

Abstract
Alzheimer's disease (AD), with a typical pathological hallmark of amyloid-beta (Aβ)-containing plaques and neurofibrillary tangles, is one of the most common types of chronic neurodegenerative diseases. Aβ oligomers serve a crucial role in the pathogenesis of AD, and lead to neuronal loss. However, the precise mechanism of Aβ oligomers in AD remains to be elucidated. The present study demonstrated that 10 µM Aβ-42 activated the caspase signaling pathway, and induced significant apoptosis in primary cultured mouse cerebral cortical neurons. The results of reverse transcription-quantitative polymerase chain reaction and western blotting demonstrated that Aβ-42 (10 µM) also significantly upregulated the transcription and expression of the mitochondrial fission protein dynamin-related protein 1 (Drp1), and downregulated the transcription and expression of mitochondrial fusion proteins, including mitofusin 1/2 (Mfn1/2) and mitochondrial dynamin like GTPase (OPA-1). Neurons were transfected with pDsRed2-Mito for mitochondrial imaging, which revealed that 10 µM Aβ-42 induced mitochondrial fission in cortical neurons. In addition, 2′,7′-dichlorodihydrofluorescein diacetate and tetramethylrhodamine ethyl ester staining indicated that Aβ-42 increased the reactive oxygen species (ROS) level and reduced mitochondrial membrane potential in neurons. Inhibition of Drp1 activity by Mdivi-1 efficiently prevented Aβ-42-induced ROS production and disruption of mitochondrial membrane potential. Loss of mitochondrial membrane potential may activate PTEN-induced putative kinase 1 (Pink1), the prominent sensor for mitochondrial damage, and trigger the process of mitophagy to remove the damaged mitochondria. In the present study, western blotting revealed that the levels of autophagy marker microtubule-associated proteins 1A/1B light chain 3B (LC3B) and Pink1 were upregulated after Aβ-42 stimulation. In conclusion, these data indicated that Aβ-42 induces neuronal apoptosis by targeting mitochondria, including promotion of mitochondrial fission, disruption of mitochondrial membrane potential, increasing intracellular ROS level and activation of the process of mitophagy. Therefore, mitochondria may represent a potential therapeutic target for AD in the future.
Keywords: Amyloid β-42, mitochondria, reactive oxygen species, mitophagy, apoptosis
Introduction
Alzheimer's disease (AD) is one of the most common irreversible neurodegenerative disorders. The typical clinical features in AD are progressive cognitive and memory impairment (1). With the development of medicine, the average life span of the population continues to increase. However, AD is an age-associated disease; it was reported that in developed countries, it affects 7% of individuals aged >65, and 40% of individuals aged >80 (2,3). The estimated prevalence of AD in 2015 was 44 million people throughout the world, and it is estimated that this figure will double by 2050 (4). Unfortunately, there is still no effective therapy or vaccine for AD patients, despite decades of AD research. As a result, AD has become a serious health issue in the world (2). Therefore, it is urgent to investigate the underlying mechanism in the pathogenesis of AD. The neuropathological features of AD are the formation of amyloid β (Aβ) plaques, neurofibrillary tangles, and the progressive loss of synapses and neurons (5,6). Among these, Aβ plaques is most widely accepted as the root cause of AD. In the amyloid theory, the extracellular amyloid plaques damage synapses, induces neuronal cell death, and eventually leads to AD (6). Aβ plaques primarily consist of Aβ peptides yielded through the amyloidogenic pathway. Following the sequential enzymatic cleavage of β- and γ-secretases on amyloid precursor protein (APP), amyloid peptides of 39–43 amino acids and p3 are released from APP distributed in the neuronal membrane (1,7). Although p3 is not neurotoxic, it aggregates into fibers (8). Three main isoforms of Aβ are produced after β- and γ-secretase cleavage, including Aβ-40, Aβ-42 and Aβ-43. Of these, Aβ-42 and Aβ-43 are considered neurotoxic (9,10). Aβ-42 has been most studied, and is also widely accepted as the primary amyloidogenic form in AD (2).
Aβ peptides are hydrophobic with an intrinsic tendency to self-assemble to form aggregates. In aqueous solution, monomeric forms of Aβ peptide exhibit a random coil structure (11). However, Aβ peptides form different sizes and conformations of homodimers or oligomers with β-pleated sheet structures after incubation at 37°C for several days in a nucleation-dependent process, and Aβ-42 becomes the ‘seed’ in Aβ oligomers (12,13). It was reported that the mixture of monomeric and Aβ-42 oligomers is more toxic than monomeric, protofibrillar fractions or fibril (14). On the other hand, several studies have indicated that Aβ oligomers can directly incorporate into membranes and form pore-like structures (11,15,16). From observations in nuclear magnetic resonance, Aβ-42 pores consist of tetrameric and hexameric β-sheet subunits (16). The Aβ pore-like structures are selectively permeable to Ca2+. Thus, these pore-like structures formed by Aβ oligomeres are termed as ‘amyloid channels’. With high selective permeability for Ca2+, these ‘amyloid channels’ cause a rapid increase in [Ca2+]i, and disrupt the calcium homeostasis in neurons (17). Calcium homeostasis serves an important role in maintenance of neuronal synaptic plasticity, which is crucial for the process of memory formation (18). In the early stages of AD, the ‘amyloid channels’ constructed by Aβ oligomers disrupt calcium homeostasis, cause synaptic degeneration (synaptotoxicity), and lead to memory impairment. In addition, the Aβ oligomers are neurotoxic, and induce neuronal cell death in the later stage of AD (11). Although Aβ oligomers serve a crucial role in the pathogenesis of AD, there are some debates about its underlying mechanism. Therefore, the present study investigated whether Aβ-42 induces neuronal damage by targeting mitochondria.
Mitochondria serve an essential function in cells through the production of energy and the ability to regulate intracellular Ca2+. As such, they are involved in a variety of cellular processes, including survival, proliferation and apoptosis (19–21). Neurons with unique cellular processes heavily rely on mitochondria for energy supply, and are also sensitive to any defects in mitochondria. Mitochondria are also highly dynamic organelles and move through the cell with frequent fission and fusion events (22). Highly conserved dynamin-associated GTPases are identified as the mediator of mitochondrial dynamics. Dynamin-related protein 1 (Drp1) is involved in the process of mitochondrial fission, while mitofusin (Mfn)1/2 and mitochondrial dynamin like GTPase (OPA-1) are required for mitochondrial fusion in mammalian cells (23). Mitochondrial trafficking to pre- and postsynaptic sites serves an important role in control of basal synaptic transmission and plasticity (24,25). A decline in mitochondrial function is well recognized in neurodegenerative diseases and aging. Hirai et al (26) and Wang et al (27) observed that the quantity of mitochondria with normal morphology was decreased in AD neurons, whereas mitochondria with abnormal cristae was increased. On the other hand, there is an important quality control system, mitophagy, to remove those damaged mitochondria in cells. PTEN-induced putative kinase 1 (Pink1)/Parkin have been identified as crucial components of mitophagy (28). Pink1/Parkin acts as a sensor for mitochondrial quality, and is activated after the loss of matrix metalloproteinase (29,30). Furthermore, specific removal of individual damaged mitochondria seems to be supported by a fission process that separates healthy from defective organelles (31). However, whether Aβ oligomers induce neuronal apoptosis through promoting mitochondrial fission, disrupting MMP and activating mitophagy in AD remains to be further elucidated.
The present study established a primary culture of mouse cerebral cortical neurons, and stimulated neurons with 10 µM Aβ-42. It was demonstrated that Aβ-42 not only induced neuronal apoptosis by activating the caspase pathway, but also promoted mitochondrial fission, increased reactive oxygen species (ROS) production, disrupted MMP, and may activate the process of mitophagy by upregulation of microtubule-associated proteins 1A/1B light chain 3B (LC3B) and Pink1. Therefore, these data suggested that mitochondria may be an important potential target of Aβ-42 in the pathogenesis of AD.
Materials and methods
Primary culture of mouse cerebral cortical neurons
C57BL/6 mice (n=60; 45 female, 15 male; age, 12 weeks; 24±3 g) were purchased from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China), and ethical approval was obtained from the Shanghai Animal Institution, Chinese Academy of Sciences (Shanghai, China). All mice were maintained at 20–25°C with 45–55% humidity, a 12-h light/dark cycle and free access to food and water. The pregnant female C57BL/6 mice on day 13 or 14 were anesthetized via an intraperitoneally injection of 5 mg/100 g pentobarbital solution. The cerebral cortex from the ~13–14-embryonic day fetal C57BL/6 mice was then removed under aseptic conditions, and treated with 0.125% trypsin (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and 0.004% DNase-I (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 37°C for 15 min and mechanically dissociated. The cerebral cortical neurons were plated on poly-L-lysine (Sigma-Aldrich; Merck KGaA) and laminin (Roche Diagnostics, Indianapolis, IN, USA)-coated glass bottomed or plastic-bottomed 35-mm culture dishes. Cell density was ~25,000–30,000/35-mm dish for microscopy or ~45,000–50,000/35-mm dish for western blotting. Cultures were maintained in neurobasal plating media supplemented with B27 Supplement (1 ml/50 ml), 0.5 mM Glutamine solution, 25 µM Glutamate, 100 µg/ml penicillin-streptomycin (P/S), 1 mM HEPES and 10% fetal bovine serum (FBS) at 37°C in 5% CO2. On the second day, half the volume of culture media was replaced with same volume of neurobasal feeding media [Neurobasal Media containing B27 Supplement (1 ml/50 ml), 0.5 mM Glutamine solution, 100 µg/ml P/S and 1 mM HEPES]. The medium, B27 supplement, Glutamine, FBS and P/S were from Invitrogen; Thermo Fisher Scientific, Inc. Subsequently, neurons were fed every 4 days by replacing half of the old media with same volume of neurobasal feeding media. Cultures were used for experiments on days 9–11.
Preparation of Aβ-42 peptide
Aβ-42, a toxic peptide fragment derived from APP, was purchased from Sigma-Aldrich; Merck KGaA as previously described (32). Aβ-42 peptide was resuspended in dimethyl sulfoxide (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China) to 5 mM and then diluted to 100 µM in sterile PBS (pH 7.4). The suspension was allowed to oligomerize for 5 days at 37°C, and then diluted to the desired concentrations immediately before being added to the cell culture medium.
Neuronal apoptosis assay
Firstly, mouse cerebral cortical neuronal cultures were exposed to 0, 2.5, 5 or 10 µM Aβ-42, and the morphology of neurons was observed under microscope at 0, 12 and 24 h after treatment with Aβ-42. To detect the apoptotic neurons, Hoechst 33258 staining was conducted. In brief, the neurons were washed with PBS at 24 h after incubation with Aβ-42, and fixed with 4% paraformaldehyde for 10 min. To label neurons, immunofluorescence staining was performed with a microtubule-associated protein 2 (anti-MAP2; cat. no. 8707; 1:200; Cell Signaling Technology, Inc., Danvers, MA, USA) primary antibody for 2 h at room temperature and an Alexa Fluor 488-conjugated goat anti-rabbit secondary fluorescence antibody (cat. no. SA00006-2; 1:200; ProteinTech Group, Inc., Chicago, IL, USA) for 1 h at room temperature. Cells were stained with Hoechst 33258 (cat. no. 94403; Sigma-Aldrich; Merck KGaA) for 5 min at room temperature for nucleus staining according to the manufacturer's protocol. Fluorescence of MAP2 and Hoechst 33258 were detected and analyzed under a fluorescence microscope (Olympus Corporation, Tokyo, Japan). Neurons with condensed Hoechst 33258 were counted as dead cells.
Western blot analysis
After treatment with Aβ-42, neurons were harvested and lysed in radioimmunoprecipitation assay lysis buffer (Beijing Solarbio Science & Technology Co., Ltd.) according to the manufacturer's protocol. Protein concentrations in whole cell lysates were determined using a bicinchoninic acid assay kit (Thermo Fisher Scientific, Inc.). Whole cell lysates were mixed with an equal volume of 2X loading buffer (cat. no. P1019; Beijing Solarbio Science & Technology Co., Ltd.), sonicated, boiled for 5 min and stored at −20°C prior to use. A total of 20 µg protein for each sample was separated by 8% SDS-PAGE, and transferred onto a PVDF membrane (Millipore, Massachusetts, USA). The membrane was blocked with 5% skim milk in TBS with 0.1% Tween-20 (TBST) buffer for 1 h at room temperature, and then incubated for 2 h at room temperature with the following primary antibodies: Rabbit anti-Caspase-3 (cat. no. 9662), anti-Drp1 (cat. no. 8570), anti-Mfn2 (cat. no. 9482), anti-LC3B (cat. no. 3868) and anti-Pink1 (cat. no. 6946) (all from Cell Signaling Technology, Inc.; 1:1,000), rabbit anti-Mfn1 (cat. no. ab104274) and anti-OPA-1 (cat. no. ab42364; both from Abcam, Cambridge, UK; 1:1,000), and rabbit anti-GAPDH (cat. no. sc-25778; Santa Cruz Biotechnology, Dallas, TX, USA; 1:1,000). After three washes with TBST, the membranes were further incubated with a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (cat. no. HS101; Beijing TransGen Biotech, Co., Beijing, China; 1:2,000) for 2 h at room temperature. A chemiluminescence assay was performed with Amersham Enhanced Chemiluminescence Prime Western Blotting Detection reagents (CWBIO, Beijing, China). The immunoblot signal was detected using the Molecular Imager®Chemi DOCTXRS+ system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and the density of each band was analyzed using ImageJ software (version 2006.02.01; National Institutes of Health, Bethesda, MD, USA).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
After treatment with Aβ-42, neurons were harvested, and total RNA was extracted using an RNA simple Total RNA kit (Tiangen Biotech Co., Ltd., Beijing, China). Total RNA was reverse-transcribed using PrimeScript™ RT reagent kit with gDNA Eraser. qPCR was performed using SYBR® Premix Ex TaqTM II on a 7,500 Real Time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.). Relative quantification of gene expression was calculated using the formula 2−∆∆Cq, ∆∆Cq=(Cqtarget gene-CqGAPDH)A,β-42-(Cqtarget gene-CqGAPDH)control (33). qPCR was performed with the following primer sequences: Forward, 5′-CAGGAATTGTTACGGTTCCCTAA-3′ and reverse, 5′-CCTGAATTAACTTGTCCCGTGA-3′ for Drp1; forward, 5′-AACTTGATCGAATAGCATCCGAG-3′ and reverse, 5′-GCATTGCATTGATGACAGAGC-3′ for Mfn1; forward, 5′-CTGGGGACCGGATCTTCTTC-3′ and reverse, 5′-CTGCCTCTCGAAATTCTGAAACT-3′ for Mfn2; forward, 5′-CGACTTTGCCGAGGATAGCTT-3′ and reverse, 5′-CGTTGTGAACACACTGCTCTTG-3′ for OPA-1; and forward, 5′-AGCCAAAAGGGTCATCATCT-3′ and reverse, 5′-GGGGCCATCCACAGTCTTCT-3′ for GAPDH. The PCR thermocycling conditions were as follows: 94°C for 30 sec, followed by 40 cycles of 94°C for 5 sec, 60°C for 15 sec and 72°C 10 sec. Three independent experiments for each condition were performed.
Mitochondrial imaging
As described previously (34), pDsRed2-Mito (Clontech Laboratories, Inc., Mountainview, CA, USA) was transfected into mouse cerebral cortical neurons to label mitochondria with Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.). At 24 h after transfection, mitochondrial morphology was observed in live-cell imaging with an inverted fluorescence microscope with an excitation wavelength of 545 nm. After Aβ-42 treatment for 12 h, mitochondrial morphology in neurons was continuously detected for 1 h under a fluorescence microscope.
Detection of the intracellular ROS level
Neuronal cultures on days 9–11 were used for experiments. To examine the effect of Drp1-dependent mitochondrial fission on Aβ-42-induced intracellular ROS production, neurons were pretreated with 5 µM Mdivi-1 (Sigma-Aldrich; Merck KGaA) for 2 h prior to 10 µM Aβ-42 treatment. To detect the intracellular ROS level, cells were incubated with 10 µM of the fluorescent probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Sigma-Aldrich; Merck KGaA) for 30 min at 37°C in the dark. After incubation, the cells were washed twice with PBS. Fluorescence signals of ROS were detected with excitation and emission wavelengths at 488/525 nm under a fluorescence microscope.
Measurement of mitochondrial membrane potential (∆ψm)
The mitochondrial membrane potential (∆ψm) of cells was measured using tetramethylrhodamine ethyl ester (TMRE; Invitrogen; Thermo Fisher Scientific, Inc.), a potentiometric, cell-permeable fluorescent indicator that accumulates in the highly negatively charged interior of mitochondria. To examine the effect of Drp1-dependent mitochondrial fission on Aβ-42 induced intracellular ROS production, neurons were pretreated with 5 µM Mdivi-1 for 2 h prior to 10 µM Aβ-42 treatment. According to the manufacturer's protocol, cells were incubated with 50 nM TMRE for 20 min at 37°C. After incubation, the cells were washed twice with PBS. Fluorescence signals of MMP (∆ψm) were detected under a fluorescence microscope with excitation and emission wavelengths of 540 and 575 nm, respectively.
Statistical analysis
Data are presented as the mean ± standard deviation. Statistical analysis was performed using SPSS 17.0 (SPSS, Inc., Chicago, IL, USA). Data were analyzed by one-way analysis of variance followed by planned comparisons of multiple conditions, and P<0.05 was considered to indicate a statistically significant difference.
Results
Aβ-42 induces neuronal apoptosis by activating the caspase pathway
Firstly, the primary cultured mouse cerebral cortical neurons were treated with 0, 2.5, 5 and 10 µM of Aβ-42. The morphology of neurons was observed under microscope at 0, 12 and 24 h after Aβ-42 treatment. As presented in Fig. 1, there were no significant changes in neuronal shape after 0, 2.5 and 5 µM Aβ-42, whereas obvious damage in the neuronal shape occurred at 24 h after 10 µM Aβ-42. Neuronal apoptosis was also detected using Hoechst 33258 and MAP2 staining. It was observed that there was little detectable neuronal death 24 h after 0 and 5 µM Aβ-42. In contrast, treatment with 10 µM Aβ-42 resulted in significant neuronal death (Fig. 2A and B). In addition, Caspase-3 is a critical executioner of apoptosis, and proteolytic cleavage of its zymogen is important for activation of Caspase-3. From western blotting, it was demonstrated that treatment with 10 µM Aβ-42 also led to significantly cleaved Caspase-3 in neurons (Fig. 2C and D). These results indicated that 10 µM Aβ-42 induces neuronal apoptosis by activating the Caspase pathway.
Figure 1.
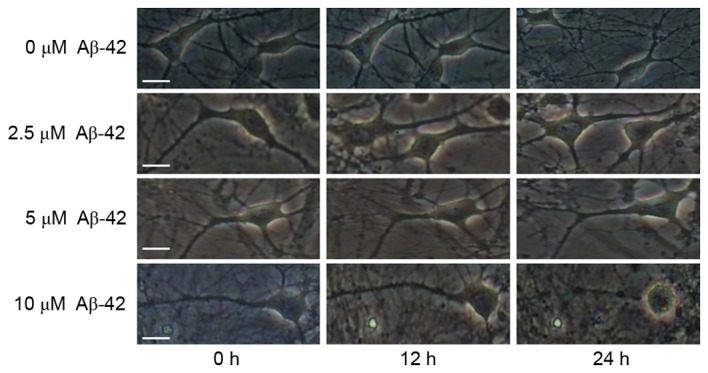
Effect of Aβ-42 at different concentrations on mouse cerebral cortical neurons. Representative fluorescence microscopy images of mouse cerebral cortical neurons (10 days in vitro) exposed to Aβ-42. Scale bar=20 µm. Aβ-42, amyloid β-42.
Figure 2.

Aβ-42-induced cortical neuronal apoptosis and activated caspase pathway. (A) Representative fluorescence microscopy images and (B) quantification of neuronal apoptosis of mouse cerebral cortical neurons (10 days in vitro) exposed to Aβ-42 and stained with Hoechst 33258 and MAP2. Scale bar=50 µm. (C) Representative western blot images and (D) quantification of caspase 3 protein expression levels. Data are presented as the mean ± standard deviation of at least three independent experiments. *P<0.05 vs. 0 h, **P<0.01 vs. Cont or 0 h. Cont, control; Aβ-42, amyloid β-42.
Effect of Aβ-42 on transcription and expression of mitochondrial fission and fusion proteins in neurons
To investigate the possible role of mitochondrial dynamics in Aβ-42-induced neuronal apoptosis, the transcription and expression level of mitochondrial fission and fusion proteins in neurons was examined by RT-qPCR and western blotting assays. The mRNA and protein expression levels of the mitochondrial fission protein, Drp1, were upregulated from 6 to 36 h post-treatment with 10 µM Aβ-42. In contrast, the transcription and expression of both mitochondrial outer membrane fusion proteins (Mfn1 and Mfn2) and the inner membrane fusion protein OPA1 was down-regulated at 12~36 h after 10 µM Aβ-42 (Fig. 3). These results suggested that mitochondrial fission or fusion events might be influenced by Aβ-42 stimulation, and implicate the possible role of mitochondrial dynamics in Aβ-42-induced neuronal apoptosis.
Figure 3.

Transcription and expression of mitochondrial fission and fusion proteins after Aβ-42 treatment. mRNA expression levels of (A) Drp1 and (B) Mfn1, Mfn2 and OPA-1. (C) Representative western blot images and (D) quantification of Drp1, Mfn1, Mfn2 and OPN-1 protein expression levels. GAPDH served as an endogenous control. Data are presented as the mean ± standard deviation of at least three independent experiments. *P<0.05, **P<0.01 vs. 0 h. Aβ-42, amyloid β-42; Drp1, dynamin-related protein 1; Mfn, mitofusin; OPA-1, mitochondrial dynamin like GTPase.
Aβ-42 promotes mitochondrial fission and intracellular ROS production in neurons
Based on the alteration in expression of mitochondrial fission and fusion proteins, mitochondrial morphology in neurons was further examined pre- and post-Aβ-42 treatment. Cultured cortical neurons were transfected with pDsRed2-Mito to label mitochondria. The morphology of mitochondria was monitored using live-cell imaging. As previously demonstrated, mitochondria are localized to neuronal axons and dendrites, and exhibit an elongated, tubular and threadlike shape (35). After treatment with 10 µM Aβ-42, elongated mitochondria became much more fragmented and shorter in morphology (Fig. 4A), indicating that 10 µM Aβ-42 induced mitochondrial fission in neurons. Although the sources of intracellular ROS include the mitochondrial electron transport chain (ETC), NADPH oxidases, xanthine oxidases, cyclooxygenases and lipoxygenases, electron leak from mitochondrial ETC is the major source of ROS. To examine the effect of Aβ-42 on mitochondrial function, the ROS level in neurons was measured using DCFH-DA staining. As presented in Fig. 4B, 10 µM Aβ-42 markedly increased the intracellular ROS level. Inhibition of Drp1 activity with Mdivi-1 efficiently attenuated Aβ-42-induced ROS production. These results suggested that Aβ-42 might disturb mitochondrial respiration through promoting excessive mitochondrial fission.
Figure 4.

Effect of Aβ-42 on mitochondrial fission and intracellular ROS production. (A) Mitochondrial morphology in neurons pre- and post-treatment with Aβ-42. Scale bar=5 µm. (B) Effect of Aβ-42 on intracellular ROS production. Scale bar=20 µm. ROS, reactive oxygen species; Aβ-42, amyloid β-42.
Aβ-42 disrupts mitochondrial membrane potential and upregulates LC3B and Pink1 expression levels
The effect of Aβ-42 on mitochondrial membrane potential was measured using TMRE staining. Compared with the control, 10 µM Aβ-42 markedly decreased mitochondrial membrane potential in neurons. In contrast, inhibition of Drp1 activity by pre-treatment with Mdivi-1 efficiently restored mitochondrial membrane potential after Aβ-42 (Fig. 5A). As loss of mitochondrial membrane potential is the crucial initiating signal to trigger the process of mitophagy, the protein expression levels of the autophagy marker LC3B and the mitophagy sensor Pink1 were examined after Aβ-42 treatment by western blotting assay. As presented in Fig. 5B and C, the expression levels of LC3B and Pink1 in neurons was dramatically up-regulated at 12~36 h post-treatment with 10 µM Aβ-42. These results suggested that Aβ-42 stimulation may disrupt mitochondrial membrane potential, and possibly activate the process of mitophagy to remove the damaged and defective mitochondria in neurons.
Figure 5.

Aβ-42 disrupts mitochondrial membrane potential and upregulates expression of LC3B and Pink1. (A) Effect of Aβ-42 on mitochondrial membrane potential in neurons, as assessed by tetramethylrhodamine ethyl ester staining. Scale bar=20 µm. (B) Representative western blot images and (C) quantification of protein expression levels of LC3B and Pink1 after Aβ-42 treatment. GAPDH was used as an endogenous control. Data are presented as the mean ± standard deviation of at least three independent experiments. *P<0.05 vs. 0 h. Aβ-42, amyloid β-42; LC3B, microtubule-associated proteins 1A/1B light chain 3B.
Discussion
AD, a neurodegenerative disorder, remains the most common cause of dementia. The prevalence of has AD become a serious health issue in the ageing population worldwide (4). It is well recognized that amyloid peptides serve a crucial role in the formation of Aβ plaques, neurofibrillary tangles and progressive loss of synapses and neurons. Among these main amyloid peptides, Aβ-42 has been identified as the most neurotoxic, especially the Aβ-42 oligomers. The soluble Aβ oligomers incorporate into cellular membrane to construct ‘amyloid channels’ which are permeable to Ca2+. As a result, ‘amyloid channels’ disrupt the calcium homeostasis in neurons (36). Calcium dyshomeostasis by these channels leads to impairment of synaptic plasticity and eventually loss of neurons. In addition, it has been reported that Aβ-42 can induce mitochondrial mis-localization. After Aβ-42 treatment, mitochondria are reduced in axons and dendrites, and accumulate in the soma of neurons (37). Oligomeric and fibrillar Aβ-42 also induces mitochondrial dysfunctions in cortical neurons, including a reduction in mitochondrial membrane potential, state 3 respiration, respiratory control ratio and uncoupled respiration (38). Consistent with a previous study (39), the present study demonstrated that Aβ-42 induced significant neuronal apoptosis, and activated the Caspase pathway. Ca2+ is an active second messenger, and is involved in many cellular processes. Previous studies have indicated that calcium impacts on mitochondria, including mitochondrial functions and its dynamics (23,34). To investigate whether Aβ-42 induces damage to neurons by targeting mitochondria, mitochondrial morphology and the expression of mitochondrial fission and fusion proteins were examined. It was demonstrated that expression of the mitochondrial fission protein Drp1 was upregulated after Aβ-42 treatment. Mitochondrial fusion proteins Mfn1/2 and OPA1 were downregulated after Aβ-42 treatment. Consistent with the alteration in expression of mitochondrial fission and fusion proteins, Aβ-42 induced obvious mitochondrial fission in neurons. These results imply the possibility that Aβ-42 might induce neuronal apoptosis through disrupting mitochondrial dynamics, promoting mitochondrial fission in neurons.
The most important functions of mitochondria include ATP synthesis through oxidative phosphorylation, regulation of redox, calcium buffering and the control of apoptosis in response to extracellular and intracellular stimulation or stress (40). In all cell types, neurons have high energy consumption for maintenance of their functions. Mitochondria are often recruited to subcellular regions with high metabolic requirements, for example in the active or growth cones. In healthy neurons, mitochondria are abundant at neuronal processes, including synapses and axons, and are involved in regulation of neuronal activity, synaptic plasticity and neurotransmitter release (41). As a result, neurons are particularly vulnerable to mitochondrial defects. Neuronal apoptosis is often accompanied by disruption in mitochondrial dynamics and functions. Mitochondrial fission is an early event during apoptosis, and inhibition of mitochondrial fission by silencing Drp1, and overexpression of dominant negative mutant Drp1-K38A or chemical Drp1 inhibitor Mdivi-1 efficiently reduces apoptosis (42). In contrast, mitochondrial fusion protects cells against apoptosis. It has been reported that OPA-1, the inner mitochondrial membrane fusion protein, serves a crucial role in regulation of the structure and tightness of mitochondrial cristae junctions. Most cytochrome C molecules are encapsulated within the folds of the mitochondrial intermembrane space. Cristae junctions sealed by the complex of OPA-1 oligomers prevent leakage of cytochrome C from mitochondria. Thus, knocking down OPA-1 remodels cristae junctions, increases cytochrome C release and sensitizes cells to apoptosis (42). In addition, it has also been reported that outer mitochondrial membrane fusion protein Mfn1 and 2 interact with Bcl-2 X-associated protein and Bcl-2 homologous antagonist killer in regulation of mitochondrial morphology and apoptosis (43). This is consistent with the results of the present study; Aβ-42 stimulation upregulated the expression of Drp1, decreased the level of OPA-1, Mfn1 and 2, promoted mitochondrial fission and increased neuronal apoptosis.
It has been reported that excessive fission and cristae remodeling constantly coincide with mitochondrial membrane permeability (44,45). Alteration of mitochondrial membrane permeability leads to certain defects in mitochondrial functions. One of them is the loss of the electric potential across the inner mitochondrial membrane. The present study demonstrated that the mitochondrial membrane potential in neurons was decreased after treatment with Aβ-42. Inhibition of Drp1 activity by Mdivi-1 efficiently attenuated Aβ-42-induced disruption of mitochondrial membrane potential. These results suggested that Drp1-dependent mitochondrial fission is involved in the disruption of mitochondrial membrane potential after Aβ-42. Loss of mitochondrial membrane potential results in serious damage to mitochondrial functions, and is also an important initiating signal to trigger mitophagy. Mitophagy is a variant of autophagy to remove the damaged mitochondria. In case of specific elimination of damaged mitochondria, mitophagy requires a specific labeling and autophagosome recruitment system, involving Pink1/Parkin. Pink1 is the prominent sensor for mitochondria, and accumulates at the surface of damaged mitochondria, especially at potential-deficient mitochondria. Pink1 subsequently recruits and activates Parkin, and conjugates ubiquitin to various outer mitochondrial membrane proteins (29,30,46). Ubiquitin and autophagic protein LC3 binding with some adaptor proteins sequestrate the damaged mitochondria in the autophagosomal membrane, and then fuses with a lysosome for degradation (28). In the present study, it was demonstrated that Aβ-42 upregulated the expression of both Pink1 and LC3B. Although the underlying mechanism remains to be elucidated, these data imply that Aβ-42 stimulation may trigger the process of mitophagy to remove the damaged mitochondria in neurons.
In conclusion, the present study demonstrated that Aβ-42 treatment activated the Caspase signaling pathway, and induced obvious neuronal apoptosis. Although the role of Aβ-42 in neuronal survival has been described (14,39), to the best of our knowledge, these data demonstrated for the first time that Aβ-42 increases mitochondrial fission by changing the expression of mitochondrial fission and fusion proteins. Inhibition of Drp1-dependent mitochondrial fission efficiently suppressed Aβ-42-induced intracellular ROS production and loss of mitochondrial membrane potential in neurons. Furthermore, Aβ-42 may trigger the process of mitophagy to remove the damaged mitochondria through upregulating expression of the autophagy marker LC3B and the mitophagy sensor Pink1. Although the underlying mechanism of how Aβ-42 targets mitochondria remains to be elucidated, the present study suggested mitochondria may represent a potential therapeutic target for AD in the future.
Acknowledgements
The present study was supported by the National Natural Science Foundation of China (grant no. 31360241), Natural Science Foundation of Jiangxi (grant no. 20161ACB20019), the Postgraduate Student Foundation for New Teacher from the Ministry of Education of China (grant no. 20123601120001) and the Foundation from Education Department of Jiangxi Province (grant no. GJJ13162).
References
- 1.Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G. Therapies for prevention and treatment of Alzheimer's disease. Biomed Res Int. 2016;2016:2589276. doi: 10.1155/2016/2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arbor SC, LaFontaine M, Cumbay M. Amyloid-beta Alzheimer targets-protein processing, lipid rafts, and amyloid-beta pores. Yale J Biol Med. 2016;89:5–21. [PMC free article] [PubMed] [Google Scholar]
- 3.Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–934. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: Clinical implications and perspectivs. Genet Med. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.WNL.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, Liu Y, Sun M. Autophagy and Alzheimer's Disease. Cell Mol Neurobiol. 2017;37:377–388. doi: 10.1007/s10571-016-0386-8. [DOI] [PubMed] [Google Scholar]
- 7.Barage SH, Sonawane KD. Amyloid cascade hypothesis: Pathogenesis and therapeutic strategies in Alzheimer's disease. Neuropeptides. 2015;52:1–18. doi: 10.1016/j.npep.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 8.Dulin F, Léveillé F, Ortega JB, Mornon JP, Buisson A, Callebaut I, Colloc'h N. P3 peptide, a truncated form of A beta devoid of synaptotoxic effect, does not assemble into soluble oligomers. FEBS Lett. 2008;582:1865–1870. doi: 10.1016/j.febslet.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 9.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: Follow-up of arandomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 10.Saito T, Suemoto T, Brouwers N, Sleegers K, Funamoto S, Mihira N, Matsuba Y, Yamada K, Nilsson P, Takano J, et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci. 2011;14:1023–1032. doi: 10.1038/nn.2858. [DOI] [PubMed] [Google Scholar]
- 11.Kawahara M, Ohtsuka I, Yokoyama S, Kato-Negishi M, Sadakane Y. Membrane incorporation, channel formation and disruption of calcium homeostasis by alzheimer's β-amyloid protein. Int J Alzheimers Dis. 2011;2011:304583. doi: 10.4061/2011/304583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawahara M. Neurotoxicity of β-amyloid protein: Oligomerization, channel formation and calcium dyshomeostasis. Curr Pharm Des. 2010;16:2779–2789. doi: 10.2174/138161210793176545. [DOI] [PubMed] [Google Scholar]
- 13.Jarrett JT, Lansbury PT., Jr Seeding ‘one-dimensional crystallization’ of amyloid: A pathogenic mechanism in Alzheimer's disease andscrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 14.Jan A, Adolfsson O, Allaman I, Buccarello AL, Magistretti PJ, Pfeifer A, Muhs A, Lashuel HA. Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J Biol Chem. 2011;286:8585–8596. doi: 10.1074/jbc.M110.172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mirzabekov T, Lin MC, Yuan WL, Marshall PJ, Carman M, Tomaselli K, Lieberburg I, Kagan BL. Channel formation in planar lipid bilayers by a neurotoxic fragment of the beta-amyloid peptide. Biochem Biophys Res Commun. 1994;202:1142–1148. doi: 10.1006/bbrc.1994.2047. [DOI] [PubMed] [Google Scholar]
- 16.Strodel B, Lee JW, Whittleston CS, Wales DJ. Transmembrane structures for Alzheimer's Aβ(1–42) oligomers. J Am Chem Soc. 2010;132:13300–13312. doi: 10.1021/ja103725c. [DOI] [PubMed] [Google Scholar]
- 17.Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism ofsoluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- 18.Foster TC. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell. 2007;6:319–325. doi: 10.1111/j.1474-9726.2007.00283.x. [DOI] [PubMed] [Google Scholar]
- 19.Oakes SA, Korsmeyer SJ. Untangling the web: Mitochondrial fission and apoptosis. Dev Cell. 2004;7:460–462. doi: 10.1016/j.devcel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell. 2004;16:59–68. doi: 10.1016/j.molcel.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 21.Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 2002;12:178–184. doi: 10.1016/S0962-8924(01)02246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wan YY, Zhang JF, Yang ZJ, Jiang LP, Wei YF, Lai QN, Wang JB, Xin HB, Han XJ. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol Rep. 2014;32:619–626. doi: 10.3892/or.2014.3235. [DOI] [PubMed] [Google Scholar]
- 23.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 24.Tang Y, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/S0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- 25.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 26.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, et al. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Su B, Zheng L, Perry G, Smith MA, Zhu X. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem. 2009;109(Suppl 1):S153–S159. doi: 10.1111/j.1471-4159.2009.05867.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rüb C, Wilkening A, Voos W. Mitochondrial quality control by the Pink1/Parkin system. Cell Tissue Res. 2017;367:111–123. doi: 10.1007/s00441-016-2485-8. [DOI] [PubMed] [Google Scholar]
- 29.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Camilleri A, Zarb C, Caruana M, Ostermeier U, Ghio S, Högen T, Schmidt F, Giese A, Vassallo N. Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim Biophys Acta. 2013;1828:2532–2543. doi: 10.1016/j.bbamem.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 33.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 34.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. 2008;182:573–585. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han XJ, Lu YF, Li SA, Tomizawa K, Takei K, Matsushita M, Matsui H. Involvement of calcineurin in glutamate-induced mitochondrial dynamics in neurons. Neurosci Res. 2008;60:114–119. doi: 10.1016/j.neures.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 36.Lin H, Bhatia R, Lal R. Amyloid beta protein forms ion channels: Implications for Alzheimer's disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 37.Iijima-Ando K, Hearn SA, Shenton C, Gatt A, Zhao L, Iijima K. Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer's disease. PLoS One. 2009;4:e8310. doi: 10.1371/journal.pone.0008310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckert A, Hauptmann S, Scherping I, Meinhardt J, Rhein V, Dröse S, Brandt U, Fändrich M, Müller WE, Götz J. Oligomeric and fibrillar species of beta-amyloid (A beta 42) both impair mitochondrial function in P301 L tau transgenic mice. J Mol Med (Berl) 2008;86:1255–1267. doi: 10.1007/s00109-008-0391-6. [DOI] [PubMed] [Google Scholar]
- 39.Wang ZJ, Zhou B, Mao WW, Yin M. Overexpression of 5-lipoxygenase increases the neuronal vulnerability of PC12 cells to Aβ42. Yakugaku Zasshi. 2011;131:1843–1853. doi: 10.1248/yakushi.131.1843. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura T, Lipton SA. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: Potential implications for Alzheimer's and Parkinson's diseases. Apoptosis. 2010;15:1354–1363. doi: 10.1007/s10495-010-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Büeler H. Mitochondrial dynamics, cell death and the pathogenesis of Parkinson's disease. Apoptosis. 2010;15:1336–1353. doi: 10.1007/s10495-010-0465-0. [DOI] [PubMed] [Google Scholar]
- 42.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1 and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie ZJ, Dong Z. Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA. 2007;104:11649–11654. doi: 10.1073/pnas.0703976104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Landes T, Martinou JC. Mitochondrial outer membrane permeabilization during apoptosis: The role of mitochondrial fission. Biochim Biophys Acta. 2011;1813:540–545. doi: 10.1016/j.bbamcr.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 45.Cosentino K, García-Sáez AJ. Mitochondrial alterations in apoptosis. Chem Phys Lipids. 2014;181:62–75. doi: 10.1016/j.chemphyslip.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496:372–376. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]