

Abstract
Purpose
Checkpoint inhibitors demonstrate salutary anti-cancer effects including long-term remissions. PD-L1 expression/amplification, high mutational burden and mismatch repair-deficiency correlate with response. We have, however, observed a subset of patients who appear to be “hyper-progressors,” with a greatly accelerated rate of tumor growth and clinical deterioration compared to pre-therapy, which was also recently reported by Institut Gustave Roussy. The current study investigated potential genomic markers associated with “hyper-progression” after immunotherapy.
Method
Consecutive stage IV cancer patients who received immunotherapies (CTLA-4, PD-1/PD-L1 inhibitors or other [investigational] agents) and had their tumor evaluated by next-generation sequencing were analyzed (N=155). We defined hyper-progression as time-to-treatment failure (TTF) <2 months, >50% increase in tumor burden compared to pre-immunotherapy imaging, and >2-fold increase in progression pace.
Results
Amongst 155 patients, TTF <2 months was seen in all six individuals with MDM2/MDM4 amplification. After anti-PD1/PDL1 monotherapy, four of these patients showed remarkable increases in existing tumor size (55% to 258%), new large masses, and significantly accelerated progression pace (2.3-, 7.1-, 7.2- and 42.3-fold compared to the two months before immunotherapy). In multivariate analysis, MDM2/MDM4 and EGFR alterations correlated with TTF<2 months. Two of 10 patients with EGFR alterations were also hyper-progressors (53.6% and 125% increase in tumor size; 35.7- and 41.7-fold increase).
Conclusion
Some patients with MDM2 family amplification or EGFR aberrations had poor clinical outcome and significantly increased rate of tumor growth after single-agent checkpoint (PD-1/PD-L1) inhibitors. Genomic profiles may help to identify patients at risk for progression on immunotherapy. Further investigation is urgently needed.
Keywords: Genomic alteration, hyper-progressor, immunotherapy, checkpoint inhibitor, MDM2 amplification
INTRODUCTION
Immune checkpoint inhibitors have become a standard of care for multiple cancer types. These agents are considered transformative, at least in part because of the phenomenon of hyper-responders that refers to a subset of patients with long-term complete remission. Overall, however, response rates for single agent immune checkpoint inhibitors in solid malignancies range from 20 to 40% (1–3). There are now several biomarkers partially capable of predicting response: PD-L1 expression/amplification, high tumor mutational burden, and mismatch repair gene defects (4–6). Recently, potential markers for acquired resistance have also been reported: loss-of-function mutations in the Janus kinase 1 (JAK1) or JAK2 and beta-2-microglobulin truncation (7). Immunotherapy may also result in a unique response pattern known as pseudo-progression where tumors initially appear larger on imaging, but subsequently regress (8). Importantly, Champiat et al, also documented “hyper-progressive disease,” which they noted in 9% of patients treated with immune checkpoint inhibitors (9). We also recently identified a unique subset of patients whose disease paradoxically accelerated on immunotherapy. Herein, we describe our cohort of hyper-progressors and the genomic profiles that they harbor.
CASE REPORTS
We describe all six patients with stage IV cancer and documented MDM2 gene family amplification who received immunotherapy (see genomic profiles in Supplemental Table 1)
Case #1
A 73 year-old man with bladder cancer metastatic to the liver and lymph nodes (high tumor mutational burden with multiple alterations including MDM2 amplification) was started on the anti-PD-L1 agent atezolizumab (10). Prior therapies included gemcitabine/cisplatinum, as well as a trial of lenvatinib and olaparib, on which he had shown slow progression. Re-staging imaging done 1.9 months after starting atezolizumab showed a 258% increase in size of the liver masses from pre-immunotherapy imaging as well as new liver metastases that were highly positron emission tomography (PET)- fluorodeoxyglucose (FDG) avid (Figure 1A and 2). Repeat imaging one month later confirmed progression. Patient lost weight, developed tumor fevers and progressive syndrome of inappropriate antidiuretic hormone (SIADH) and died soon afterwards.
Figure 1.
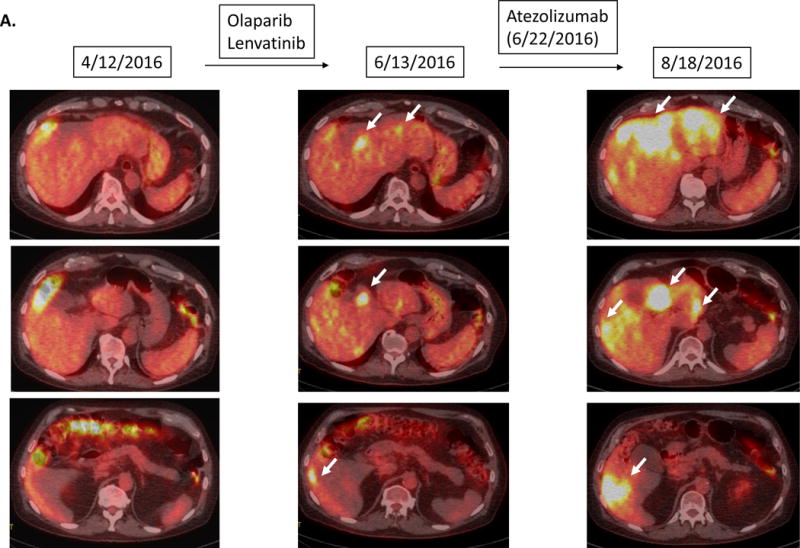
Serial imaging before and after immunotherapy among patients with MDM2/4 amplifications (N=6). Baseline imaging refers to images about 2 months before immunotherapy. Pre-immunotherapy imaging refers to imaging immediately before immunotherapy.
A.
Case #1:
Patient with bladder carcinoma. Tumor showed gradual progression over several months prior to atezolizumab. Restaging 1.9 months after atezolizumab showed a 258% increase in tumor size compared to pre-immunotherapy accompanied by a dramatic increase in PET FDG avidity and new liver masses. Follow up imaging 2.8 months after the initiation of atezolizumab confirmed the progression (imaging not shown) and the patient died soon afterwards.
B.
Case #2:
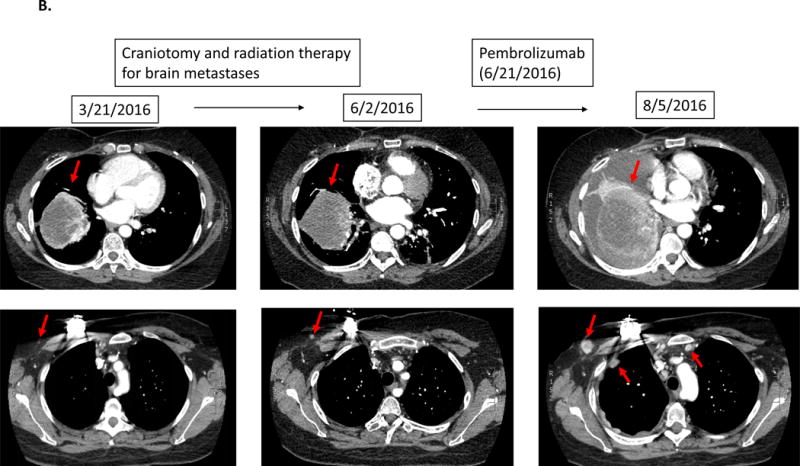
Patient with triple-negative breast cancer. While receiving local therapy against brain metastases, left lung metastasis was overall stable. However 1.5 months after the initiation of pembrolizumab, a CT scan revealed a 55% increase of the left lung mass as well as new chest wall masses and lymphadenopathy.
C.
Case #3:
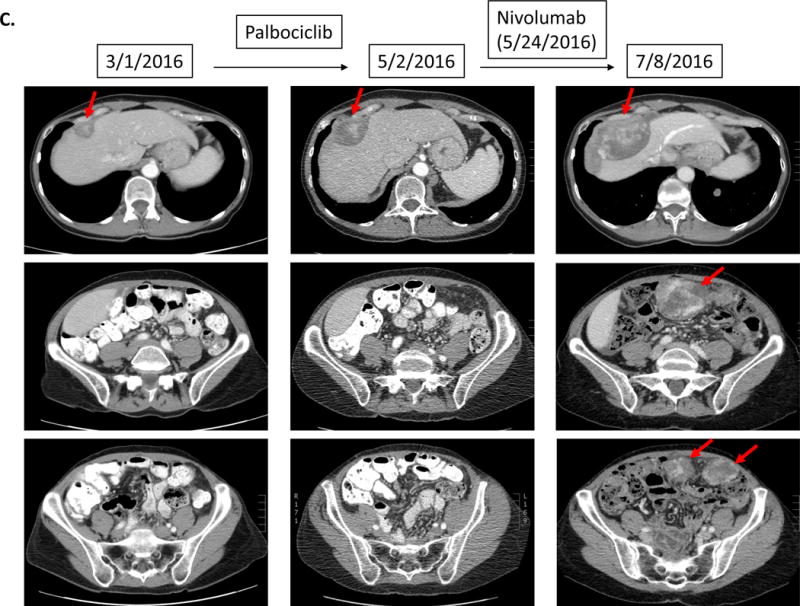
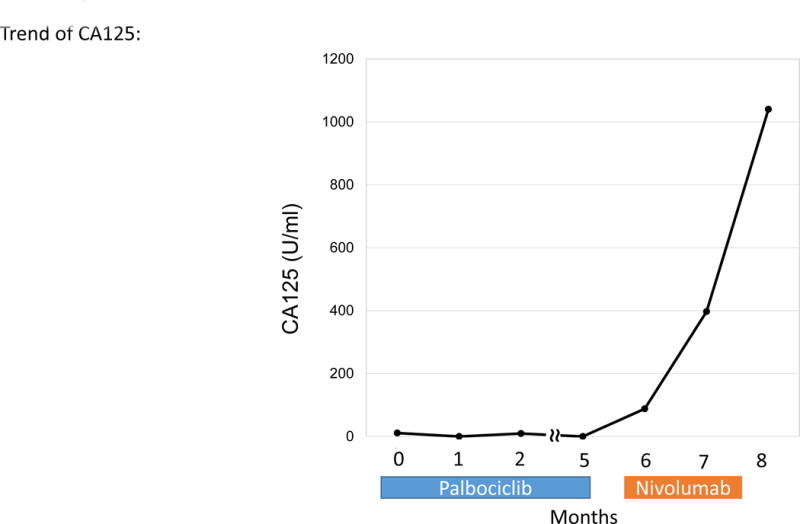
Patient with endometrial stromal sarcoma. Patient had shown increase in tumor size and CA125 (11 to 33 U/mL) over six months. On 1.5 months of nivolumab, CT imaging demonstrated rapid progression of liver metastases and new bulky abdominal masses (overall 242% increase from pre-immunotherapy imaging) (upper panel). CA125 also increased from 33 to 1040 (U/mL) (lower panel).
D.
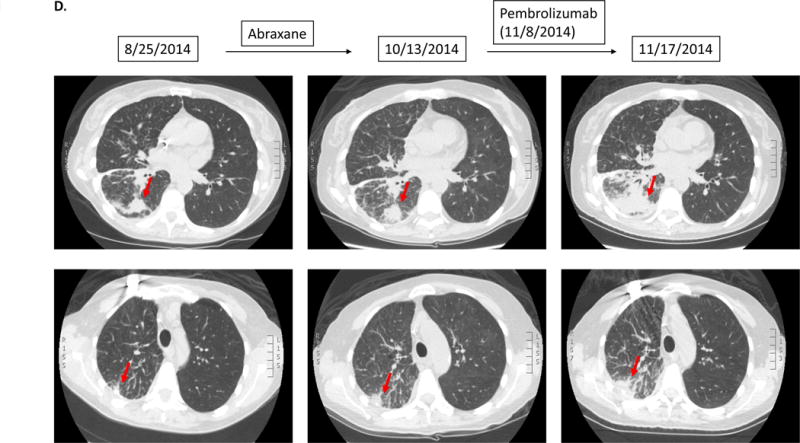
Case #4:
Patient with adenocarcinoma of lung. Patient had gradual progression on Abraxane. Soon after starting pembrolizumab, patient noted severe fatigue/malaise, which prompted the physician to obtain repeat CT imaging. The scan showed rapid progression of known lung metastases (135% increase from pre-immunotherapy).
E.
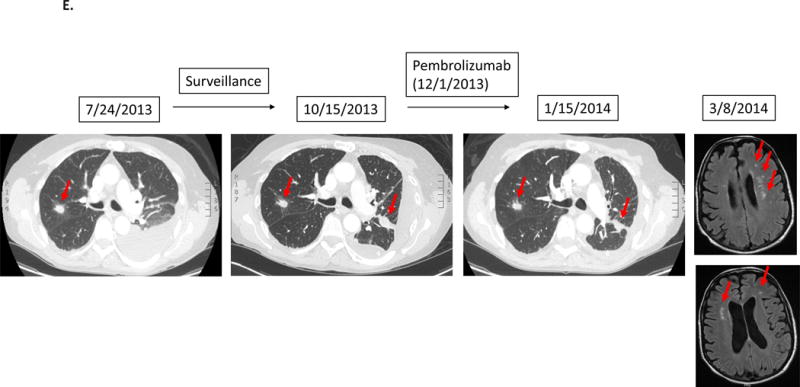
Case #5:
Patient with adenocarcinoma of lung. After first-line chemotherapy, imaging detected new lung disease. Patient was then started on pembrolizumab. However, patient noticed rapidly worsening shortness of breath and severe generalized fatigue. Although CT of the chest showed stable disease, patient was taken off therapy for clinical progression about 1.5 months after the initiation of pembrolizumab. Subsequent MRI of the brain showed multiple new brain metastases.
F.
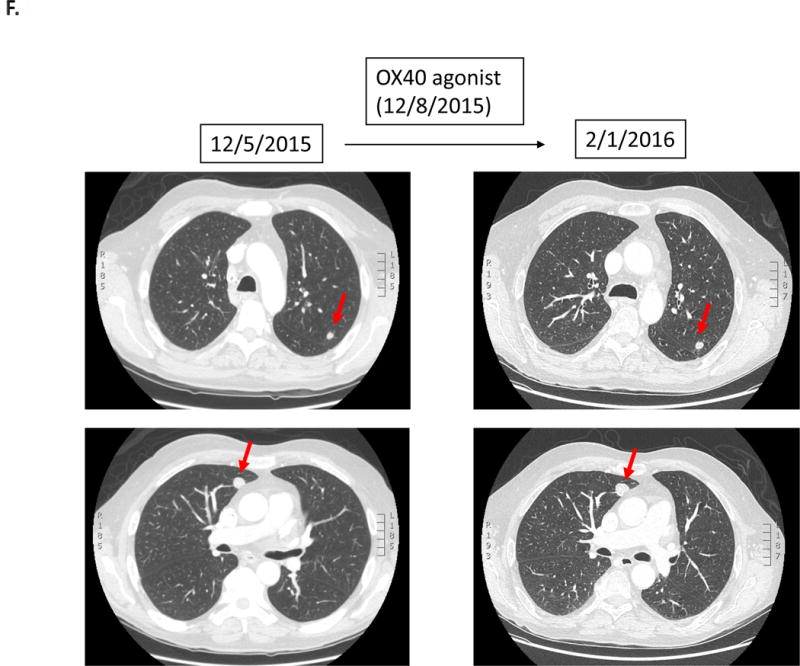
Case #6:
Patient with squamous cell carcinoma of the hypopharynx was treated with an OX40 agonist (third-line therapy). Within 1.4 months, patient was taken off study due to progressive altered mental status secondary to worsening hyponatremia attributed to tumor-associated SIADH. Imaging at the time was stable. The patient died three months later.
Figure 2.
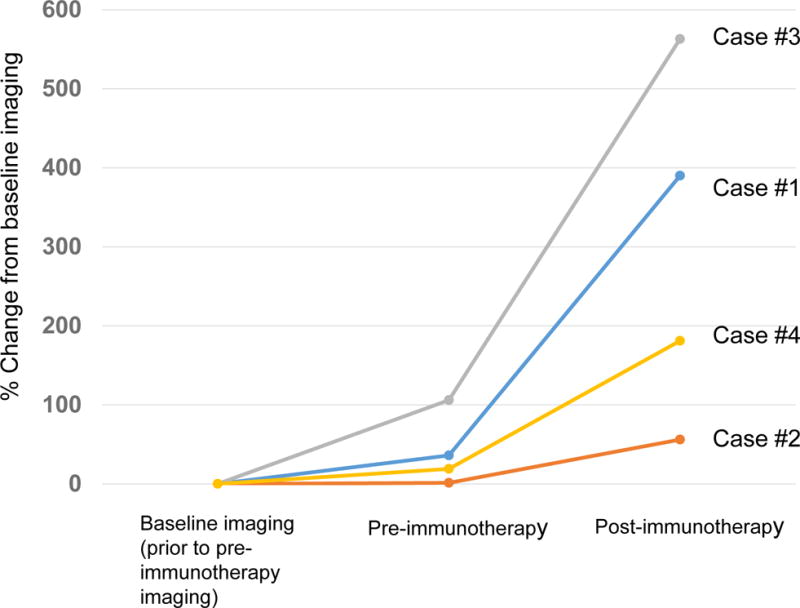
Rate of change in growth pattern in four cases with MDM2 amplification that progressed rapidly while on immunotherapy. Rate of progression is compared from about 2 months prior to immunotherapy (baseline) to image immediately before immunotherapy (pre-immunotherapy), and then to first imaging after immunotherapy. Percent change was evaluated with immune-related response criteria (18).
Case #1: Pre-immunotherapy imaging showed ~36% increase in size of tumors when compared to baseline imaging. After immunotherapy, tumor progressed with 390% increase in lesions when compared to baseline imaging (258% increase from pre-immunotherapy) (7.2-fold increase in progression pace compared to ~2 months before immunotherapy). New liver masses also appeared.
Case #2: Pre-immunotherapy imaging showed 1.3% increase in size of tumors when compared to baseline imaging. After immunotherapy, tumor progressed with 56% increase when compared to baseline imaging (55% increase from pre-immunotherapy) (42.3-fold increase in pace of progression compared to the ~2 months before immunotherapy). New masses also appeared.
Case #3: Pre-immunotherapy imaging showed 106% increase in size of tumors when compared to baseline imaging. After immunotherapy, patient’s tumor progressed with 563% increase compared to baseline (242% increase compared to pre-immunotherapy) (~2.3 fold increase in rate of progression compared to the 2 months before immunotherapy). Multiple new large masses were seen.
Case #4: Pre-immunotherapy imaging showed a 19% increase in size of tumors when compared to baseline imaging. After immunotherapy, patient’s tumor progressed with 181% increase from baseline imaging (135% increase from pre-immunotherapy) (7.1-fold increase in progression pace compared to 2 months before therapy).
Case #2
A 44 year-old woman with triple-negative breast cancer and MDM2 amplification showed stable lung metastases while receiving only local radiation therapy for brain metastases. However, 1.5 months after starting pembrolizumab, CT scan revealed a 55% increase in the left lung mass as well as new chest wall masses and lymphadenopathy (Figure 1B).
Case #3
A 65 year-old woman with endometrial stromal sarcoma had progression in liver metastases on targeted therapy over six months (CA125 increase from 11 to 33 U/mL). Therapy was switched to a trial of nivolumab combined with stereotactic body radiation therapy (SBRT). Soon thereafter, she experienced abdominal pain and new palpable masses. CT imaging (1.5 months post nivolumab) showed rapid progression of liver metastasis plus new bulky abdominal masses (242% increase from pre-immunotherapy) (Figure 1C). CA125 also increased from 33 to 1040 U/ml. Tumor biopsy at the time of progression did not reveal signs of pseudoprogression including lymphocyte infiltration or tumor necrosis (Supplemental Figure 1). Two tumor biopsies (2 weeks and 2 months after the initiation of nivolumab) both showed MDM2 amplification.
Case #4
A 50 year-old woman with lung adenocarcinoma harboring KIF5B-RET fusion and MDM2 amplification had gradual progression on Abraxane. This therapy was changed to pembrolizumab. Nine days later, patient presented with severe fatigue, which prompted the physician to obtain CT imaging. Scans showed rapid progression of lung metastases (135% increase from pre-immunotherapy) (Figure 1D).
Case #5
A 61 year-old man with lung adenocarcinoma had a genomic profile that demonstrated MDM2 and CDK4 amplification. He completed first-line therapy with carboplatin, paclitaxel and bevacizumab. Surveillance imaging demonstrated new lung disease and the therapy was changed to pembrolizumab. The patient then developed worsening dyspnea and severe generalized fatigue. Although CT scan of the chest showed stable disease, therapy was discontinued due to clinical progression 1.5 months after starting pembrolizumab. Subsequent MRI showed multiple new brain metastases (Figure 1E).
Case #6
A 62 year-old man with squamous cell carcinoma (hypopharynx) was treated with an OX40 agonist (third-line therapy). However, 1.4 months afterwards, the patient was taken off study due to progressive altered mental status attributed to hyponatremia from tumor-associated SIADH. Although imaging was stable at the time (Figure 1.F), the patient died three months later. NGS showed several alterations including MDM4 amplification.
METHODS
Patients
Two patients (Cases #1 and #3) were seen in the authors’ clinic. Observations of rapid progression and genomic commonalities prompted further investigation via database and chart review. Consequently, we analyzed all patients with stage IV cancers who received immunotherapies (CTLA-4, PD-1/PD-L1 inhibitors or other [investigational] agents) (March 2011 through July 2016) and had tumor evaluated by NGS (Foundation Medicine, Cambridge MA) at Moores UCSD Cancer Center (N=155). When available, clinical information including lymphocyte count, albumin, lactate dehydrogenase (LDH), number of organ metastases and Eastern Cooperative Oncology Group Performance Status (ECOG PS) were collected (laboratory, radiographic and ECOG PS information was collected within 4 weeks of the initiation of immunotherapy). Royal Marsden Hospital score (11) and MD Anderson Cancer Center prognostic score (12) were evaluated when available. This study was performed in accordance with the guidelines of the UCSD Internal Review Board (PREDICT protocol; NCT02478931) and the investigational studies for which the patients gave consent.
Comprehensive Genomic Profiling
NGS was performed with assay panels of 182, 236 or 315 genes according to previously reported methods in a CLIA-certified, CAP-accredited laboratory (https://www.foundationmedicine.com) (13–15). Average sequencing depth of coverage was greater than 250x, with >100x at >99% of exons. This method of sequencing allows for detection of copy number alterations, gene rearrangements, and somatic mutations with 99% specificity and >99% sensitivity for base substitutions at ≥5 mutant allele frequency and >95% sensitivity for copy number alterations. A threshold of ≥8 copies was used for gene amplification.
Statistical analysis
Fisher’s exact test was used to assess the association between categorical variables. Exact conditional logistic regression analysis was used for the multivariate analysis (16). Bootstrapping was performed using random sampling with replacement to create a large number (N = 982) of “phantom samples” known as bootstrap samples. The sample summary is then computed on each of the bootstrap samples. This method can be superior to approaches relying on the asymptotic distribution of the tests that assumes the data come from a normal distribution, allowing the data of the sample study at hand to be utilized as a surrogate for a larger population. Although cross validation remains the preferred approach for validating predictive models, bootstrapping can be used when the sample size is too small to be split into a training and a validation set and/or there is no independent cross-validation cohort (17) (as is the case in our study).
All tests were 2-sided. P values ≤ 0.05 were considered significant. Fisher’s exact test was performed using Graph-Pad Prism version 7.0 (San Diego, CA, USA). Multivariate exact conditional logistic regression was performed with SAS software, version 9.4 (Cary, NC, USA). Bootstrapping with multiple logistic regression analysis was performed with SPSS version 24.0 (Chicago, IL, USA).
RESULTS
Analysis of all patients who received immunotherapy and had molecular profiling (N=155 patients):
PATIENT CHARACTERISTICS (Table 1)
Table 1.
Patient characteristics (N = 155)1
| Variables | All (N=155) |
TTF <2 months (N=49) |
TTF ≥2 months (N=106) |
Odds- ratio * (95% CI) (Univariate) |
p-value * (Univariate) |
Odds- ratio # (95% CI) (Multivariate) |
p-value # (Multivariate) |
p-value § (Bootstrap) |
|---|---|---|---|---|---|---|---|---|
| Age ≤ 65 years | 104 (67.1%) | 32 (65.3%) | 72 (67.9%) | 0.89 (0.44–1.80) | 0.85 | |||
| Age > 65 years | 51 (32.9%) | 17 (34.7%) | 34 (32.1%) | |||||
| Cancer diagnosis | ||||||||
| Melanoma | 51 (32.9%) | 6 (12.2%) | 45 (42.5%) | 0.19 (0.08–0.48) | 0.0002 | 0.12 (0.01–0.74) | 0.02 | 0.004 |
| Non-small cell lung cancer | 38 (24.5%) | 18 (36.7%) | 20 (18.9%) | 2.50 (1.17–5.32) | 0.03 | 1.33 (0.44–4.00) | 0.75 | 0.56 |
| Squamous cell carcinoma of head and Neck | 11 (7.1%) | 4 (8.2%) | 7 (6.6%) | 1.26 (0.39–4.25) | 0.74 | |||
| Cutaneous squamous cell carcinoma | 9 (5.8%) | 0 (0) | 9 (8.5%) | <0.26 (0.02–1.77) ¶ | 0.06 | 0.85 (0–7.36) | 0.91 | 0.001 |
| Renal cell carcinoma | 6 (3.9%) | 3 (6.1%) | 3 (2.8%) | 2.24 (0.50–9.82) | 0.38 | |||
| Colorectal cancer | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| Types of immunotherapy | ||||||||
| Anti-PD-1 or PD-L1 | 102 (65.8%) | 39 (79.6%) | 63 (59.4%) | 2.66 (1.19–5.72) | 0.02 | 0.37 (0.05–2.50) | 0.39 | 0.19 |
| Anti-CTLA-4 alone or in combination with anti-PD-1 † | 35 (22.6%) | 5 (10.2%) | 30 (28.3%) | 0.29 (0.12–0.76) | 0.01 | 0.84 (0.12–7.40) | 1.00 | 0.74 |
| High-dose IL2 | 10 (6.5%) | 3 (6.1%) | 7 (6.6%) | 0.92 (0.25–3.66) | >0.9999 | |||
| Investigational agents | 8 (5.2%) | 2 (4.1%) | 6 (5.7%) | 0.71 (0.14–2.95) | >0.9999 | |||
| Genomic alterations | ||||||||
| TP53 | 65 (41.9%) | 24 (49.0%) | 41 (38.7%) | 1.52 (0.78–2.99) | 0.29 | |||
| CDKN2A/B | 46 (29.7%) | 12 (24.5%) | 34 (32.1%) | 0.69 (0.31–1.50) | 0.45 | |||
| TERT | 37 (23.9%) | 7 (14.3%) | 30 (28.3%) | 0.42 (0.16–1.05) | 0.07 | 1.68 (0.43–6.62) | 0.57 | 0.47 |
| BRAF | 28 (18.1%) | 8 (16.3%) | 20 (18.9%) | 0.84 (0.35–1.99) | 0.82 | |||
| LRP1B | 19 (12.3%) | 4 (8.2%) | 15 (14.2%) | 0.54 (0.19–1.67) | 0.43 | |||
| PTEN | 16 (10.3%) | 2 (4.1%) | 14 (13.2%) | 0.28 (0.06–1.14) | 0.10 | 0.26 (0.02–1.54) | 0.19 | 0.06 |
| MYC | 15 (9.7%) | 5 (10.2%) | 10 (9.4%) | 1.09 (0.40–3.20) | >0.9999 | |||
| NF1 | 14 (9.0%) | 1 (2.0%) | 13 (12.3%) | 0.15 (0.01–0.85) | 0.07 | 0.26 (0.01–2.27) | 0.37 | 0.17 |
| PIK3CA | 14 (9.0%) | 6 (12.2%) | 8 (7.5%) | 1.71 (0.54–5.50) | 0.37 | |||
| KRAS | 12 (7.7%) | 3 (6.1%) | 9 (8.5%) | 0.70 (0.20–2.79) | 0.75 | |||
| MLL2 | 12 (7.7%) | 2 (4.1%) | 10 (9.4%) | 0.41 (0.09–1.66) | 0.34 | |||
| NOTCH1 | 11 (7.1%) | 0 (0) | 11 (10.4%) | <0.19 (0.02–1.22) ¶ | 0.02 | 0.55 (0–3.67) | 0.63 | 0.003 |
| NRAS | 11 (7.1%) | 1 (2.0%) | 10 (9.4%) | 0.20 (0.02–1.24) | 0.18 | |||
| ARID2 | 10 (6.5%) | 1 (2.0%) | 9 (8.5%) | 0.22 (0.02–1.46) | 0.17 | |||
| EGFR | 10 (6.5%) | 8 (16.3%) | 2 (1.9%) | 10.2 (2.28–48.3) | 0.002 | 8.36 (1.28–98.5) | 0.02 | 0.004 |
| CTNNB1 | 9 (5.8%) | 3 (6.1%) | 6 (5.7%) | 1.09 (0.29–4.08) | >0.9999 | |||
| APC | 8 (5.2%) | 3 (6.1%) | 5 (4.7%) | 1.32 (0.34–5.29) | 0.71 | |||
| ARID1A | 8 (5.2%) | 2 (4.1%) | 6 (5.7%) | 0.71 (0.14–2.95) | >0.9999 | |||
| ASXL1 | 8 (5.2%) | 1 (2.0%) | 7 (6.6%) | 0.29 (0.03–1.74) | 0.44 | |||
| BRCA2 | 8 (5.2%) | 2 (4.1%) | 6 (5.7%) | 0.71 (0.14–2.95) | >0.9999 | |||
| CCND1 | 8 (5.2%) | 2 (4.1%) | 6 (5.7%) | 0.71 (0.14–2.95) | >0.9999 | |||
| FGFR1 | 8 (5.2%) | 4 (8.2%) | 4 (3.8%) | 2.27 (0.63–8.02) | 0.26 | |||
| PTCH1 | 8 (5.2%) | 1 (2.0%) | 7 (6.6%) | 0.29 (0.03–1.74) | 0.44 | |||
| SETD2 | 8 (5.2%) | 2 (4.1%) | 6 (5.7%) | 0.71 (0.14–2.95) | >0.9999 | |||
| ATM | 7 (4.5%) | 2 (4.1%) | 5 (4.7%) | 0.86 (0.17–4.25) | >0.9999 | |||
| NOTCH2 | 7 (4.5%) | 2 (4.1%) | 5 (4.7%) | 0.86 (0.17–4.25) | >0.9999 | |||
| RB1 | 7 (4.5%) | 3 (6.1%) | 4 (3.8%) | 1.66 (0.40–6.38) | 0.68 | |||
| RET | 7 (4.5%) | 3 (6.1%) | 4 (3.8%) | 1.66 (0.40–6.38) | 0.68 | |||
| SMAD4 | 7 (4.5%) | 2 (4.1%) | 5 (4.7%) | 0.86 (0.17–4.25) | >0.9999 | |||
| BAP1 | 6 (3.9%) | 3 (6.1%) | 3 (2.8%) | 2.24 (0.50–9.82) | 0.38 | |||
| CREBBP | 6 (3.9%) | 0 (0) | 6 (5.7%) | <0.42 (0.04–3.24) ¶ | 0.18 | |||
| ERBB2 | 6 (3.9%) | 2 (4.1%) | 4 (3.8%) | 1.09 (0.20–4.79) | >0.9999 | |||
| FAT1 | 6 (3.9%) | 1 (2.0%) | 5 (4.7%) | 0.42 (0.04–3.24) | 0.67 | |||
| GNAS | 6 (3.9%) | 1 (2.0%) | 5 (4.7%) | 0.42 (0.04–3.24) | 0.67 | |||
| MCL1 | 6 (3.9%) | 2 (4.1%) | 4 (3.8%) | 1.09 (0.20–4.79) | >0.9999 | |||
| MDM2/4 | 6 (3.9%) | 6 (12.2%) | 0 (0) | >11.9 (1.53–141.6) ¶ | 0.001 | 10.8 (1.88-infinity) | 0.02 | 0.001 |
| PDGFRA | 6 (3.9%) | 1 (2.0%) | 5 (4.7%) | 0.42 (0.04–3.24) | 0.67 | |||
| SOX2 | 6 (3.9%) | 4 (8.2%) | 2 (1.9%) | 4.62 (1.04–24.7) | 0.08 | 1.56 (0.14–22.0) | 1.00 | 0.49 |
| STK11 | 6 (3.9%) | 1 (2.0%) | 5 (4.7%) | 0.42 (0.04–3.24) | 0.67 | |||
| CD274 (PD-L1) | 5 (3.2%) | 0 (0) | 5 (4.7%) | <0.53 (0.04–3.36) ¶ | 0.18 | |||
| DNMT3A | 5 (3.2%) | 4 (8.2%) | 1 (0.9%) | 9.33 (1.46–115.1) | 0.03 | 13.9 (1.11–783.2) | 0.04 | 0.04 |
| ERBB4 | 5 (3.2%) | 0 (0) | 5 (4.7%) | <0.53 (0.04–3.36) ¶ | 0.18 | |||
| FBXW7 | 5 (3.2%) | 3 (6.1%) | 2 (1.9%) | 3.39 (0.67–19.4) | 0.33 | |||
| FGF19 | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| FGF3 | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| FGF4 | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| FGF6 | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| JAK2 | 5 (3.2%) | 0 (0) | 5 (4.7%) | <0.53 (0.04–3.36) ¶ | 0.18 | |||
| KDM6A | 5 (3.2%) | 0 (0) | 5 (4.7%) | <0.53 (0.04–3.36) ¶ | 0.18 | |||
| MET | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| PBRM1 | 5 (3.2%) | 2 (4.1%) | 3 (2.8%) | 1.46 (0.25–7.33) | 0.65 | |||
| PDCD1LG2 | 5 (3.2%) | 0 (0) | 5 (4.7%) | <0.53 (0.04–3.36) ¶ | 0.18 | |||
| ZNF217 | 5 (3.2%) | 0 (0) | 5 (4.7%) | <0.53 (0.04–3.36) ¶ | 0.18 | |||
| Absolute lymphocyte count (mm3) (N=152) ¥ | ||||||||
| All (N=152) | TTF < 2 months (N=48) | TTF ≥2 months (N=104) | Odds- ratio* | p-value* (Univariate) | ||||
| < 1,000 | 48 (31.6%) | 18 (37.5%) | 30 (28.8%) | 1.48 (0.73–3.09) | 0.35 | |||
| ≥ 1,000 | 104 (68.4%) | 30 (62.5%) | 74 (71.2%) | |||||
| Royal Marsden Hospital score (N=133) ** | ||||||||
| All (N=133) | TTF < 2 months (N=40) | TTF ≥2 months (N=93) | Odds- ratio* | p-value* (Univariate) | ||||
| 0 or 1 | 86 (64.7%) | 22 (55%) | 64 (68.8%) | 0.55 (0.26–1.20) | 0.17 | |||
| 2 or 1 | 47 (35.3%) | 18 (45%) | 29 (31.2%) | |||||
| MD Anderson Cancer Center prognostic score (N=133) ** | ||||||||
| All (N=133) | TTF < 2 months (N=40) | TTF ≥2 months (N=93) | Odds- ratio* | p-value* (Univariate) | ||||
| 0 or 1 | 31 (23.3%) | 7 (17.5%) | 24 (25.8%) | 0.61 (0.24–1.51) | 0.37 | |||
| 2 to 5 | 102 (76.7%) | 33 (82.5%) | 69 (74.2%) | |||||
Odds-ratio and p-value by Fisher’s exact test.
Odds-ratio and p-value by exact conditional logistic regression (multivariate) analysis. Included characteristics with p-value ≤ 0.1 from univariate analysis.
Bootstrapping with multiple logistic regression analysis was conducted on characteristics with p-value ≤ 0.1 from univariate analysis. p-value based on 982 bootstrap samples.
Anti-CTLA-4 alone; N=19, anti-CTLA-4 in combination with ant-PD-1; N=16. Among patients who received anti-CTLA-4 alone, 3 patients had TTF <2 months. Among patients receiving combination of anti-CTLA-4/PD-1, 2 patients had TTF <2 months.
If a variable dichotomized as N versus zero, and the odds ratio was thus zero or infinity, we adjusted the events to be 1 (instead of zero) versus N-1. This produces a numerical odds ratio, which is less than the actual infinite odds ratio. For example, for MDM2/4, where there were N = 6 versus zero patients with TTF<2 versus ≥2 months, the actual odds ratio is infinity. Using the adjustment above, the numeric odds ratio for N = 5 versus one patient is 11.9 and we list it as “>11.9.” The p-value shown is the actual p-value for the unadjusted numbers.
Total of N=152 were tested for lymphocyte count within 4 weeks of starting the immunotherapy.
N=133 were evaluable for Royal Marsden Hospital score and MD Anderson Cancer Center prognostic score. For Royal Marsden Hospital score, each point was counted when albumin was <3.5 (g/dl), LDH > upper limit of normal (175 U/L) and number of organ metastases > 2. For MD Anderson Cancer Center prognostic score, each point was counted when albumin was <3.5 (g/dl), LDH > upper limit of normal (175 U/L), number of organ metastases > 2, gastrointestinal tumor type, and ECOG PS ≥ 1.
Included variables with N ≥ 5.
Abbreviations: CI = confidence interval; IL2 = interleukin-2; TTF = time-to-treatment failure.
Amongst the 155 patients with diverse cancers who received immunotherapy and had molecular profiling, 49 (31.6%) had a time-to-treatment failure (TTF) <2 months; 106 (68.4%), TTF ≥2 months. The most common cancers were melanoma (32.9% [51/155]) and NSCLC (24.5% [38/155]). Most patients received anti-PD-1/PD-L1 agents (65.8% [102/155]) followed by anti-CTLA-4 alone or in combination with anti-PD-1 (22.6% [35/155]). The most commonly altered gene was TP53 (41.9% [65/155]) followed by CDKN2A/B (29.7% [46/155]) and TERT (23.9% [37/155]). MDM2/MDM4 amplifications were found in 6 patients (4%).
CHARACTERISTICS ASSOCIATED WITH OUTCOME AFTER IMMUNOTHERAPIES
Univariate analysis
Patients with melanoma were more likely to have TTF≥2 months (odds-ratio [OR]: 0.19, p = 0.0002); NSCLC, TTF<2 months (OR: 2.5, p = 0.03). Among types of immunotherapy, patients who received anti-PD-1 or PD-L1 antibodies tended to have TTF<2 months (OR: 2.66, p = 0.02); patients who received anti-CTLA-4 alone or in combination with anti-PD-1 tended to have TTF≥2 months (OR: 0.29, p =0.01) (Table 1). Thirty-nine of 102 patients (38%) treated with anti-PD-1 or PD-L1 monotherapy had TTF<2 months versus 10 of 53 patients (19%) who received other immunotherapy agents (p = 0.02) (Table 1). Absolute lymphocyte count and prognostic scores including Royal Marsden Hospital score and MD Anderson Cancer Center prognostic score were not associated with duration of TTF from immunotherapies (Table 1).
Alterations in several genes were associated with favorable clinical outcomes (TTF≥2 months): TERT (OR: 0.42, p = 0.07), PTEN (OR: 0.28, p = 0.10), NF1 (OR: 0.15, p = 0.07) and NOTCH1 (OR: <0.19, p = 0.02). In contrast, EGFR (OR: 10.2, p = 0.002), MDM2/4 (OR: >11.9, p = 0.001) and DNMT3A (OR: 9.33, p = 0.03) alterations were associated with poorer clinical outcomes (TTF<2 months) (Table 1).
Multivariate analysis
Variables with p ≤ 0.1 were included in multivariate analysis. A diagnosis of melanoma was significantly associated with TTF≥2 months (p = 0.02). EGFR, MDM2/4 and DNMT3A alterations remained independent predictors of poor clinical outcome (TTF<2 months) with immunotherapies (all p ≤ 0.04) (Table 1).
Analysis with bootstrapping method
Bootstrapping was conducted on characteristics with p-value ≤ 0.1 from univariate analysis (Table 1). Diagnoses of melanoma and cutaneous squamous cell carcinoma as well as genomic alteration with NOTCH1 were significantly associated with TTF≥2 months (p = 0.004, p = 0.001 and p = 0.003 respectively) (Table 1). In contrast, EGFR, MDM2/4 and DNMT3A alterations were all significant for TTF<2 months (p = 0.004, p = 0.001 and p = 0.04 respectively) (Table 1).
Analysis of patients treated with anti-PD-1/PD-L1 monotherapy (N = 102 patients)
Seven of 8 (87.5%) patients harboring EGFR alterations had TTF <2 months; 1 of 8 (12.5%) had TTF ≥2 months (p = 0.005 by univariate analysis). Among five patients with MDM2 amplifications, all had TTF < 2 months (p = 0.007 by univariate analysis) (Supplemental Table 2). Three of 4 patients (75%) harboring DNMT3A alteration had TTF <2 months; however this was not statistically significant (p = 0.15 by univariate analysis). After multivariate analysis, EGFR alterations and MDM2 amplification continue to show significance for correlation with TTF <2 months (p = 0.02). With the bootstrap analysis, both EGFR and MDM2 alterations remained statistically significant (p = 0.014 and 0.001 respectively) (Supplemental Table 2). Altogether, six of 13 patients (46%) with EGFR or MDM2 alterations demonstrated hyper-progression (Supplemental Tables 1 and 3).
HYPER-PROGRESSORS AND POOR-RISK GENOMIC ALTERATOINS (EGFR, MDM2/4 AND DNMT3A)
We defined hyper-progression as TTF<2 months, >50% increase in tumor burden compared to pre-immunotherapy imaging that was obtained within 2 months of the initiation of immunotherapy, and >2-fold increase in progression pace. Immune-related response criteria (18) were used for the evaluation of response.
Among six patients with MDM2 family amplifications (N = 5, MDM2; N = 1, MDM4), all had TTF<2 months. Four of these patients (67%) demonstrated hyper-progression (all with MDM2 amplification) with increases in lesions compared to pre-immunotherapy as follows: Case #1, 258%; Case #2, 55%; Case #3, 242%; Case #4, 135% (increase in pace of progression ranging from 2.3-fold to 42.3-fold) (Table 1, Figure 1.A–D, Figure 2 and Supplemental Table 1). Of note, Case #3 also had a precipitous rise in cancer antigen 125 (CA125) (Figure 1.C). Two additional patients with MDM2/4 alterations had TTF<2 months, but showed only minor progression of target lesions. Both were taken off study early for clinical symptoms suggesting progression. Case #5 (MDM2 amplification) subsequently demonstrated new brain metastases (Figure 1.E). The other patient (MDM4 amplification) showed worsening SIADH despite stable imaging resulting in stopping drug; patient expired three months later (Case #6, Figure 1.F).
Among 10 patients with EGFR alterations, eight had a TTF<2 months (Table 1). Two (20%) demonstrated hyper-progression (increase in lesions of 53.6% and 125% until first re-staging; increase in progression pace of 35.7- and 41.7-fold) (Supplemental Table 3 and Supplemental Figure 2, Cases #11 and #13). Four of 5 patients with DNMT3A alterations had a TTF <2 months (Table 1). Only one patient was evaluable with serial imaging and did not manifest hyper-progression (Supplemental Table 4).
DISCUSSION
We report that patients with MDM2 family amplifications appear to be at risk of accelerated progression after immunotherapy. Although immunotherapy-induced hyper-responses are a well-known phenomenon, hyper-progression has only recently been described (9, 19, 20). Champiat et al. demonstrated that 9% of patients (12/131) showed a ≥two-fold increase in tumor burden when compared to pre-immunotherapy imaging (9). Older age was associated with hyper-progression in that study, but not in ours (Table 1). Genomic profiling was not reported.
In our 155 patients, all six (4%) with MDM2/MDM4 amplification were taken off immunotherapy in less than two months, and four showed a clearly accelerated rate of tumor growth compared to that before treatment (~2.3 to 42.3-fold increase in progression rate) (Figures 1 and 2). One of our hyper-progressors had a high tumor mutational burden, which is usually associated with response. Another two MDM2/MDM4-amplified patients demonstrated rapid clinical deterioration, with new brain metastases and quickly worsening SIADH, respectively.
A TTF less than two months was also documented in 8 of 10 patients with EGFR alterations and 4 of 5 with DNMT3A alterations. In two patients with EGFR alterations (Supplemental Tables 3 and Supplemental Figure 2, Cases #11 and #13), imaging data documented hyper-progression (~36-fold and 42-fold increase in progression pace compared to the two months before immunotherapy). The other patients did not have available serial imaging or showed evidence of progression compatible with standard resistance.
MDM2 amplification is found in about 7% of cancers; it inhibits the p53 tumor suppressor (MDM4 is a homolog of MDM2 that interacts with it and also inhibits p53) (21). The exact mechanism linking MDM2 amplification and hyper-progression is unclear (Figure 1. A–D and Figure 2). However, immune checkpoint inhibitors can lead to elevated interferon (IFN)-γ (22), which in turn activates JAK-STAT signaling (23) resulting in an increase in interferon regulatory factor (IRF)-8 expression (24). IRF8 binds to the MDM2 promoter inducing MDM2 expression (25, 26). It is conceivable that this cascade may not have significant impact when MDM2 is not amplified; however, in the presence of MDM2 amplification, hyper-expression could occur. Other hypotheses are also plausible, including the involvement of a gene that sits on the MDM2 amplicon and is co-amplified with it. Further investigation is crucial. Of note, MDM2 inhibitors are currently in clinical development (27) raising the possibility that a combination strategy could limit hyper-progression on immunotherapy.
EGFR activation is associated with upregulation of PD-1, PD-L1 and CTLA-1, which can drive immune escape (28). This may explain resistance in EGFR-mutated tumors, though it does not explain the hyper-progression seen in two of our patients. Other resistance alterations that have been described include JAK1 or JAK2 inactivating alterations and beta-2-microglobulin truncation (7).
The possibility of hyper-progression has considerable importance for affected patients. Here we report that specific genomic alterations may be associated with accelerated progression, i.e., the presence of MDM2 family amplification or EGFR aberrations. Importantly, however, a preliminary report by Tawbi et al, (29) suggests that patients with liposarcomas, a disease that commonly harbors MDM2 amplification, occasionally demonstrated clinical benefit (partial response rate of 11% [1/9]) after immunotherapy. The latter observation indicates that, not unexpectedly, the hyper-progression response pattern does not apply universally to patients with this genomic alteration. It is also plausible that hyper-progression may be more likely in some histologies than in others. Indeed, our patients with MDM2/4 amplification and hyper-progresssion had bladder, breast, endometrial stromal sarcoma, lung and hypopharyngeal cancers, and the two patients with EGFR alterations and hyper-progression had adenocarcinoma of the lung. Of note, all of our hyper-progressors were treated with anti-PD1/PDL1 monotherapy, and it remains unknown if other immunotherapy drugs would exhibit similar phenomena. However, in univariate analysis, patients who received an anti-CTLA-4 (alone or combined with antiPD1/PDL1) were significantly less likely to have a TTF less than two months, and none were hyper-progressors. It is also plausible that MDM2 family amplification is not the marker for hyper-progression but rather there is another gene that resides nearby and is co-amplified, leading to hyper-progression from immunotherapy, or that there is some other co-factor present.
Finally, there were several limitations to our study. For instance, an increased pace of progression in our individual patients was based on comparing progression rate in the first two months after immunotherapy treatment to that in the preceding two months. However, a prospective randomized study of checkpoint inhibitors versus standard therapy would be required to have an absolutely unbiased estimate of comparative progression rates.
In summary, our observations suggest that patients for whom anti-PD1/PDL1 monotherapy is planned may require genomic testing to determine if their tumors harbor specific alterations associated with hyper-progression. Individuals with these alterations, if treated with anti-PD1/PDL1 agents, should be closely monitored. Larger studies, validation cohorts, and translational research are urgently needed.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Unique immunotherapy-induced response patterns have been observed, including a report of hyper-progression. Our patients showed remarkably accelerated tumor growth rate after anti-PD1/PDL1 monotherapy. Hyper-progressors harbored MDM2/4 or EGFR alterations, which independently correlated with time-to-treatment failure <2 months, suggesting the need for caution in the presence of these genomic profiles.
Acknowledgments
Funded in part by the Joan and Irwin Jacobs fund and by National Cancer Institute grant P30 CA016672 (RK)
Footnotes
Disclosures: Razelle Kurzrock receives consultant fees from X-biotech and from Actuate Therapeutics, as well as research funds from Genentech, Pfizer, Sequenom, Guardant, Foundation Medicine and Merck Serono, and has an ownership interest in CureMatch Inc
References
- 1.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 3.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 4.Goodman A, Patel SP, Kurzrock R. PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat Rev Clin Oncol. 2016 doi: 10.1038/nrclinonc.2016.168. [DOI] [PubMed] [Google Scholar]
- 5.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel SP, Kurzrock R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther. 2015;14:847–56. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 7.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiou VL, Burotto M. Pseudoprogression and Immune-Related Response in Solid Tumors. J Clin Oncol. 2015;33:3541–3. doi: 10.1200/JCO.2015.61.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive disease (HPD) is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1741. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–20. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkenau HT, Barriuso J, Olmos D, Ang JE, de Bono J, Judson I, et al. Prospective validation of a prognostic score to improve patient selection for oncology phase I trials. J Clin Oncol. 2009;27:2692–6. doi: 10.1200/JCO.2008.19.5081. [DOI] [PubMed] [Google Scholar]
- 12.Wheler J, Tsimberidou AM, Hong D, Naing A, Falchook G, Piha-Paul S, et al. Survival of 1,181 patients in a phase I clinic: the MD Anderson Clinical Center for targeted therapy experience. Clin Cancer Res. 2012;18:2922–9. doi: 10.1158/1078-0432.CCR-11-2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomas RK, Nickerson E, Simons JF, Janne PA, Tengs T, Yuza Y, et al. Sensitive mutation detection in heterogeneous cancer specimens by massively parallel picoliter reactor sequencing. Nat Med. 2006;12:852–5. doi: 10.1038/nm1437. [DOI] [PubMed] [Google Scholar]
- 15.Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta CR, Patel N, Senchaudhuri P. Exact stratified linear rank tests for ordered categorical and binary data. Journal of Computational and Graphical Statistics. 1992;1:21–40. [Google Scholar]
- 17.Steyerberg EW, Harrell FE, Jr, Borsboom GJ, Eijkemans MJ, Vergouwe Y, Habbema JD. Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J Clin Epidemiol. 2001;54:774–81. doi: 10.1016/s0895-4356(01)00341-9. [DOI] [PubMed] [Google Scholar]
- 18.Wolchok JD, Hoos A, O’Day S, Weber JS, Hamid O, Lebbe C, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15:7412–20. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 19.Lahmar J, Facchinetti F, Koscielny S, Ferte C, Mezquita L, Bluthgen MV. Effect of tumor growth rate (TGR) on response patterns of checkpoint inhibitors in non-small cell lung cancer (NSCLC) J Clin Oncol. 2016;34(suppl) abstr 9034. [Google Scholar]
- 20.Saada-Bouzid E, Defaucheux C, Karabajakian A, Palomar Coloma V, Servois V, Paoletti X. Tumor’s flare-up and patterns of recurrence in patients (pts) with recurrent and/or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC) treated with anti-PD-1/PD-L1 inhibitors. J Clin Oncol. 2016;34(suppl) abstr 6072. [Google Scholar]
- 21.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012;72:5209–18. doi: 10.1158/0008-5472.CAN-12-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–63. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 24.Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123:4464–78. doi: 10.1172/JCI68189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao Y, Yu H, Hu W. The regulation of MDM2 oncogene and its impact on human cancers. Acta Biochim Biophys Sin (Shanghai) 2014;46:180–9. doi: 10.1093/abbs/gmt147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou JX, Lee CH, Qi CF, Wang H, Naghashfar Z, Abbasi S, et al. IFN regulatory factor 8 regulates MDM2 in germinal center B cells. J Immunol. 2009;183:3188–94. doi: 10.4049/jimmunol.0803693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tawbi HA-H, Burgess MA, Crowley J, Van Tine BA, Hu J, Schuetze S, et al. Safety and efficacy of PD-1 blockade using pembrolizumab in patients with advanced soft tissue (STS) and bone sarcomas (BS): Results of SARC028–A multicenter phase II study. ASCO Annual Meeting Proceedings. 2016;2016:11006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.