

A comprehensive design offers global pneumococcal vaccine coverage.
Abstract
Commensal organisms with the potential to cause disease pose a challenge in developing treatment options. Using the example featured in this study, pneumococcal disease begins with Streptococcus pneumoniae colonization, followed by triggering events that prompt the release of a virulent subpopulation of bacteria. Current vaccines focus on colonization prevention, which poses unintended consequences of serotype niche replacement. In this study, noncovalent colocalization of two classes of complementary antigens, one to prevent the colonization of the most aggressive S. pneumoniae serotypes and another to restrict virulence transition, provides complete vaccine effectiveness in animal subjects and the most comprehensive coverage of disease reported to date. As a result, the proposed vaccine formulation offers universal pneumococcal disease prevention with the prospect of effectively managing a disease that afflicts tens to hundreds of millions globally. The approach more generally puts forth a balanced prophylactic treatment strategy in response to complex commensal-host dynamics.
INTRODUCTION
Commensal microorganisms generally serve a symbiotic relationship with the inhabited host. However, under certain circumstances, a transition to virulence can prompt disease from a normally benign commensal. This poses a question of whether a therapy should prevent initial colonization, thus not only eliminating any potential for future disease but also removing any benefit (either direct or indirect) provided by commensal establishment, or whether a therapy should retain colonization (and any associated benefit) while targeting the process of virulence transition. This dilemma is well represented by ongoing vaccination efforts to address pneumococcal disease.
Pneumococcal disease is a major global infectious disease threat that affects millions worldwide, particularly the young, the elderly, and the resource-limited (1–3). Disease outcomes include pneumonia, bacteremia, meningitis, and otitis media (middle ear infection) (4–6). As introduced generally above, treating this disease is challenging because of the scope of infection and the steps of disease progression.
More specifically, human colonization with the Streptococcus pneumoniae bacteria responsible for pneumococcal disease is nearly universal (7). For example, colonization occurs in more than 95% of children in the first few years of life (8, 9). The composition of colonization is complicated by >95 different serotypes of S. pneumoniae, differentiated by the polysaccharide content coating the bacterial cell (10). Current vaccines harvest, purify, and combine the surface capsular polysaccharides (CPSs) of S. pneumoniae serotypes associated with aggressive invasive infection (11). The approach has been effective at minimizing initial colonization, enabling herd immunity, and reducing the disease (12).
However, the progression of pneumococcal disease requires more than colonization. Upon acquisition, S. pneumoniae establishes residence in the nasopharynx as an asymptomatic biofilm (13, 14). In other words, S. pneumoniae represents an upper respiratory tract commensal. Disease occurs upon biofilm dissemination, often prompted by disruptive events (such as viral infection) that spur a subset of bacteria to become virulent (fig. S1) (15). Upon transition, these bacteria break free from the biofilm, spread to the lungs, blood, middle ear, and other locations, and cause the aforementioned disease types (16). When viewed from this perspective, preventing colonization through current vaccine options serves as an upstream route to eliminating subsequent disease progression.
However, limiting colonization is only possible for the 13 to 23 serotypes covered by current vaccines, which fall well short of the >95 serotypes thus far identified (17). Hence, eliminating colonization prevents disease from certain serotypes; but it also creates the potential for the unintended consequence of an expanded niche in the nasopharynx for the remaining serotypes to establish residence and progress to virulence (18, 19). Thus, according to this strategy, the only hypothetical vaccine capable of providing universal coverage would incorporate polysaccharide surface antigens from every known serotype of S. pneumoniae. Such an objective is prohibitively expensive from a manufacturing perspective and still does not eliminate the danger of niche replacement with as yet undetected S. pneumoniae serotypes or with other upper respiratory tract commensals and pathogens (19–23).
Here, a dual-functioning liposomal formulation allowed the colocalization of complementary antigen types, each associated with a separate state in commensal disease progression. From a manufacturing perspective, the liposomal design allows a cost-effective and scalable formulation, in stark contrast to the production of current vaccine options. From a potency perspective, the liposomal formulation capacity and the broad and distinct nature of the enclosed antigens enable the most robust and comprehensive immune response reported to date, as measured against Prevnar and Pneumovax controls, the current standards in commercial vaccine options for pneumococcal disease.
RESULTS AND DISCUSSION
Best-in-class vaccination comparison
Our initial experiments sought to establish an alternative to the Prevnar vaccine family, which rely on covalently attaching S. pneumoniae polysaccharide from specific serotypes to the immunogenic CRM197 protein (an inactivated mutant of the diphtheria toxin capable of amplifying immune reactivity of the conjugated polysaccharide components), resulting in so-called glycoconjugate vaccines (for example, Prevnar 13 features 13 different serotype polysaccharides covalently attached to CRM197). As opposed to covalent polysaccharide protein coupling, we used liposomal technology to encapsulate the required polysaccharide content and to noncovalently attach and surface-display the CRM197 protein [termed liposomal encapsulation of polysaccharides (LEPS); figs. S2 and S3]. By doing so, we achieved an in vivo antibody shift pattern from immunoglobulin M (IgM) to IgG and effectiveness in total antibody titers and protection against S. pneumoniae bacterial challenge comparable to Prevnar 13 (Fig. 1, A to C). In these comparisons, the Pneumovax 23 vaccine (which contains purified CPSs from 23 serotypes) was also tested and showed reduced effectiveness (antibody class shift and titer and bacterial challenge protection) relative to Prevnar 13 and the LEPS formulation. LEPS achieves glycoconjugate vaccine effectiveness through noncovalent colocalization of polysaccharide and CRM197. The option to scalably encapsulate additional polysaccharides in the LEPS formulation further highlights differences to current glycoconjugate vaccine options, which require successive effort and manufacturing cost with each covalent attachment of a new serotype CPS to CRM197, thus limiting both broad vaccine coverage and global access (24).
Fig. 1. LEPS vaccine effectiveness compared to commercial standards.
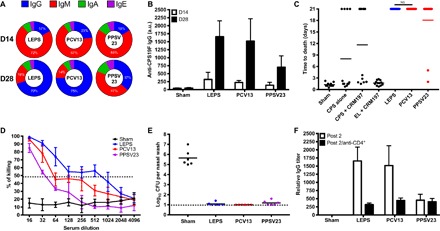
Mice were vaccinated at 0 and 14 days with (i) LEPS containing CPS from the S. pneumoniae serotype 19F, (ii) Prevnar 13 (PCV13), (iii) Pneumovax 23 (PPSV23), (iv) PBS (sham), and (v) various controls [CPS alone, CPS + CRM197, and empty LEPS (EL) + CRM197]. Serum was collected at 14 and 28 days. (A and B) Antibody class shifting of LEPS, PCV13, and PPSV23 (A) and total IgG titers (B). a.u., arbitrary units. (C) Protection against lethal challenge of serotype 19F in a murine sepsis model. (D) Functional antibody activity assessment of select vaccination strategies using the OPA assay. (E) Nasopharynx bacterial burden assessed in unimmunized or immunized mice measured at 5 days after colonization. CFU, colony-forming units. (F) 19F CPS antibody titers measured at 28 days (post 2) in mice serum without (white) or with (black) CD4+ T cell depletion. NS, not significant.
Additional assays provided insight into the immune response afforded by the LEPS platform. Namely, Fig. 1D presents data from an opsonophagocytic activity (OPA) assay in which serum from immunized mice is added to the 19F S. pneumoniae serotype (recognized by current Prevnar and Pneumovax formulations) before coculture with HL60 human phagocytes. Bacterial killing is readily observed for all three vaccine formulations, with LEPS again performing at levels comparable to current standards. Table S1 presents a more thorough comparison of serotypes assessed via the OPA assay across the three vaccine systems. Similarly, fig. S4 highlights the effectiveness in protection against the serotypes covered by Prevnar 7 and Prevnar 13 in addition to the seven nonvaccine serotypes included in the LEPS formulation. Nasal wash experiments indicate that the 19F S. pneumoniae serotype does not establish residence in the mouse nasopharynx because of the corresponding polysaccharide included in the three vaccine systems being compared in this case (Fig. 1E). Insight into the immune response mechanism was provided by an anti-CD4+ T cell depletion assay in which antibody titers were significantly reduced, indicating antigen processing through a thymus-dependent pathway (Fig. 1F). Finally, antibody titers and OPA activity in rabbit subjects demonstrated consistent in vivo trends compared to those observed in mice subjects (fig. S5).
Expansive protein antigen serotype assessment and the prospect of universal coverage
The same liposomal technology that allowed an alternative pneumococcal disease vaccine has the interesting potential to address complementary aspects of commensal-based disease progression. Namely, the formulation offers the simultaneous encapsulation of polysaccharides (colonization immune targets) that serve as the basis for current vaccines in addition to the liposomal surface localization of protein antigens [GlpO and PncO; identified through an antigen discovery and validation model (25)] that selectively target pneumococci virulence transition (fig. S1 and table S3). Before testing this possibility, the OPA assay enabled a more careful assessment of the GlpO and PncO protein antigens as a correlate of protection for future immunization experiments. In this case, however, the OPA assay assessed both colonization (planktonic cells containing surface CPS) and dispersion (biofilm-released cells displaying surface protein antigen targets) aspects of commensal disease progression in response to the corresponding polysaccharide or protein antigen component. The resulting insight contrasts traditional OPA assays that only assess planktonic CPS cellular targets and, hence, have an innate bias toward colonization prevention. As shown in table S2 and fig. S6, Prevnar 13, Pneumovax 23, and the GlpO/PncO protein antigens demonstrate specific activity for their respective cellular targets. Notably, the GlpO/PncO tandem is active for >70 biofilm-released serotypes of S. pneumoniae. This is the largest and most comprehensive assessment of advanced antigens for pneumococcal disease and, together with the high-sequence conservation of the glpO/pncO genes across S. pneumoniae serotypes (26), emphasizes universal protection potential for any virulence-transitioned S. pneumoniae cell. The protein antigens were also tested more thoroughly for immune response mechanism (either in or separate from the LEPS formulation), showing predominant T helper 2 cell (TH2) response with contributing TH1 and TH17 activity in mice (Fig. 2, A to E) and corresponding humoral response in rabbits (Fig. 2F). Supporting the complementary aspect of the antigen types, the PncO and GlpO proteins were tested in both coadministration and add-on format with Prevnar 7, demonstrating protective capabilities by addressing commensal colonization, virulence transition, or both (fig. S7).
Fig. 2. Immune response assessment of the PncO and GlpO protein antigens.
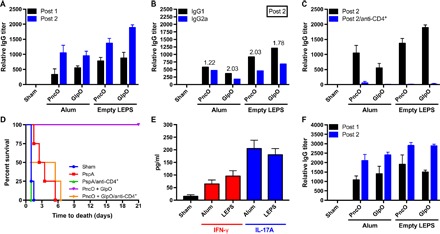
(A) Anti-PncO and anti-GlpO antibody titers in mice serum after initial (post 1) and booster (post 2) vaccination with either alum adjuvant or empty LEPS. (B) IgG1 (TH1 corresponding) and IgG2a (TH1 corresponding) titers in mice serum after a booster vaccination. IgG1/IgG2a ratio is shown above the bars. (C) Anti-PncO and anti-GlpO antibody titers in mice serum with or without CD4+ T cell depletion. (D) Murine challenge protection sepsis model after vaccination with either PspA (a commonly used pneumococcal protein antigen) (26) or PncO + GlpO with or without CD4+ cell depletion; S. pneumoniae serotype 19F was the challenge strain, and four mice were used per formulation. (E) Titers of interferon-γ (IFN-γ) (TH1 corresponding) and interleukin-17A (IL-17A) (TH17 corresponding) in mice serum after PncO + GlpO vaccination. (F) Anti-PncO and anti-GlpO antibody titers in rabbit serum after initial and booster vaccination.
A comprehensive vaccine platform through LEPS technology
Next, coupling the PncO and GlpO protein antigens to the liposomal containment of polysaccharide resulted in the completed LEPS vehicle (LEPS20/PncO + GlpO; Fig. 3), thus allowing for a definitive assessment of the commensal disease progression vaccination paradigm. From data in Figs. 1 and 2, the complete LEPS vehicle was expected to have ideal immune response characteristics (class switching, memory, and broad T cell activation). A humoral response resulted for each of the antigen types in the completed vehicle (Fig. 3A) in addition to comprehensive and directed protection when tested in the in vivo influenza A virus (IAV)–prompted pneumonia model of pneumococcal disease transition. The challenge assays across strategic serotypes [19F (fig. S8), 11A, and 35C) confirm the protective limits of Prevnar 13 and Pneumovax 23 and highlight the full protective capabilities of the complete LEPS system (Fig. 3B). Emphasizing antigen complementarity, the LEPS20/PncO + GlpO formulation provides full protection even without the requisite CPS needed to prevent the colonization of the 11A and 35C serotypes. The associated anatomical profiling further confirms the ability of the final LEPS formulation to inhibit disease dispersion of nonvaccine serotypes. Similar results are found for pneumonia and sepsis disease models (fig. S9).
Fig. 3. Assessment of the full LEPS platform featuring CPS encapsulation across 20 serotypes and surface localization of GlpO and PncO.
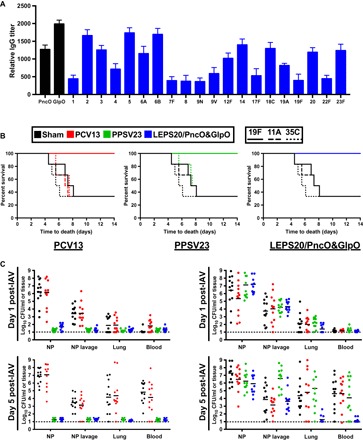
(A) IgG titers against PncO, GlpO, and encapsulated CPS from indicated S. pneumoniae serotypes in mice serum after vaccination. (B) Mouse challenge protection data comparison between PCV13, PPSV23, and the complete LEPS system (LEPS20/PncO + GlpO) using a colonization model and in vivo perturbation with influenza A administration to prompt pneumonia development. Survival is indicated across vaccination options when challenged with the indicated serotypes, which are, in part, covered by PCV13 (19F) and PPSV23 (19F and 11A) and fully covered by LEPS20/PncO + GlpO. (C) Bacterial counts from the nasopharynx surface (NP), nasopharynx wash (lavage), lungs, and blood 1 and 5 days after IAV administration when challenged with 11A (left) and 35C (right) serotypes.
Although the effectiveness of the LEPS vaccine was a key result, a broader implication is the dual potential for eliminating the colonization of the most invasive serotypes of S. pneumoniae while simultaneously safeguarding against virulence transition of niche-replacement serotypes not covered by current vaccine formulations. This is possible because of the universal protection potential of the GlpO and PncO antigens (tables S2 and S3), aided by sequence conservation across S. pneumoniae strains and the minimization of antigenic drift across multiple antigens (26). Figure 4 provides a current and predicted time line of invasive pneumococcal disease (IPD) cases, which emphasizes the recalcitrance of nonvaccine serotypes and the potential of the LEPS formulation to address these concerns. Notably, the LEPS platform also provides a technically feasible vision for universality through the prospect of encapsulating all known S. pneumoniae serotype polysaccharides (in addition to surface-displaying the GlpO and PncO protein antigens), an option that is economically infeasible with polysaccharide-protein coupling chemistry accompanying current glycoconjugate vaccine products. Hence, the LEPS platform combines the best aspects of current vaccines while anticipating the basic tenets of commensal disease progression.
Fig. 4.
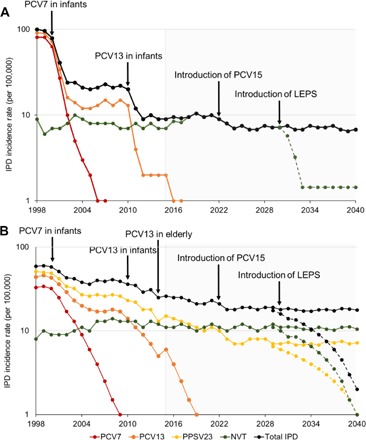
IPD incidence rates in children (age, <5) (A) and the elderly (age, ≥65) (B) from 1998 to 2040. The total incidence rate of IPD was segmented into disease caused by Prevnar 7 (PCV7), PCV13, PPSV23, and nonvaccine-type (NVT) serotypes. Nonvaccine-type serotypes are defined as those not contained in PCV13 (A) or PPSV23 (B) vaccines. Incidence rates for PCV7 serotypes from 1998 to 2007 were obtained from Pilishvili et al. (30). Total incidence rates and incidence rates for PCV13 serotypes were obtained from the Centers for Disease Control and Prevention’s Active Bacterial Core surveillance program for S. pneumoniae (www.cdc.gov/abcs/reports-findings/survreports/spneu-types.html). Incidence rates beyond 2007 (PCV7 serotypes) and 2015 (PCV13 serotypes) were predicted from trends observed in previous years. Reduction in the total IPD after Prevnar 15 (PCV15) introduction was projected on the basis of the serotype invasiveness (31, 32) and previous trends for PCV7 and PCV13 with the rates stabilizing for 4 years after the introduction of the vaccine. Projections for IPD with (dotted lines) or without (solid lines) LEPS introduction were made beyond 2030. A reduction of IPD by 80% over 4 years (A) or 90% over 10 years (B) after the introduction of LEPS was based on the 98% sequence coverage of the GlpO and PncO protein antigens (25) and activity demonstrated against 70 S. pneumoniae serotypes (fig. S2).
In summary, we report on the colocalization of two complementary antigens as a next-generation vaccine for pneumococcal disease. Without compromising the effectiveness of current vaccine designs, we introduce a modification that enables universality in vaccine response to disease. The resulting LEPS platform thus minimizes invasive serotype colonization while also safeguarding against virulence transition for all colonizing serotypes. As a result, the full disease progression pathway has guided the development of a vaccine that offers a comprehensive answer to the challenging aspects of addressing pneumococcal disease. More broadly, the concept outlined in this study seeks to balance the benefits and drawbacks to microbial commensalism, with an approach adjustable to eliminate or retain residence as a function of maximum benefit to the host.
MATERIALS AND METHODS
Experimental design
Complementary antigens derived from the understanding and analysis of commensal-based disease progression were colocalized using a liposomal carrier platform in the context of pneumococcal disease vaccination. Forms of assessment included liposomal vector characterization; tissue-specific disease progression; antibody, cytokine, and T cell depletion analysis; end point and time-course challenge protection assays; comprehensive opsonophagocytic activity assays; and tests conducted in mouse and rabbit models. Data uniformly support a potent vaccination strategy capable of directing an immune response across the stages of commensal-based disease, with the potential for universal coverage in the case of pneumococcal disease.
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee at the University at Buffalo. All bacterial inoculations and treatments were performed under conditions designed to minimize any potential suffering of the animals.
Reagents
Bacterial and cell culture media [including chemically defined bacterial growth medium (CDM)] and reagents were purchased from Fisher Chemical and Sigma-Aldrich. Sheep blood was purchased from Hemostat Laboratories. Lipids DOPG-Na and DSPE-PEG2000 were obtained from NOF. DOGS-NTA-Ni was purchased from Avanti Polar Lipids. Dioleoylphosphatidylcholine (DOPC), cholesterol, alum (as aluminum phosphate), and polysaccharides were purchased from Sigma-Aldrich. LL-37 was purchased from InvivoGen. Prevnar 7 and Prevnar 13 and Pneumovax 23 vaccines were obtained from Pfizer and Merck, respectively.
Antigen preparation
All proteins [CRM197, green fluorescent protein (GFP), PspA, GlpO, and PncO] were recombinantly produced with polyhistidine tags through Escherichia coli. Plasmids containing the genes for GFP, CRM197, and PspA were obtained through collaborative exchange (26, 27). Remaining genes were polymerase chain reaction (PCR)–amplified from S. pneumoniae genomic DNA and cloned into separate pET21c vectors using restriction sites Sac I/Xho I (glpO) and Nde I/Xho I (pncO) introduced to the amplified products by the PCR primers (25). After plasmids were confirmed by restriction digestion and colony PCR, final constructs were chemically transformed into E. coli BL21(DE3) to confirm individual expression via induction of 3 ml of lysogeny broth cultures at OD600 (optical density at 600 nm) values of 0.4 to 0.6 using 1 mM isopropyl β-d-1-thiogalactopyranoside before analysis by SDS–polysaccharide gel electrophoresis. Confirmed expression prompted 1-liter-scale cultures for expression and protein purification through disruption of cells using a French press and by passing cell lysate over a fast protein liquid chromatography column (GE Healthcare HisTrap HP, 1 × 1 ml). Final protein products were quantified using the Pierce Micro BCA Protein Assay kit.
LEPS preparation and characterization
Proteins and polysaccharide components were colocalized through a liposomal delivery system. LEPS liposomal carriers were composed of DOPC/DOPG/DOGS-NTA-Ni/cholesterol/DSPE-PEG2000 at a molar ratio of 3:3:1:4:0.1 to a total lipid mass of 500 μg. After dissolving lipids in chloroform, the solution was sonicated for 1 min using a Branson 450D Sonifier (at 20% amplitude using a tapered tip) and then evaporated using a rotary evaporator to form a film. Lipids were then rehydrated with phosphate-buffered saline (PBS) containing the polysaccharide antigens to form liposomes, which were then passed 10 to 12 times through a handheld extruder (Avanti Polar Lipids) with a pore size of 200 nm. On ice, the background liposome solution was passed twice through a filter with 50-nm pore size and replaced each time with PBS. Next, proteins were incubated with liposomes for 30 min at 4°C with surface attachment mediated via polyhistidine tag–Ni chelation. CRM197 was included in the LEPS formulations used for Fig. 1, figs. S4, S5, and S10, and table S1; CRM197 was not included (replaced by GlpO/PncO) in the LEPS formulations used for Figs. 2 and 3 and figs. S6, S8, and S9. At this stage, any unbound protein and polysaccharide were separated from the final LEPS construct via overnight dialysis (molecular weight cutoff, 100 kDa) at 4°C. Throughout the study, final formulations used during vaccinations were composed of mixtures of LEPS particles containing individual polysaccharides. For example, the final 20 valent CPS LEPS formulation resulted from mixing 20 individually prepared samples. Alternatively, a coformulation approach was also tested (with a limited number of serotype polysaccharides) and compared to the mixing approach described directly above with no differences in final vaccine protection (fig. S10). All final LEPS formulations were prepared to deliver the same amount of polysaccharide content (and CRM197 protein, as needed) used in Prevnar and Pneumovax formulations, which were diluted 1:10 in PBS before immunizations.
Characterization of the LEPS particles began by analyzing polysaccharide encapsulation and surface protein binding efficiency. Quantification was completed over a range of polysaccharide (using 19F)– or protein (using GFP)–to–lipid ratios to identify the crossing point of efficiency (fig. S3). For polysaccharide evaluation, 150 μl of concentrated sulfuric acid and 30 μl of phenol [5% (w/v)] were added to 50 μl of liposome solution, followed by incubation at 80°C for 30 min and 22°C for 30 min before colorimetric analysis at 495 nm. Protein content was measured via fluorescence analysis at an excitation wavelength of 359 nm and an emission wavelength of 508 nm. Both analyses were conducted using a Synergy 4 Multi-Mode Microplate Reader (BioTek Instruments Inc.), and measured values were compared to standards (using either glucose or GFP) and ratioed to initial amounts of polysaccharide and protein introduced to the LEPS production process. Dynamic light scattering on a Zetasizer Nano ZS90 (Malvern) was used to evaluate the particle diameter and zeta potential of liposomes at 25°C. All experiments were conducted using a 4-mW, 633-nm HeNe laser as the light source at a fixed measuring angle of 90° to the incident laser beam. Images of the LEPS particles were obtained through JEOL JSM-CXII transmission electron microscopy analysis at 100 kV, with samples prepared by dip-coating a 200-mesh formvar and a carbon-coated grid (FCF-200-Cu-TB, Electron Microscopy Sciences), followed by negative staining using a 1% solution of uranyl acetate.
Vaccine immunization
Outbred 6-week-old female CD-1 mice (Harlan Laboratories) were used in immunization experiments. Mice were immunized by subcutaneous injection (200 μl). The background solution used for the formulations was PBS (“sham” negative controls were background solutions administered without antigen components). Final protein antigen levels were 12.5 μg in either naked or liposomal formulations (GlpO, PncO, CRM197, and PspA). When combined, PncO and GlpO were administered at 6.25 μg each. Unless indicated otherwise, alum adjuvant was added to protein samples according to the manufacturer’s instructions. After 14 days, mice were boosted with the same formulations. Serum samples were collected on days 14 and 28 by retro-orbital bleeding for antibody and OPA analyses. Four-month-old New Zealand white rabbits (Cocalico Biologicals) were immunized through intramuscular administration of 500 μl of respective samples at days 0 and 14. Peripheral blood samples were collected on days 14 and 28 for antibody and OPA analyses.
Bacterial preparation and biofilm release
Bacterial strains used in this study were initially grown on Todd Hewitt agar plates supplemented with 0.5% yeast extract and 5% sheep blood and incubated overnight at 37°C. Single colonies were used to inoculate 5 ml of Todd Hewitt broth containing 0.5% yeast extract and incubated at 37°C to an OD600 of 0.6. At this point, bacteria were collected by centrifugation, washed once with and resuspended in PBS, and quantified by OD600 measurement for experiments requiring planktonic cells. Planktonic cells were introduced to the in vitro biofilm model and were used in all OPA assays (unless specifically indicated otherwise), mouse colonization analyses, and in vivo influenza-induced pneumonia models.
To establish biofilm conditions, NCI-H292 epithelial cells [CRL-1849, American Type Culture Collection (ATCC)] were first cultured in RPMI 1640 with the addition of fetal bovine serum in T75 flasks at 37°C and 5% CO2. After reaching 100% confluency, H292 cells were prefixed in 4% buffered paraformaldehyde at 34°C for 48 hours, followed by three washes with PBS. CDM-grown pneumococci were then seeded onto fixed H292 cells with change of medium occurring every 12 hours. Formed biofilms were exposed to heat (38.5°C) for 4 hours, and released cells were then collected by centrifugation, washed once with and resuspended in PBS, and quantified by OD600 measurement. Experiments using biofilm-released cells included in vivo sepsis and pneumonia models.
Challenge models
Mice were challenged with 1 × 104 (sepsis model) or 1 × 106 (pneumonia model) CFU of pneumococci strains through intraperitoneal or intranasal (with isoflurane) administration, respectively. To induce colonization, mice were administered 1 × 106 CFU bacteria intranasally without isoflurane. To prompt influenza-induced pneumonia, pneumococci colonization was followed by intranasal inoculation with 40 plaque-forming units of IAV [strain A/PR/8/34 (H1N1); ATCC VR-95]; titers were determined by plaque assays. Mice were monitored every 4 hours for signs of morbidity (huddling, ruffled fur, lethargy, and abdominal surface temperature). Mice found to be moribund were euthanized via CO2 asphyxiation and cervical dislocation.
Antibody analysis
Antigen antibody titer analysis was conducted as described previously (26), with the method extended to include 20 polysaccharides from associated serotypes and the GlpO and PncO protein antigens. Thus, an analysis was extended to all antigens used in the study. Cytokine measurements were accomplished using IFN-γ and IL-17A ELISA kits (R&D Systems).
Tissue bacterial count
At 5 days after colonization, samples were analyzed as presented in Fig. 1E and fig. S7 (B and C). After influenza-induced pneumonia, mice were analyzed at 1 and 5 days after IAV administration, as indicated, or upon becoming moribund; a combination of nasopharynx tissue, nasopharyngeal lavage fluid, lung, and blood samples was collected, and bacterial burden was determined as described previously (28). Briefly, tissue and organ homogenate, lavage fluid, and blood were homogenized to ensure dissociation of bacterial aggregates and then serially diluted on tryptic soy and 5% blood agar plates before enumeration.
CD4+ depletion
For in vivo CD4+ T cell depletion, 0.5 mg of anti-CD4+ monoclonal antibody (Invitrogen) was injected intraperitoneally into the mice for 3 consecutive days. After day 6, a T cell depletion of ≥95% was confirmed using flow cytometry, and these mice subjects were then used in the indicated experiments.
OPA analysis
Extending upon a previous protocol (29), human HL60 cells were differentiated with dimethlyformamide to quantify antibody-mediated opsonophagocytosis and killing of S. pneumoniae exposed to dilutions of sera collected from immunized mice subjects to identify the 50% killing end point (quantified by CFU counts). HL60 cells and pneumococci were incubated for 75 min.
Statistical analysis
Comparisons were analyzed for statistical significance using a two-tailed Student’s t test for unpaired data through the GraphPad Prism software (version 6.0h.283, GraphPad Software Inc.). All data resulted from a minimum of three samples, with animal experiments using between 4 and 12 subjects.
Supplementary Material
Acknowledgments
We thank D. Briles (University of Alabama at Birmingham) for the provision of S. pneumoniae strains and plasmid pUAB055 (containing pspA), A. Hochkoeppler (University of Bologna) for the CRM197 genetic construct, and S. Andreadis (University at Buffalo) for the GFP genetic construct. Funding: The authors recognize the support from NIH awards AI088485 and AI117309 (to B.A.P.) and a SUNY-Buffalo Schomburg fellowship (to C.H.J.). Author contributions: C.H.J. designed and executed the experiments related to vaccine formulations and immunizations. G.Z. completed the LEPS characterization experiments. R.N. assisted with the liposomal formulations. M.B. assisted with the protein binding analyses. Y.L. assisted with the animal experiments. A.H. and P.R. conducted the analysis supporting Fig. 4. B.A.D. and P.K. assisted with the viral administration procedures. C.H.J. and B.A.P. supervised the study and wrote the manuscript. Competing interests: C.H.J. and B.A.P. are drafting a provisional patent application related to this work, which will be submitted to the U.S. Patent and Trademark Office through the University at Buffalo. C.H.J., A.H., and B.A.P. are cofounders of Abcombi Biosciences Inc., a company focused on vaccine design. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/e1701797/DC1
fig. S1. Commensal disease progression model featuring pneumococcal disease.
fig. S2. The LEPS platform.
fig. S3. LEPS characterization.
fig. S4. Protective capabilities of LEPS immunizations when challenged with serotypes that span the current Prevnar 7 and Prevnar 13 treatment options.
fig. S5. LEPS vaccine strategy in rabbits.
fig. S6. OPA assay for GlpO and PncO directed against specific S. pneumoniae cell types.
fig. S7. Evaluation of immunogenicity of GlpO and PncO administered either jointly (coadministration) or as a booster (add-on) with Prevnar 7.
fig. S8. Additional assessment of LEPS20/PncO and GlpO when using a murine IAV-induced pneumonia model with serotype 19F.
fig. S9. Additional murine disease model assessment of LEPS20/PncO and GlpO.
fig. S10. Alternative LEPS formulation procedures and comparison in murine challenge protection assays.
table S1. OPA comparison between Prevnar 13, Pneumovax 23, and a LEPS formulation containing 20 polysaccharides (that is, 20 valent).
table S2. OPA comparison between Prevnar 13, Pneumovax 23, and the PncO + GlpO protein antigens (administered with alum adjuvant).
table S3. GlpO and PncO summary.
REFERENCES AND NOTES
- 1.O’Brien K. L., Wolfson L. J., Watt J. P., Henkle E., Deloria-Knoll M., McCall N., Lee E., Mulholland K., Levine O. S., Cherian T. ; Hib and Pneumococcal Global Burden of Disease Study Team , Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 374, 893–902 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Saokaew S., Rayanakorn A., Wu D. B.-C., Chaiyakunapruk N., Cost effectiveness of pneumococcal vaccination in children in low- and middle-income countries: A systematic review. Pharmacoeconomics 34, 1211–1225 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Papadatou I., Spoulou V., Pneumococcal vaccination in high-risk individuals: Are we doing it right? Clin. Vaccine Immunol. 23, 388–395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vergison A., Dagan R., Arguedas A., Bonhoeffer J., Cohen R., DHooge I., Hoberman A., Liese J., Marchisio P., Palmu A. A., Ray G. T., Sanders E. A. M., Simões E. A. F., Uhari M., van Eldere J., Pelton S. I., Otitis media and its consequences: Beyond the earache. Lancet Infect. Dis. 10, 195–203 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Walker C. L. F., Rudan I., Liu L., Nair H., Theodoratou E., Bhutta Z. A., O’Brien K. L., Campbell H., Black R. E., Global burden of childhood pneumonia and diarrhoea. Lancet 381, 1405–1416 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black R. E., Cousens S., Johnson H. L., Lawn J. E., Rudan I., Bassani D. G., Jha P., Campbell H., Walker C. F., Cibulskis R., Eisele T., Liu L., Mathers C. ; Child Health Epidemiology Reference Group of WHO and UNICEF , Global, regional, and national causes of child mortality in 2008: A systematic analysis. Lancet 375, 1969–1987 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Bogaert D., de Groot R., Hermans P. W. M., Streptococcus pneumoniae colonisation: The key to pneumococcal disease. Lancet Infect. Dis. 4, 144–154 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Gray B. M., Converse G. M. III, Dillon H. C. Jr, Epidemiologic studies of Streptococcus pneumoniae in infants: Acquisition, carriage, and infection during the first 24 months of life. J. Infect. Dis. 142, 923–933 (1980). [DOI] [PubMed] [Google Scholar]
- 9.Huebner R. E., Dagan R., Porath N., Wasas A. D., Klugman K. P., Lack of utility of serotyping multiple colonies for detection of simultaneous nasopharyngeal carriage of different pneumococcal serotypes. Pediatr. Infect. Dis. J. 19, 1017–1020 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Daniels C. C., Rogers P. D., Shelton C. M., A review of pneumococcal vaccines: Current polysaccharide vaccine recommendations and future protein antigens. J. Pediatr. Pharmacol. Ther. 21, 27–35 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feldman C., Anderson R., Review: Current and new generation pneumococcal vaccines. J. Infect. 69, 309–325 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Loo J. D., Conklin L., Fleming-Dutra K. E., Deloria Knoll M., Park D. E., Kirk J., Goldblatt D., O’Brien K. L., Whitney C. G., Systematic review of the effect of pneumococcal conjugate vaccine dosing schedules on prevention of pneumonia. Pediatr. Infect. Dis. J. 33 (suppl. 2), S140–S151 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bakaletz L. O., Bacterial biofilms in the upper airway - Evidence for role in pathology and implications for treatment of otitis media. Paediatr. Respir. Rev. 13, 154–159 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muñoz-Elías E. J., Marcano J., Camilli A., Isolation of Streptococcus pneumoniae biofilm mutants and their characterization during nasopharyngeal colonization. Infect. Immun. 76, 5049–5061 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks L. R., Davidson B. A., Knight P. R., Hakansson A. P., Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4, e00438-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marks L. R., Parameswaran G. I., Hakansson A. P., Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect. Immun. 80, 2744–2760 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tin Tin Htar M., Christopoulou D., Schmitt H.-J., Pneumococcal serotype evolution in Western Europe. BMC Infect. Dis. 15, 419 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richter S. S., Heilmann K. P., Dohrn C. L., Riahi F., Diekema D. J., Doern G. V., Pneumococcal serotypes before and after introduction of conjugate vaccines, United States, 1999–2011. Emerg. Infect. Dis. 19, 1074–1083 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weinberger D. M., Malley R., Lipsitch M., Serotype replacement in disease after pneumococcal vaccination. Lancet 378, 1962–1973 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hausdorff W. P., Hoet B., Adegbola R. A., Predicting the impact of new pneumococcal conjugate vaccines: Serotype composition is not enough. Expert Rev. Vaccines 14, 413–428 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Madhi S. A., Adrian P., Kuwanda L., Cutland C., Albrich W. C., Klugman K. P., Long-term effect of pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumoniae—and associated interactions with Staphylococcus aureus and Haemophilus influenzae colonization—in HIV-infected and HIV-uninfected children. J. Infect. Dis. 196, 1662–1666 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Keller L. E., Robinson D. A., McDaniel L. S., Nonencapsulated Streptococcus pneumoniae: Emergence and pathogenesis. mBio 7, e01792-e15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spijkerman J., Prevaes S. M. P. J., van Gils E. J. M., Veenhoven R. H., Bruin J. P., Bogaert D., Wijmenga-Monsuur A. J., van den Dobbelsteen G. P. J. M., Sanders E. A. M., Long-term effects of pneumococcal conjugate vaccine on nasopharyngeal carriage of S. pneumoniae, S. aureus, H. influenzae and M. catarrhalis. PLOS ONE 7, e39730 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Gregorio E., Rappuoli R., From empiricism to rational design: A personal perspective of the evolution of vaccine development. Nat. Rev. Immunol. 14, 505–514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Hill A., Beitelshees M., Shao S., Lovell J. F., Davidson B. A., Knight P. R. III, Hakansson A. P., Pfeifer B. A., Jones C. H., Directed vaccination against pneumococcal disease. Proc. Natl. Acad. Sci. U.S.A. 113, 6898–6903 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y., Beitelshees M., Fang L., Hill A., Ahmadi M. K., Chen M., Davidson B. A., Knight P. III, Smith R. J. Jr, Andreadis S. T., Hakansson A. P., Jones C. H., Pfeifer B. A., In situ pneumococcal vaccine production and delivery through a hybrid biological-biomaterial vector. Sci. Adv. 2, e1600264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefan A., Conti M., Rubboli D., Ravagli L., Presta E., Hochkoeppler A., Overexpression and purification of the recombinant diphtheria toxin variant CRM197 in Escherichia coli. J. Biotechnol. 156, 245–252 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Tyx R. E., Roche-Hakansson H., Hakansson A. P., Role of dihydrolipoamide dehydrogenase in regulation of raffinose transport in Streptococcus pneumoniae. J. Bacteriol. 193, 3512–3524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero-Steiner S., Frasch C. E., Carlone G., Fleck R. A., Goldblatt D., Nahm M. H., Use of opsonophagocytosis for serological evaluation of pneumococcal vaccines. Clin. Vaccine Immunol. 13, 165–169 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pilishvili T., Lexau C., Farley M. M., Hadler J., Harrison L. H., Bennett N. M., Reingold A., Thomas A., Schaffner W., Craig A. S., Smith P. J., Beall B. W., Whitney C. G., Moore M. R., Sustained reductions in invasive pneumococcal disease in the era of conjugate vaccine. J. Infect. Dis. 201, 32–41 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Brueggemann A. B., Peto T. E. A., Crook D. W., Butler J. C., Kristinsson K. G., Spratt B. G., Temporal and geographic stability of the serogroup-specific invasive disease potential of Streptococcus pneumoniae in children. J. Infect. Dis. 190, 1203–1211 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Sleeman K. L., Griffiths D., Shackley F., Diggle L., Gupta S., Maiden M. C., Moxon E. R., Crook D. W., Peto T. E. A., Capsular serotype–specific attack rates and duration of carriage of Streptococcus pneumoniae in a population of children. J. Infect. Dis. 194, 682–688 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/10/e1701797/DC1
fig. S1. Commensal disease progression model featuring pneumococcal disease.
fig. S2. The LEPS platform.
fig. S3. LEPS characterization.
fig. S4. Protective capabilities of LEPS immunizations when challenged with serotypes that span the current Prevnar 7 and Prevnar 13 treatment options.
fig. S5. LEPS vaccine strategy in rabbits.
fig. S6. OPA assay for GlpO and PncO directed against specific S. pneumoniae cell types.
fig. S7. Evaluation of immunogenicity of GlpO and PncO administered either jointly (coadministration) or as a booster (add-on) with Prevnar 7.
fig. S8. Additional assessment of LEPS20/PncO and GlpO when using a murine IAV-induced pneumonia model with serotype 19F.
fig. S9. Additional murine disease model assessment of LEPS20/PncO and GlpO.
fig. S10. Alternative LEPS formulation procedures and comparison in murine challenge protection assays.
table S1. OPA comparison between Prevnar 13, Pneumovax 23, and a LEPS formulation containing 20 polysaccharides (that is, 20 valent).
table S2. OPA comparison between Prevnar 13, Pneumovax 23, and the PncO + GlpO protein antigens (administered with alum adjuvant).
table S3. GlpO and PncO summary.