

Abstract
A culmination of large-scale ideas and efforts has truly allowed for the use of large genomic DNA clones housed in Bacterial Artificial Chromosome (BAC) vectors for biological research. Fundamental advances that have allowed this to happen include (1) the completion of genome sequencing projects and the establishment of highly annotated web-accessible databases allowing for the rapid identity and purchase of BAC clones containing genes of interest. (2) The generation of methodologies to modify BACs genetically, allowing for the rapid creation of gene targeting constructs or transgenic reporter gene constructs using homologous recombination in bacteria.
Recent efforts on our part have capitalized on these advances by using BACs and bacterial recombination methods to generate fluorescent protein reporter transgenic mice to study skeletal biology. The rationale for using BAC genomic DNA clones to engineer reporter gene constructs is based on their much larger size, thus increasing the likelihood that most, if not all, of a gene’s respective cis regulator elements are present, giving a truer representation of the endogenous gene’s expression. In a relatively short amount of time, we have become extremely proficient at generating BAC reporters. Contrary to the widely perceived notion that working with BACs is complex and difficult, we decided to write this chapter to encourage laboratories that are currently using traditional molecular cloning methods to engineer transgenic DNA constructs to strongly consider learning BAC methodologies. As an example, we walk through the steps we took to generate the transgenic reporter mouse line, Tenascin C (TNC)-mCherry.
Keywords: BAC, Bacterial recombination, GFP, Transgenic, Reporter gene
1. Introduction
The DNA sequencing of genomic DNA libraries and the careful annotation of Bacterial Artificial Chromosome (BAC) clones with open access to the research community has allowed for the rapid identity and procurement of BAC clones, thus transforming the field of molecular genetics. BACs are vectors capable of holding very large pieces of DNA typically 100–300 kb in size and are commonly used to contain genomic DNA libraries. For the mouse, several BAC genomic DNA libraries exist, with 17 murine genomic DNA BAC libraries derived from a variety of different mouse strains being available from the Children’s Hospital Oakland Research Institute (CHORI). However, high-resolution, large-scale genome DNA sequencing has thus far been carried out only on the C57BL/6J strain (1), whose two libraries were constructed by the Roswell Park Cancer Institute (RPCI) and aptly named RPCI-23 and RPCI-24 (2). The mapping of BAC clones against genetic loci can be conveniently viewed by using a web browser at a few different web sites, including Map Viewer from the National Center for Biotechnology Information (3) and the Genome Browser from the University of California, Santa Cruz (4).
In conjunction with the annotation and accessibility of BAC clones, equally important has been the evolution of methodologies to modify large pieces of DNA genetically. Given the large size of genomic DNA inserts, conventional cloning methodologies are not possible. An alternative strategy is to modify the genomic DNA BAC clone genetically while still in the bacteria using homologous recombination. At least two different recombination systems are in wide use, including those based on RecA recombinase and the bacteriophage l Red recombinase system. We have been successful using both systems, and each system offers different advantages based on the experimental context (5). To create conditional knock-out gene targeting DNA constructs, a number of plasmids and bacterial strains have been generated using the Red recombinase system (6). To create transgenic reporter gene animals, both the one and two vector systems utilizing RecA recombinase work well (7–9).
This chapter will focus on methodologies used to modify BAC clones genetically for the purposes of building reporter gene constructs based on the RecA system. The rationale for using BAC genomic DNA clones to engineer reporter gene constructs is based on their much larger size, thus increasing the likelihood that most, if not all, of a gene’s respective cis regulator elements are present, giving a more faithful representation of the endogenous gene’s expression. This methodology cannot be applied to all genes, given that some genes are very large and extend over multiple BAC clones. Large-scale, BAC-based transgenesis projects such as GENSAT indicate that BAC reporters can be generated from a very high percentage of genes (~85%), with the average transcriptional unit estimated to be ~100 kb in size (10). To provide a comprehensive review of our methodology, we have organized the engineering of a BAC reporter into six consecutive steps (Fig. 1). As an aid to this chapter, we have included some data on the recent construction of a new reporter gene animal model for the gene Tenascin C.
Fig. 1.
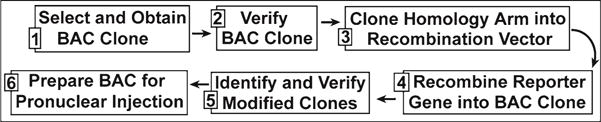
Diagram organizing the process of generating a BAC reporter in six defined steps. Once all the necessary reagents are obtained, the process typically takes only 2 weeks.
2. Materials
2.1. Growing and Storing BACs
LB Media: 10 g peptone from casein, 5 g yeast extract, 10 g NaCl, up to 1 L dH2O, adjust pH to 7.0, and sterilize by autoclaving.
Antibiotics: chloramphenicol: 2,000×; 25 mg/ml in ethanol, ampicillin: 1,000×; 50 mg/ml in water, and tetracycline: 1,000×; 10 mg/ml in water.
Glycerol stock solution: 65% (v/v) glycerol (sterile), 0.1 M MgSO4, 25 mM Tris-Cl, pH 8.0.
2.2. BAC Purification
2.2.1. BAC Mini-prep
P1 Solution: 50 mM Tris, pH 8.0, 10 mM EDTA, 100 μg/ml RNase A.
P2 Solution: 200 mM NaOH, 1% SDS.
EB Solution: 10 mM Tris-HCl, pH 8.0.
2 M potassium acetate, pH 5.5.
Phenol/chloroform.
Chloroform.
2-Propanol.
2.2.2. Preparing BAC for Pronuclear Injection
Qiagen Maxi kit.
2 M potassium acetate, pH 5.5.
Whatman 1 circular filter paper (Fisher Scientific).
Phenol/chloroform.
Chloroform.
2-Propanol.
CL-4B Sepharose (Sigma).
Poly-Prep columns (BioRad).
Restriction enzyme solutions.
Injection buffer: 10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 100 mM NaCl.
1,000× polyamine solution: 30 mM spermine (tetrahydrochloride), 70 mM spermidine (trihydrochloride) (Sigma) dissolved in ddH2O and sterile-filtered through a 0.2-μm filter.
2.3. Field Inversion Gel Electrophoresis
FIGE mapper (BioRad).
Cooling module (BioRad).
Variable speed pump (BioRad).
Agarose.
1× TBE: 89 mM Tris base, 89 mM boric acid, 1 mM EDTA.
Ethidium bromide (10 mg/ml in water).
Low-range pulse-field gel marker (New England Biolabs).
6× Loading dye: 12%glycerol, 60 mM Na2EDTA, pH 8.0, 0.003% bromophenol blue, and 0.003% xylene cyanol.
2.4. Bacterial Recombination
pLD53.SC2 and pSV1 vectors (generously provided by Shiaoching Gong).
Pir2+ cells (Invitrogen).
BAC clone suppliers (BAC-PAC Resource Center, CHORI, Invitrogen).
Shaker incubators set at 30°C and 37°C.
Incubator set at 42°C.
Qiagen Midi Kit.
2.5. Making and Transforming Electro-Competent Bacteria
Electroporator.
Cuvette with 0.1-cm gap.
Sterile dH2O.
Sterile 10% (v/v) glycerol.
SOB media: 20 g peptone from casein, 5 g yeast extract, 0.5 g NaCl, up to 1 L with dH2O, adjust pH to 7.0, sterilize by autoclaving. Just before use, add 2.5 mM KCl, 10 mM Mg2Cl, and 10 mM Mg2SO4.
2.6. Reagents for Cloning Homology Arm and Colony PCR
Thermocycler.
Taq polymerase.
PFX polymerase (Invitrogen).
PCR purification kit (Qiagen).
1× TAE (40 mM Tris acetate, 1 mM EDTA).
Zymoclean gel DNA recovery kit (Zymo Research).
Calf intestinal alkaline phosphatase.
T4 DNA ligase.
2.7. Websites and Softwares
Genome Bioinformatics: http://genome.ucsc.edu/.
NCBI Map Viewer: http://www.ncbi.nlm.nih.gov/projects/mapview/;
CHORI: http://bacpac.chori.org/.
Vector NTI (Invitrogen).
3. Methods
3.1. Selecting and Obtaining a BAC Clone
The first step of this process is selecting and obtaining a BAC clone containing the gene of interest. Unfortunately, there is no one simple answer to which BAC clone one should choose that will guarantee faithful readout of the reporter gene to the endogenous gene. Gene regulation is complex, with the key cis regulatory elements being potentially present far upstream and downstream or within the intron regions of a gene. In our own work, we tend to favor BACs, where the gene is centrally located with perhaps some favoritism toward the upstream region of the gene. As an example, the BAC clone RP24-79K20 we chose for Tenascin C (Fig. 2) has 68.5 kb of DNA sequence upstream of the translational start site and 54.8 kb of DNA sequence downstream of the stop codon. BAC clones from RPCI-23 and RPCI-24 are available from the BAC/PAC Resource Center at CHORI. Below we list instructions on how to view BAC clones for a gene of interest and how to download annotated DNA sequence information.
Fig. 2.
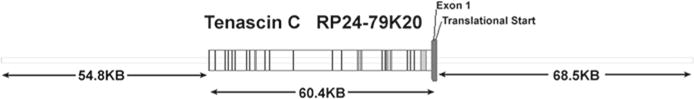
Schematic of Tenascin C BAC clone RP24-79K20. While the initial selection of BAC clones is gene dependent, we typically favor clones where the exons, shown as gray vertical bands, are located in the central region of DNA fragment, leaving plenty of upstream and downstream regulatory sequences. This BAC clone was used to generate a Tenascin C-mCherry reporter mouse shown in Fig. 5.
3.2. Viewing BAC Clones Across a Gene of Interest (Contig Map)
Go to the UCSC Genome Bioinformatics web site (http://genome.ucsc.edu/).
Select: Genome Browser
-
Make sure the appropriate information is selected, in our case:
clade: Mammal; genome: Mouse; assembly: (use most recent); position or search term: (Type in Gene Name Here; i.e. Tenascin), and select submit.
A list will appear, select the appropriate gene. A Contig Map should appear. You should see a list of BAC clone ID’s starting with RP23 or RP24 on the left (see Note 1).
You can zoom in and out by selecting 1.5×, 3×, or 10× toward the top of the browser page.
You can click on the desired Clone ID to find more detailed information about that clone. For example, for TNC, we picked RP24-79K20: Chromosome 4; Start: 63566320; End: 63750190; Length: 183871.
3.3. Downloading and Viewing an Annotated DNA Sequence for a BAC Clone
Open the NCBI Map Viewer: http://www.ncbi.nlm.nih.gov/projects/mapview/.
Under Search, select Mus musculus, type in your Clone ID (i.e., RP24-79K20), and hit Go.
Select the Clone ID again under map element.
On the left under Region Shown, type in the Start and End coordinates of your Clone ID (For TNC, Start: 63566320, End: 63750190) and hit Go (see Note 2).
Toward the top right of the browser, select Download/View Sequence Evidence.
Change sequence format from FASTA to Genbank and hit Save to Disk.
This downloaded file is a Genbank DNA sequence file with all the annotations, which is extremely useful for visualizing the details of your BAC clone. At this point, we work with the BAC clone in Vector NTI (see Note 3).
3.4. Storing BAC Clones
From a bacterial stab, inoculate 2 ml of LB media containing the appropriate antibiotic and grow overnight at 37°C.
Place 1 ml of culture into a 1.7-ml screw cap tube. Add 0.5 ml of glycerol stock solution and mix by inverting. Freeze on dry ice and store at −70°C.
3.5. Verifying BAC Clones
Upon receiving the BAC clone, it is important to verify that you received the correct piece of genomic DNA. From the downloaded sequence, identify rare-cutting restriction enzymes that will cut the genomic insert two to four times. Pick three to four restriction enzymes and carry out a diagnostic restriction enzyme digest. Some restriction sites may be present in the BAC vector, so it is worthwhile to obtain annotated DNA sequence information of the BAC vector used for your clone, which is available from CHORI in the form of a downloadable Genbank file or map (http://bacpac.chori.org/vectorsdet.htm). In our experience, most BAC clones cut in a predicted fashion. At the same time, it is not entirely unusual to see some differences from the predicted DNA fragment size. If you are focused on a single gene, I would recommend obtaining two different BAC clones against your gene of interest, allowing you to compare one with the other. Future PCR amplification of homology arm(s) will further verify the identity of your BAC clone as well.
3.6. BAC Mini-prep
Inoculate LB media containing the appropriate antibiotic with a frozen glycerol stock, and grow overnight in a shaker incubator at 37°C and ~200 rpm. (Estimate 1 ml of culture per restriction enzyme digestion reaction.)
Aliquot 1 ml of culture into 1.5-ml microfuge tubes, centrifuge, and aspirate off LB medium.
Carryout alkaline lysis: (see Note 4) thoroughly resuspend bacterial pellet in 300 μl of P1 solution. Then, add 300 μl of P2 buffer, invert two to three times to mix, and incubate for 5 min. Finally, add 300 μl of 2 M potassium acetate and invert two to three times to mix. Chill on ice for 15 min followed by centrifuging at > 13,000 × G in a microfuge. Transfer the supernatant into a new microfuge tube.
Carryout phenol/cholorform extractions: Add 500 μl of a 1:1 mixture of phenol/chloroform to each tube and mix by inverting. Separate into two phases by centrifuging at >13,000 × G in a microfuge for 5 min. Transfer the upper aqueous phase into a new tube and add 500 μl of chloroform. Mix again by inverting and centrifuge at > 13,000 × G in a microfuge for 5 min. Transfer the upper aqueous phase into a new tube.
Precipitate BAC by adding an equal volume of 100% 2-propanol. Place on ice for 15 min followed by centrifuging for 30 min at > 13,000 × G in a microfuge. Wash the pellet with 750 μl of 70% ethanol and centrifuge for 5 min. Remove ethanol and let the pellet air dry for 5 min.
Add 10 μl of EB solution to the pellet. Allow 2–5 min for the pellet to go into the solution and store on ice.
3.7. Diagnostic Restriction Enzyme Digestion
Select restriction enzymes that cut two to four times according to the BAC sequence. Set up digestion: 10 μl of BAC DNA from mini-prep, 0.5 μl of enzyme, 2 μl of 10× buffer, and 8 μl of H2O. Incubate at 37°C for 30 min to 1 h.
3.8. Field Inversion Gel Electrophoresis
Prepare a 1% agarose gel in 1× TBE buffer and extra 1× TBE buffer for the electrophoresis chamber. Pre-chill the buffer and gel in the refrigerator for at least 1 h before use.
Load the gel with a DNA marker and samples: Cut a thin slice of the Low-Range PFG Marker and slide it into one well of the gel. Immerse the gel into the electrophoresis chamber containing pre-chilled 1× TBE buffer. Add 5 μl of loading dye to each sample and load onto the gel.
Run the gel: Turn on the FIGE mapper, select program 4, and hit Start. Initially, run the samples into the gel for 20 min with a cooling module and pump off. After 20 min, turn on the cooling module (set to 18°C) and pump (set to 70). Run overnight for ~16 h (see Note 5).
Stain the gel with ethidium bromide: Stop the gel the next morning and stain the gel in 300 ml of 1× TBE buffer containing ~200 μl of ethidium bromide (10 mg/ml).
3.9. Homologous Recombination in Bacteria and Homology Arm Design
For the purposes of modifying large DNA fragments cloned into BAC vectors, homologous recombination systems such as those that utilize the function of the bacterial recombinase A (RecA) have been developed. Both one vector and two vector RecA systems have been developed by Shiaoching Gong and colleagues. The major distinction between these two systems is that with the two vector system, pLD53.SC2 and pSV1, the vector carrying the reporter gene, pLD53.SC2, remains integrated into the BAC clone, while the one vector system, pLD53-SCAEB, allows for the removal of unwanted vector sequences using a resolution step involving the product of the SacB gene (7). While the resolution of unwanted vector sequences is preferable, the one vector system is less high throughput because the process involves cloning two separate homology arms and carrying out two consecutive recombination steps. Moreover, some BAC vectors contain a SacB gene, requiring its removal prior to using the pLD53-SCAEB system (8). In contrast, the two vector system is high throughput involving the cloning of only one homology arm and carrying out one recombination step. The two vector system has been extensively used in the large-scale BAC transgenesis project, GENSAT. Additionally, the vector backbone of pLD53.SC2 does not appear to contribute to or alter the gene expression of its targets. Therefore, for the rapid generation of BAC reporters, we favor the two vector system and will focus on this recombination system.
The two vector RecA recombinase system includes the vectors pLD53.SC2 and pSV1 (Fig. 3a, b). The original pLD53.SC2 contains a multiple cloning site (MCS) upstream of a cassette containing a Kozak sequence, EGFP reporter, and bovine growth hormone polyadenylation signal. This vector confers ampicillin resistance and has an R6kγ replication origin. Plasmids that contain the R6kγ replication origin require the replication protein p, a product of the pir gene, to replicate (7, 11, 12). Pir + cells, PIR1 and PIR2, are commercially available from Invitrogen. The pSV1 plasmid carries the RecA recombinase, tetracycline resistance gene, and a temperature-sensitive replication origin allowing it to replicate at 30°C and stop replicating at 42°C (9).
Fig. 3.
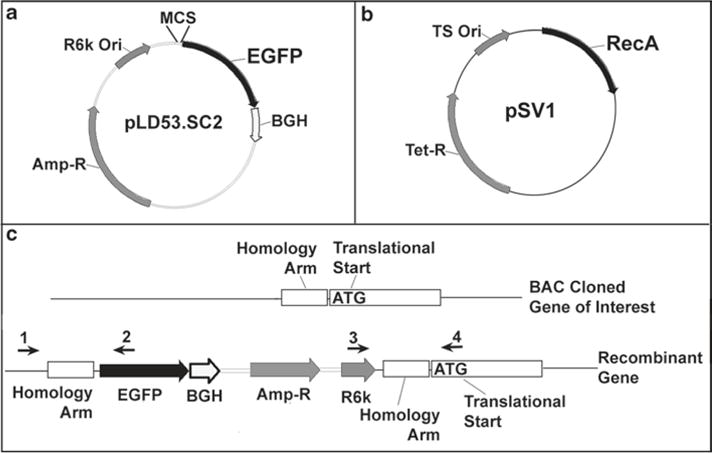
The two vector recombination system using RecA. (a) The pLD53.SC2 vector contains an EGFP reporter gene downstream of a multiple cloning site (MCS), ampicillin resistance, and an R6kγ replication origin. (b) The pSV1 vector contains a RecA recombinase, tetracycline resistance, and a temperature-sensitive replication origin (replicates at 30°C, does not replicate at 42°C). pSV1 is transformed into the bacteria containing the BAC clone of interest to introduce RecA. (c) A homology arm, typically located a few nucleotides upstream of the translational start site, is cloned into the MCS of pLD53.SC2. Recombination is carried out by transforming pLD53.SC2 (with homology arm) into the bacteria containing your BAC clone and pSV1. Recombinants are initially selected for by colony PCR using primers ((1&2) or (3&4)) that flank the homology arm. Final recombinants are verified by diagnostic restriction enzyme digestion and FIGE (see Fig. 4).
To generate a BAC reporter, a homology arm located a few nucleotides upstream of your gene of interest translational start site is PCR cloned into the MCS of pLD53.SC2. pSV1 is transformed into the bacteria containing your BAC clone of interest, introducing the RecA recombinase into these cells. Then, pLD53. SC2 (containing your cloned homology arm) is transformed into the pSV1/BAC clone bacteria cells. Recombinants are selected for by antibiotic selection and screened first by colony PCR, followed by diagnostic restriction digest. For colony PCR, primers flanking the homology arm should be designed to identify recombinants (Fig. 3c). This protocol generally works with very high efficiency and typically, we only have to pick five to eight bacterial colonies to screen and often identify multiple recombinants in the screening process.
The design of homology arms is context dependent, based largely on the particular gene’s DNA sequence. As a general rule, homology arms should be 300–500 bp in size and located a few nucleotides upstream of the translational start site. However, one should not blindly choose this region without first viewing the DNA sequence for repetitive elements or long single nucleotide stretches. Our past experience has shown that homology arms that include repetitive elements do not typically recombine into our region of interest, but perhaps target elsewhere in the BAC insert or within the bacterial genome. Therefore, under difficult circumstances, where repetitive elements or long single nucleotide stretches are present, we favor designing smaller, more selective homology arms. Homology arm size is a flexible variable and we have successfully targeted BACs with homology arms that are only 200 bp in size. Other groups have reported recombining into BACs with as little as 25-bp regions of homology. At the same time, using very small homology arms is typically met with mixed success.
3.10. Making Electro-Competent Bacteria
Inoculate 2 ml of LB media containing the appropriate antibiotics with your BAC clone of interest and grow overnight in a shaker incubator at 37°C – 200 rpm. (If bacteria contain pSV1, remember to grow at 30°C.)
In the morning, add the 2 ml culture to 50 ml of SOB media (containing the appropriate antibiotics) and grow in the shaker incubator again until OD600 = 0.6–0.8 (estimate for ~3–4 h at 37°C and longer if grown at 30°C).
Transfer the 50 ml culture to a 50-ml conical tube and chill on an ice water bath for 15 min. (It is essential that bacteria stay ice cold for all subsequent steps).
Centrifuge bacteria in a pre-chilled centrifuge (4°C) for 10 min at 3,220 × g.
Pour off supernatant and suspend pellet in 5–10 ml of ice-cold ddH2O by swirling. After the pellet is resuspended, add more ice-cold ddH2O up to 40 ml.
Centrifuge again as before and repeat step 5.
Centrifuge again as before and repeat step 5, except use ice-cold 10% glycerol solution instead of ddH2O.
Centrifuge again as before, decant most of the 10% glycerol solution (try to leave ~200–300 μl). Resuspend pellet by swirling and tapping on the side of the ice bath bucket. Pre-chill 4–6 microfuge tubes on dry ice and pipet 50 μl of competent bacteria into each tube. Store competent bacteria at −70°C.
3.11. Cloning Homology Arm into the pLD53.SC2 Shuttle Vector (see Note 6)
Design and order PCR primers to amplify your homology arm containing the appropriate restriction enzyme sites.
Carryout a BAC Mini-prep as described above, except resuspend the DNA pellet in 50 μl of EB buffer.
Pipet and run a PCR: Use 4 μl of BAC DNA from the Mini-prep as your template in a 100-μl PCR. When possible, use a high fidelity DNA polymerase for this amplification. Our standard reaction mix contains 10 μl of 10× PFX buffer, 2 μl of 50 mM MgSO4, 8 μl of 2.5 mM dNTPs, 2 μl of sense & antisense primers (25 μM), 65 μl of water, 4 μl of BAC DNA, and 2 μl of PFX DNA polymerase. Amplify the homology arm using the appropriate thermocycling conditions. Try to keep the cycle numbers between 21 and 23 to minimize polymerase errors in the amplified product.
Verify PCR amplification: Run 4 μl of the PCR product on an agarose gel. Purify the rest of the PCR product using a QIAquick PCR purification kit (elution volume: 40 μl).
Restriction enzyme digest: Incubate 20 μl of the purified PCR product and 5 μg of pLD53.SC2 vector separately with the appropriate restriction enzyme(s) at 37°C for 3 h.
Gel purify the restriction enzyme-digested PCR product using Zymoclean gel DNA recovery kit and elute with 10 μl of water.
Add 2 μl of alkaline phosphatase (CIP) to the restriction enzyme-digested vector and incubate at 37°C for 10 min to prevent self-ligating.
Purify vector: Use DNA Clean and Concentrator kit and elute DNA with 10 μl of water.
- Ligate vector and insert: Set up the following ligation reaction and incubate for 5 min.
- Test: Homology arm: 5 μl, Vector: 5 μl, 2× ligase buffer: 10 μl, Quick T4 Ligase: 1 μl.
- Control: Water: 5 μl, Vector: 5 μl, 2× ligase buffer: 10 μl, Quick T4 Ligase: 1 μl.
Electroporate 1 μl of each ligation reaction into 50 μl of Pir + competent Escherichia coli.
Plate onto LB/ampicillin resistant plates and incubate at 37°C overnight.
3.12. Colony PCR to Confirm Subcloning
Pick colonies in the morning and grow in 2 ml of LB/ampicillin (50 μg/ml) medium for ~3–4 h.
Take 100 μl of each culture and transfer to microfuge tubes. Centrifuge and decant LB media and resuspend bacteria pellet in 20 μl of H2O.
Carryout a PCR with the primers used to amplify the homology arm. Use BAC DNA as positive control. Include a 95°C – 5 min initial step in the thermocycling parameters to break apart the bacteria and liberate the BAC DNA into the reaction. (Reaction recipe: 10× buffer (with MgCl2) 1.25 μl, 2.5 mM dNTPs 1.25 μl, 20uM sense and antisense oligos 0.5 μl each, water 6.9 μl, (5 μ/μl) Taq polymerase 0.1 μl, and 2 μl of resuspended bacteria).
3.13. Homologous Recombination in Bacteria
Transform bacterial cells containing your verified BAC clone of interest with 50 ng of pSV1 vector.
Grow in a shaker incubator with 1 ml of SOC without antibiotic selection for 1 h at 30°C – 200 rpm.
Spread 200 μl of culture onto a chloramphenicol (12.5 μg/ml)–tetracycline (10 μg/ml) containing LB agar plate and incubate at 30°C overnight.
Prepare electro-competent bacteria as described in Sub-heading 3.10.
Electroporate 1–2 μl (about 1 μg) of homology arm-pLD53. SC2 DNA into competent BAC clone – pSV1 cells.
Grow in a shaker incubator with 1 ml of SOC without antibiotic selection for 1 h at 30°C – 200 rpm.
Add 5 ml of LB media containing chloramphenicol (12.5 μg/ml)–ampicillin (50 μg/ml)–tetracycline (10 μg/ml), and grow overnight at 30°C – 200 rpm.
Spread 200 μl of culture onto a chloramphenicol (12.5 μg/ml)–ampicillin (50 μg/ml) plate, and incubate at 42°C overnight.
Pick colonies and grow in 2 ml of chloramphenicol (12.5 μg/ml)–ampicillin (50 μg/ml) LB media at 37°C for 3 or more hours.
Carryout a preliminary screen for recombinants using a colony PCR strategy using primers that flank your homology arm. A standard strategy is to have your sense primer upstream of the homology arm present in the BAC clone and your antisense primer in the reporter gene (Fig. 3).
Verify desired recombinants by carrying out a diagnostic restriction digest and FIGE. You need to identify a restriction enzyme that is present in the pLD53.SC2 vector and is also a rare cutter in your BAC clone (we frequently use Pvu1) (Fig. 4).
Fig. 4.
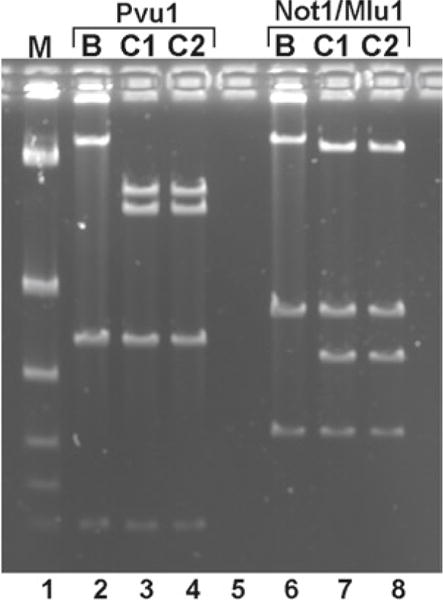
Confirmation of modified BAC clones. Diagnostic restriction enzyme digestion and FIGE were carried out to confirm candidate TNC-mCherry BAC clones identified by colony PCR. Wild-type BAC (lane B) and two BAC integrate clones (lanes C1 and C2) were digested with Pvu1 or with Not1 + Mlu1. Pvu1 cuts one time on the wild-type BAC, generating two bands of 33 kb and 150 kb. PLD53.sc2 vector brought in another Pvu1 site, splitting the 150 kb band to 69 kb and 80 kb bands. Mlu1 cuts one time on the wild-type BAC, generating 41 kb and 142 kb bands. PLD53.sc2 vector brought in a Not1 site, splitting the 142 kb band to 115 kb and 27 kb bands. The extra lower bands are from the BAC vector backbone.
3.14. Preparing the BAC for Pronuclear Injection (see Note 4)
3.14.1. Day 1: Modified Qiagen Maxi Kit Protocol
From a frozen bacterial stock, inoculate two 100 ml cultures (LB media in 500 ml bottle) containing the appropriate antibiotics. Grow overnight at 37°C with shaking at 180–200 rpm.
Pour each culture into two separate tubes and spin down cultures at 3,220 × g for 15 min.
Discard supernatant and invert the tubes over paper towels to remove extra drops of media.
Resuspend bacterial pellets by adding 20 ml of P1 solution to each tube. Gently swirl and view to make sure no clumps of bacteria are present.
Prepare fresh P2 solution. Gently swirl the culture and gradually add 20 ml of P2 to each tube. Let the tubes stand for 3–5 min.
Gradually add 20 ml of 2 M potassium acetate to each tube and incubate on ice for 20 min.
Spin down at 3,220 × g for 10 min to remove most of the heavy particulate.
While the samples are spinning down, pre-equilibrate a Qiagen Maxi column with QBT buffer. Place QF buffer in a 65°C waterbath. Also, prepare a filter paper, place it in a funnel, and pre-wet it with sterile, RNAse/DNAse-free water.
Set the funnel/filter up over the Maxi column, filter the supernatant from step 8, and let it drip into the Maxi column.
Wash the column with QC buffer.
Elute the sample with 10 ml of QF buffer and combine them for a total of 20 ml.
Carry out phenol/chloroform extractions until the interface looks clean. (Add 10 ml of phenol/chloroform for each extraction, gently mix by inverting the tube, and spin down at 3,220 × g for 10 min. On the centrifuge, remove the brake so the interface does not get disturbed.)
Carry out a chloroform extraction: add 10 ml of chloroform, mix by gently inverting the tube, and spin down at 3,220 × g for 10 min.
Precipitate BAC by adding an equal volume of 2-propanol and placing the tube on ice for 10 min. (Add 2-propanol slowly with gentle mixing of the contents.)
Spin down at 19,750 × g for 30 min. (If possible, use a swinging bucket rotor which will pellet your DNA at the very bottom of the tube, in contrast to a fixed angle which will smear your DNA over a greater surface).
Wash with 10 ml of 70% ethanol and spin for 10 min at 19,750 × g.
Discard ethanol. Remove residual ethanol by inverting the tube over a paper towel, followed by removing the extra drops by aspirating the inside of the tube, avoiding the DNA pellet area.
Resuspend the precipitated BAC by adding ~200–250 μl of EB buffer. (Do not pipet up and down to mix). Rotate the tube to move the EB buffer along the bottom surface to solubilize any precipitated BAC. Let the tube stand on ice for 10–15 min.
Transfer the BAC to a microfuge tube. First, gently mix by slowly pipetting up and down two to three times followed by transferring it to the microfuge tube.
Quantify the BAC yield by measuring on a spectrophotometer. In our hands, we typically get yields of ~30 μg of BAC DNA.
Store purified BAC at 4°C (do not freeze).
3.14.2. Day 2: BAC Linearization; Buffer Exchange: Quantification and FIGE Inspection
Linearize 10 μg of BAC by cutting with a unique restriction enzyme. If there are no obvious rare-cutting restriction enzyme sites toward the ends of the genomic DNA insert, most BAC vectors contain a rare cutter. For example, pBACe3.6 and pTARBAC1, the BAC vectors for RP23 and RP24 murine libraries, respectively, contain the rare-cutter PI-SceI in their backbone. Carry out a 200–250-μl digest for 2–3 h. Restriction digest reaction: 10 μg of BAC, 20 μl of 10× buffer, 2 μl of restriction enzyme, and make up to 200 μl with water.
Prepare 50 ml of pronuclear injection buffer. Mix and run through a 0.2-μm syringe filter.
Set up a 2-ml CL-4B sepharose column in a poly-prep chromatography column. Equilibrate the column with ~5–10 column volumes of injection buffer. Column flow rate is ~1 ml/3 min, so it will take 30 min to equilibrate the column. Prepare ten tubes for collection. Collect ~250–300 μl per fraction.
After equilibrating the column, allow the injection buffer to run down to the top surface of the sepharose resin and add the contents of your restriction digestion reaction.
Immediately start collecting fractions after the sample is applied. After the sample enters resin, gently add the injection buffer to the column ~300 μl at a time. Place the fractions on ice as you move to fill the next tube.
Determine the BAC concentration and verify the integrity and quality of the purification by running 25 μl of each fraction on a FIGE (see Notes 7 and 8).
Choose your best fraction, dilute it to a concentration of 1.5 ng/μl with injection buffer. Prepare a 3× polyamine solution in injection buffer by adding 3 μl of 1,000× polyamine solution to 997 μl of injection buffer. Then add 20 μl of 3× polyamine solution to 40 μl of your BAC reporter. Your BAC is now ready for pronuclear injection! (see Note 9) (Fig. 5).
Fig. 5.
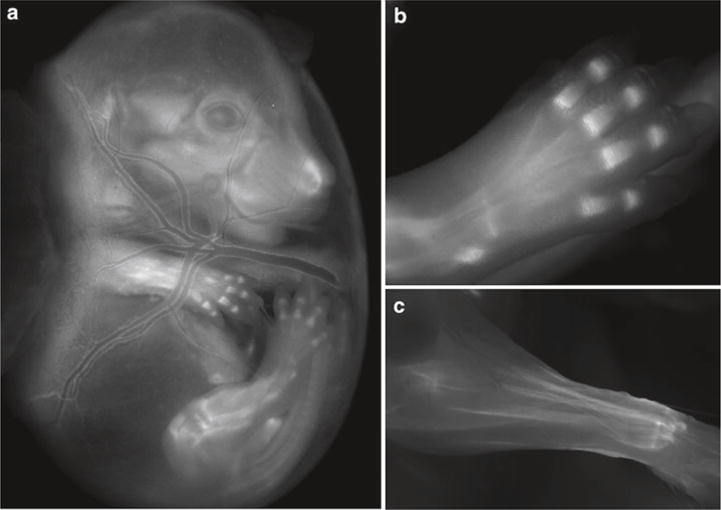
Transgenic reporter mouse generation. (a–c) Images from TNC-mCherry reporter mice. EGFP was replaced by mCherry to generate pLD53.SC2-mCherry. Three Tenascin C founder lines were generated. All lines showed comparable expression with some variation in reporter gene intensity. (a) Image of TNC-mCherry expression in a E16.5 mouse embryo while still in the yolk sac. Expression is detected in the developing craniofacial structures, developing limbs, and major blood vessels of the yolk sac. (b) Image of TNC-mCherry expression in the mouse foot at E17.5 localized within cartilage condensations. (c) Image of TNC-mCherry expression in the forearm at 2 weeks of age. TNC-mCherry reporter expression is highly expressed in tendon.
Acknowledgments
The authors would like to thank Mary Louise Stover for reviewing this manuscript and Shiaoching Gong for providing the pSV1 and pLD53.SC2 DNA constructs. This work was supported by an R21 grant from NIAMS.
Footnotes
If you do not see clone ID’s in the browser, scroll down and under Mapping and Sequencing go to BAC End Pairs and select “full” and hit Refresh.
If your coordinates are not consistent with the map viewer data, check to make sure you are using the most recent assembly in the USCS genome browser.
While we prefer to use Vector NTI, other softwares capable of reading Genbank files are commercially available.
To prevent or minimize shearing of BAC DNA and bacterial genomic DNA, do not vortex or vigorously mix at anytime during the purification procedure. With larger BAC purification preps, mix by very gently swirling. From 200 ml of culture, we typically get ~30 μg of BAC measured on a spectrophotometer. If your yields are much greater than this, I would suspect shearing of bacterial genomic DNA in your sample.
Our FIGE setup involves an electrophoresis chamber that allows buffer to circulate in and out of the unit. The buffer runs through a cooling unit set at 18°C, which dramatically improves the integrity of the BAC DNA, prevents smearing, and allows for cleaner looking gels.
For standard PCR cloning, a variety of kits and strategies can be used. The reagents and kits mentioned in this chapter are what we are currently using, but there are several other options.
Purified BAC DNA has a finite life span. Ideally, purification should start about 1 week before pronuclear injection. We typically start our culture on a Sunday night with our transgenic facility injecting on a Friday.
For quantifying the concentration of BAC DNA, we typically use the NanoDrop, which is very convenient for measuring low concentration fractions eluted from the CL-4B sepharose column. An alternative approach which we also have used in the past is a Picogreen Assay (Invitrogen).
While 1 ng/μl is a standard injection concentration for BACs, you may consider injecting at varying concentrations from 3 ng/μl down to 0.5 ng/μl.
References
- 1.Waterston RH, Lindblad-Toh K, Birney E, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420(6915):520–62. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 2.Osoegawa K, Tateno M, Woon PY, et al. Bacterial artificial chromosome libraries for mouse sequencing and functional analysis. Genome Research. 2000;10(1):116–28. [PMC free article] [PubMed] [Google Scholar]
- 3.Wheeler DL, Church DM, Lash AE, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Research. 2001;29(1):11–6. doi: 10.1093/nar/29.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Research. 2002;12(6):996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maye P, Stover ML, Liu Y, Rowe DW, Gong S, Lichtler AC. A BAC-bacterial recombination method to generate physically linked multiple gene reporter DNA constructs. BMC Biotechnology. 2009;9:20. doi: 10.1186/1472-6750-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Research. 2003;13(3):476–84. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Research. 2002;12(12):1992–8. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparwasser T, Gong S, Li JY, Eberl G. General method for the modification of different BAC types and the rapid generation of BAC transgenic mice. Genesis. 2004;38(1):39–50. doi: 10.1002/gene.10249. [DOI] [PubMed] [Google Scholar]
- 9.Yang XW, Model P, Heintz N. Homologous recombination based modification in Escherichia coli and germline transmission in transgenic mice of a bacterial artificial chromosome. Nature Biotechnology. 1997;15(9):859–65. doi: 10.1038/nbt0997-859. [DOI] [PubMed] [Google Scholar]
- 10.Gong S, Zheng C, Doughty ML, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425(6961):917–25. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 11.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid. 1996;35(1):1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 12.Stalker DM, Filutowicz M, Helinski DR. Release of initiation control by a mutational alteration in the R6K pi protein required for plasmid DNA replication. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(18):5500–4. doi: 10.1073/pnas.80.18.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]