 g/mol).40 As shown in Table 2, only a slight reduction in molecular weight was observed over a 72 h reaction period. In the absence of phosphorylation (i.e., without the addition of POCl3), the molecular weight was ~85,000 g/mol, indicating negligible acid degradation (Fig. S9). The anhydrous nature of the reaction likely mitigates degradation due to minimal hydrolysis (the primary mechanism of breaking glycosidic bonds). The consistent molecular weight also suggests that the water solubility of P-chitosan largely results from the addition of anionic phosphate groups.
Table 2.
Molecular weight and dispersity (Đ) of P-chitosan as a function of POCl3 reaction time.a
| Reaction time (h) | Molecular weight (Mn) | Đ |
|---|---|---|
| 0 | 85,100 ± 200 | 1.72 ± 0.05 |
| 24 | 78,700 ± 400 | 1.98 ± 0.05 |
| 48 | 84,300 ± 350 | 1.60 ± 0.05 |
| 72 | 83,800 ± 250 | 1.41 ± 0.09 |
Error bars represent the standard deviation from n≥3 separate preparations.
Calcium chelation efficiency
A practical application for phosphorylated biopolymers is metal ion chelation. For example, phosphorylated chitosan effectively complexes Ca2+ to enhance tooth enamel remineralization and prevent future acid-catalyzed demineralization.26,41 We examined the Ca2+ chelation properties of P-chitosan by measuring total calcium chelation as a function of phosphorylation degree (Table 3). P-chitosan chelation was compared to that observed from control chitosan (i.e., no phosphorylation). After brief exposure to a CaCl2 solution, and removal of the polymer, the supernatant was analyzed for Ca2+ as an indication of P-chitosan chelation efficiency. Chitosan lacking phosphorylation chelated approximately 24.9 μg Ca2+ per mg chitosan, an unsurprising result given previous research demonstrating chitosan’s native ability to bind divalent metal cations.42 The total amount of chelated calcium increased predictably with the P/N atomic ratio, with 46.2 μg Ca2+ per mg chitosan achieved at the greatest P/N atomic ratio (0.30). The data collected here supports the employment of P-chitosan into applications where calcium chelation is desired (e.g., oral care).
Table 3.
Calcium chelation amount of P-chitosan as a function of phosphorylation degree.
| P/N atomic ratio | Chelated calcium (μg Ca2+/mg chitosan) |
|---|---|
| 0 | 24.9 ± 0.6a |
| 0.066 | 25.1 ± 1.8 |
| 0.142 | 41.9 ± 0.8 |
| 0.232 | 43.5 ± 1.0 |
| 0.302 | 46.2 ± 0.7 |
Determined using a water-soluble 5 kDa chitosan without any phosphorylation.
Conclusions
The reaction of chitosan with POCl3 is a simple and reliable strategy for achieving monophosphorylation of chitosan’s amine and alcohols. Precise tuning of the extent of phosphorylation is possible by adjusting specific reaction conditions (e.g., chitosan concentration, reaction time). Although alginate and cellulose were not extensively studied herein, similar monophosphorylation using POCl3 should be expected. Lastly, a range of P-chitosan architectures (e.g., particles, hydrogels) could be expected given the preservation of high molecular weight throughout the reaction.
Supplementary Material
Acknowledgments
Funding for this research was provided by the National Institutes of Health (DE025207). We also thank Dr. Marc ter Horst for assistance with 31P NMR, Dr. Carrie Donley at the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) for help with XPS, and Rufai Ibrahim for collecting GPC data.
References
- 1.Klemm D, Heublein B, Fink H, Bohn A. Angew Chem Int Ed. 2005;44:3358–3393. doi: 10.1002/anie.200460587. [DOI] [PubMed] [Google Scholar]
- 2.Ummartyotin S, Manuspiya H. Renew Sust Energ Rev. 2015;41:402–412. [Google Scholar]
- 3.Chattopadhyay S, Raines RT. Biopolymers. 2014;101:821–833. doi: 10.1002/bip.22486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sell SA, McClure MJ, Garg K, Wolfe PS, Bowlin GL. Adv Drug Deliv Rev. 2009;61:1007–1019. doi: 10.1016/j.addr.2009.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Lee KY, Mooney DJ. Prog Polym Sci. 2012;37:106–126. doi: 10.1016/j.progpolymsci.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venkatesan J, Bhatnager I, Manivasag P, Kang K, Kim S. Int J Biol Macromolec. 2015;72:269–281. doi: 10.1016/j.ijbiomac.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 7.Zia K, Zia F, Zuber M, Rehman S, Ahmad M. Int J Biol Macromolec. 2015;79:377–387. doi: 10.1016/j.ijbiomac.2015.04.076. [DOI] [PubMed] [Google Scholar]
- 8.Prabharan M. J Biomater Appl. 2008;23:5–36. doi: 10.1177/0885328208091562. [DOI] [PubMed] [Google Scholar]
- 9.Kumar M, Muzzarelli R, Muzzarelli C, Sashiwa H, Domb A. Chem Rev. 2004;104:6017–6084. doi: 10.1021/cr030441b. [DOI] [PubMed] [Google Scholar]
- 10.Thakur VK, Thakur MK. ACS Sustainable Chem Eng. 2014;2:2637–2652. [Google Scholar]
- 11.Yates MR, Barlow CY. Resourc Conserv Recy. 2013;78:54–66. [Google Scholar]
- 12.Balakrishnan B, Banerjee R. Chem Rev. 2011;111:4453–4474. doi: 10.1021/cr100123h. [DOI] [PubMed] [Google Scholar]
- 13.Dang JM, Leong KW. Adv Drug Deliv Rev. 2006;58:487–499. doi: 10.1016/j.addr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Vieira M, da Silva M, dos Santos L, Beppu M. Euro Polym J. 2011;47:254–263. [Google Scholar]
- 15.Franková L, Fry SC. J Exp Bot. 2013;64:3519–3550. doi: 10.1093/jxb/ert201. [DOI] [PubMed] [Google Scholar]
- 16.Ravi Kumar M. React Funct Polym. 2000;46:1–27. [Google Scholar]
- 17.Sinha VR, Singla AK, Wadhawan S, Kaushik R, Kumria R, Bansal K, Dhawan S. Int J Pharm. 2004;274:1–33. doi: 10.1016/j.ijpharm.2003.12.026. [DOI] [PubMed] [Google Scholar]
- 18.Karra S, Gorski W. Anal Chem. 2013;85:10573–10580. doi: 10.1021/ac4026994. [DOI] [PubMed] [Google Scholar]
- 19.Ma J, Sahai Y. Carbohydr Polym. 2013;92:955–975. doi: 10.1016/j.carbpol.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Reighard KP, Hill DB, Dixon GA, Schoenfisch MH. Biofouling. 2015;31:775–787. doi: 10.1080/08927014.2015.1107548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu Y, Slomberg DL, Schoenfisch MH. Biomaterials. 2014;35:1716–1724. doi: 10.1016/j.biomaterials.2013.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishiara M. Trends Glycosci Glyc. 2002;14:331–341. [Google Scholar]
- 23.Sashiwa H, Thompson JM, Das SK, Shigemasa Y, Tripathy S, Roy R. Biomacromolecules. 2000;1:303–305. doi: 10.1021/bm005536r. [DOI] [PubMed] [Google Scholar]
- 24.Madihally SV, Matthew H. Biomaterials. 1999;20:1133–1142. doi: 10.1016/s0142-9612(99)00011-3. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Li Y, Sun X, Kishen A, Deng X, Yang X, Wang H, Cong C, Wang Y, Wu M. J Mater Sci: Mater Med. 2014;25:2619–2628. doi: 10.1007/s10856-014-5285-2. [DOI] [PubMed] [Google Scholar]
- 26.Yokogawa Y, Reyes JP, Mucalo MR, Toriyama M, Kawamoto Y, Suzuki T, Nishizawa K, Nagata F, Kamayama T. J Mater Sci Mater Med. 1997;8:407–412. doi: 10.1023/a:1018549404092. [DOI] [PubMed] [Google Scholar]
- 27.Chesnutt BM, Yuan Y, Brahmandam N, Yang Y, Ong JL, Haggard WO, Bumgardner JD. J Biomed Mater Res A. 2007;82:343–353. doi: 10.1002/jbm.a.31070. [DOI] [PubMed] [Google Scholar]
- 28.Nishi N, Ebina A, Nishimura S, Tsutsumi A, Hasegawa O, Tokura S. Int J Biol Macromol. 1986;8:311–317. [Google Scholar]
- 29.Lee YM, Shin EM. J Membr Sci. 1991;64:145–152. [Google Scholar]
- 30.Wang K, Liu Q. Carbohydr Res. 2014;386:48–56. doi: 10.1016/j.carres.2013.12.021. [DOI] [PubMed] [Google Scholar]
- 31.Masson JF. Energy Fuels. 2008;22:2637–2640. [Google Scholar]
- 32.Miao Z, Zhang J, You L, Wang J, Sheng C, Yao J, Zhang W, Feng H, Guo W, Zhou L, Zhu L, Cheng P, Che X, Wang W, Luo C, Xu Y, Dong G. Bioorg Med Chem. 2010;18:3140–3146. doi: 10.1016/j.bmc.2010.03.039. [DOI] [PubMed] [Google Scholar]
- 33.Illy N, Fache M, Ménard R, Negrell C, Caillol S, David G. Polym Chem. 2015;6:6257–6291. [Google Scholar]
- 34.Jayakumar R, Selvamurugan N, Nair SV, Tokura S, Tamura H. Int J Biol Macromolec. 2008;43:221–225. doi: 10.1016/j.ijbiomac.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Lohmar R, Sloan JW, Rist CE. J Am Chem Soc. 1950;72:5717–5720. [Google Scholar]
- 36.Reid JD, Mazzeno LW. Ind Eng Chem. 1949;41:2828–2831. [Google Scholar]
- 37.Manoi K, Rizvi SS. Carbohydr Polym. 2010;81:687–694. [Google Scholar]
- 38.Einbu A, Grasdalen H, Vårum KM. Carbohydr Res. 2007;342:1055–1062. doi: 10.1016/j.carres.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 39.Einbu A, Vårum KM. Biomacromolecules. 2007;8:309–314. doi: 10.1021/bm0608535. [DOI] [PubMed] [Google Scholar]
- 40.Sumiyoshi M, Kimura Y. J Pharm Pharmacol. 2010;58:201–207. doi: 10.1211/jpp.58.2.0007. [DOI] [PubMed] [Google Scholar]
- 41.Cao C, Mei M, Li Q, Lo E, Chu C. Int J Mol Sci. 2015;16:4615–4627. doi: 10.3390/ijms16034615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gamage A, Shahidi F. Food Chem. 2007;104:989–996. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.