

Abstract
Background and Purpose
Short‐chain fatty acids are fermentation end products produced by gut bacteria, which have been shown to ameliorate inflammatory bowel diseases and allergic asthma. However, the mechanism involved remains largely unknown. Here, we investigate the protective effects and mechanisms of sodium butyrate (SB) on LPS‐induced mastitis model.
Experimental Approach
Effects of increasing doses of SB on blood‐milk barrier function and inflammation are studied in BALB/c mice with LPS‐induced mastitis. The underlying mechanisms of anti‐inflammatory effects of SB were further investigated in LPS‐stimulated mouse mammary epithelial cells (mMECs).
Key Results
The results show that SB decreased LPS‐induced disruption in mammary tissues, infiltration of inflammatory cells and the levels of TNF‐α, IL‐6 and IL‐1β. SB up‐regulated the tight junction proteins occludin and claudin‐3 and reduced blood‐milk barrier permeability in LPS‐induced mastitis. Studies in vitro revealed that SB inhibited LPS‐induced inflammatory response by inhibition of the NF‐κB signalling pathway and histone deacetylases in LPS‐stimulated mMECs.
Conclusions and Implications
In our model, SB protected against LPS‐induced mastitis by preserving blood‐milk barrier function and depressing pro‐inflammatory responses, suggesting the potential use of SB as a prophylactic agent to protect blood‐milk barrier function in mastitis.
Abbreviations
- HDACs
histone deacetylases
- mMECs
mouse mammary epithelial cells
- MPO
myeloperoxidase
- SB
sodium butyrate
- SCFAs
short‐chain fatty acids
- TBST
Tris‐buffered saline with Tween
- TJs
tight junctions
Introduction
The mammary gland is a highly specialized organ that provides milk to the suckling infant in female mammals. Milk is secreted by alveolar mammary epithelial cells (MECs) in lactating mammary glands. However, mastitis, the inflammation of mammary glands resulting from pathogenic bacterial invasion, disrupts normal milk secretion from alveolar epithelial cells (Akers and Nickerson, 2011). The blood‐milk barrier is an important physical barrier that provides important protection for milk integrity and the health of pups (Zhang et al., 2015). However, the blood‐milk barrier formed by alveolar TJs becomes leaky in mastitis. There is some evidence that mastitis leads to losses in mammary function are directly contributing to disruption of alveolar cell integrity. Burton and Erskine (2003) have suggested that alteration of the blood‐milk barrier led to the leakage of blood constituents during the early acute stage of mammary inflammation. Furthermore, it was reported that LPS induced the weakening and disruption of the blood‐milk barrier by compositional changes of claudins in alveolar epithelial tight junctions. The tight junction complexes consist of epithelial cells and intercellular junctions (Koch and Nusrat, 2009) that are specific structures localized at the apical‐most region of the epithelial cell membranes (Tsukita and Furuse, 2000). To date, many protein components of tight junctions have been identified, such as claudins, occludin and junctional adhesion molecules (Edelblum and Turner, 2009). Patients with inflammatory bowel disease demonstrate a loss of tight junction barrier function, increased production of pro‐inflammatory cytokines and immune dysregulation. Considerable evidence suggests that maintaining tight junction networks benefits some gastrointestinal diseases (Fillon et al., 2009; Ramakrishna, 2009). In this study, we have explored the possibility that the protective effects of sodium butyrate (SB) (Figure 1) on the blood‐milk barrier are exerted through preserving tight junctions in a model of mastitis induced by LPS.
Figure 1.
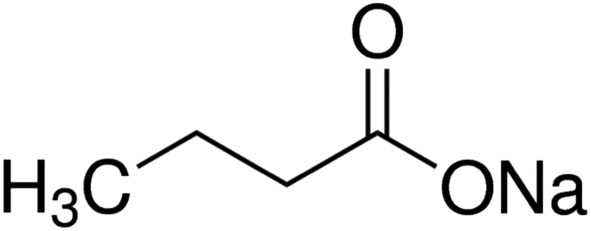
Chemical structure of sodium butyrate.
Gut commensal bacteria form the gastrointestinal immune system and have critical roles on the adaptive immune system. Recent evidence suggests that products of intestinal microbiota might have profound effects on inflammatory disease pathogenesis (Mazmanian et al., 2008; Wen et al., 2008). Germ‐free mice re‐colonized with gut microbiota showed a marked reduction in inflammation. Germ‐free mice are more susceptible to dextran sulfate sodium‐induced colonic inflammation (Maslowski et al., 2009). Short‐chain fatty acids (SCFAs) acetate (C2), propionate (C3) and butyrate (C4) are fermentation end products produced by the intestinal microbiota, especially by anaerobic bacteria (Wong et al., 2006). They have profound effects on gut function, mediated by the FFA2 and FFA3 receptors, by suppressing intestinal inflammation and maintaining intestinal homeostasis (Arpaia et al., 2013; Smith et al., 2013; Chang et al., 2014; Singh et al., 2014). SCFAs ameliorate inflammation through activating NF‐κB signalling pathways (Machado et al., 2012) although SCFAs can also suppress NF‐κB activation (Yin et al., 2001). On the other hand, butyrate acts as an inhibitor of histone deacetylases (HDACs) in a variety of cells. For instance, butyrate suppresses the expression of pro‐inflammatory cytokines in dendritic cells through inhibiting HDAC activity (Arpaia et al., 2013). Furthermore, in intestinal macrophages, butyrate decreases LPS‐induced release of pro‐inflammatory mediators via HDAC inhibition (Chang et al., 2014). Many studies have confirmed the relationship of inflammation to barrier function, but the protective effects of SB on the blood‐milk barrier and its underlying mechanism remain ill‐defined. Here, the protective effects and the molecular basis of action of SB were investigated by assessing alterations in the integrity of the blood‐milk barrier and the inflammatory response to LPS.
Methods
Mouse mammary epithelial cell culture
Mouse MECs (mMECs) were purchased from the American Type Culture Collection (ATCC, ATCC® CRL‐3063™) and cultured in DMEM‐F12 1:1 medium (Hyclone, Logan, CA, USA) containing 10% FBS (Clark), 100 U·mL‐1 penicillin and 100 μg·mL−1 streptomycin (Hyclone) at 37°C in a humidified incubator with 5% CO2.
MTT assay
The effect of SB on cell viability was determined using the MTT assay. The mMECs were treated with SB at a concentration of 100 μM to 2 mM for 18 h. Subsequently, 20 μL MTT (5 mg·mL−1) was added to each well. After 4 h, the supernatant was removed and the formation of formazan was resolved with 150 μL per well of DMSO. The absorbance (OD) was measured at 570 nm on microplate reader.
Real‐time PCR
Cells washed twice with cold PBS, and total RNA was isolated using TRIzol® Reagent (Invitrogen, Carlsbad, CA, USA). Total RNA concentration was measured with a spectrophotometer (KO, K5500), and cDNA was synthesized with RevertAid First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Waltham, MA, USA). Primers were designed based on sequences from the National Center for Biotechnology Information Database. Specificity was determined with Primer‐blast. Primer sequences are designed in Table 1. All PCR primers were synthesized by Sangon Biotech (Shanghai, China). QRT‐PCR was conducted using SYBR Green Supermix on an Applied Biosystems (AB) 7500 Real‐Time PCR System. PCR amplification conditions were as follows: 50°C (2 min) and 95°C (10 min) followed by 40 cycles of 95°C (15 s) and 60°C (1 min). The comparative Ct method was used to quantify normalized target gene expression relative to the calibrator, and the gene expression levels were analysed with the 2−△△CT method.
Table 1.
Primers used in this study
| Gene | Primer | Sequence 5′ > 3′ | Product size (bp) |
|---|---|---|---|
| TNF‐α | Sense | ACGGCATGGATCTCAAAGAC | 116 |
| Anti‐sense | GTGGGTGAGGAGCACGTAGT | ||
| IL‐1β | Sense | GCTGCTTCCAAACCTTTGAC | 121 |
| Anti‐sense | AGCTTCTCCACAGCCACAAT | ||
| IL‐6 | Sense | CCGGAGAGGAGACTTCACAG | 134 |
| Anti‐sense | CAGAATTGCCATTGCACAAC | ||
| GAPDH | Sense | TGCTGTCCCTGTATGCCTCT | 224 |
| Anti‐sense | TTTGATGTCACGCACGATTT |
Animals
All animal care and experimental procedures in the study were conducted in accordance with the animal ethics committee of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Arizona. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Pregnant BALB/c mice (10–12 weeks old, 25–30 g weight) were purchased from the Center of Experimental Animals of Baiqiuen Medical College of Jilin University (Jilin, China). The animals were housed in stainless steel cages in a room kept at 24 ± 1°C with a 12 h light/12 h dark cycle and had free access to food and water. Care conditions were adapted to facilitate access of the animals to food and water ad libitum during the experiments. Randomization was used to assign samples to the experimental groups and to collect and process data. The experiments were performed by investigators blinded to the treatment groups.
Experimental design
The mice were randomly divided into six groups (n = 12 per group): control, LPS treatment, 50 mg·kg−1 SB pretreatment followed by LPS treatment (SB + LPS), 100 mg·kg−1 SB pretreatment followed by LPS treatment (SB + LPS), 200 mg·kg−1 SB pretreatment followed by LPS treatment (SB + LPS) and 5 mg·kg−1 dexamethasone pretreatment followed by LPS treatment (DEX + LPS). On day 10 of lactation, dexamethasone or the different concentrations of SB were injected i.p., whereas the control and LPS‐treated groups were similarly injected with an equal volume of sterile saline. One hour after administration of SB, dexamethasone or saline, LPS (0.2 mg·mL−1) was injected into the fourth inguinal mammary gland. Twenty‐four hours after LPS injection, the mice were anaesthetized with ketamine (30 mg·kg−1) and xylazine (6 mg·kg−1) and the mammary glands were collected.
Histopathology
The mammary glands were fixed in 10% buffered formalin in PBS immediately after the mice were killed. Tissues were embedded in paraffin and sliced into 5 μm sections. For the evaluation of histological changes, the tissues were stained with haematoxylin–eosin and observed under a microscope.
Immunofluorescence
To visualize alveolar tight junction permeability, the animal was anaesthetised as above and 3 mg·mL−1 FITC‐conjugated albumin were used to treat the mammary glands as described (Kobayashi et al., 2013). Briefly, the mammary gland was removed and immersed in a solution containing 3 mg·mL−1 FITC‐albumin for 10 min. After treatment of the gland, it was further fixed in 4% paraformaldehyde for 10 min, embedded in optimal cutting temperature compounds and frozen at −80°C. Cryosections (6 μm) were further fixed and treated with DAPI. Images of the stained cryosections were observed using a confocal laser‐scanning microscope.
elisa and myeloperoxidase (MPO) activity assay
The levels of TNF‐α, IL‐6 and IL‐1β in mammary glands were determined using the mouse elisa kits (eBioscience, San Diego, CA, USA) according to the manufacturer's instructions. MPO activity was assessed by elisa kits (Nanjing Jiancheng Bioengineering Institute, China) according to the manufacturer's instructions.
Immunohistochemistry for MPO
For MPO immunohistochemistry of paraffin sections, the mammary glands were fixed with 10% buffered formalin and then embedded in paraffin. The embedded samples were cut into 5 μm sections. The sections were treated with 3% H2O2 for 10 min and incubated with the rabbit IgG against mouse MPO (1:200; RB‐373‐A0; ThermoFisher Scientific) overnight at 4°C after three washes with PBS. The sections were washed three times and exposed to HRP‐conjugated second antibody. All the sections were analysed after diaminobenzidine staining.
Western blotting analysis
After stimulation, mMECs and the mammary tissue samples were obtained, and the whole protein was extracted with a total protein extraction kit (ThermoFisher Scientific) according to the manufacturer's instructions. Briefly, the collected cells were washed with PBS and lysed in cell lysis buffer (Beyotime Biotechnology, Shanghai, China) and then incubated on ice for 30 min. The cell lysates were centrifuged at 12 000 × g for 10 min at 4°C, the protein concentrations were determined using the bicinchoninic acid (BCA) protein assay kit and the supernatants were mixed with one quarter volume of 4 × SDS sample buffer, boiled for 10 min. The mammary gland was homogenized with a lysis buffer with protease and phosphatase inhibitors and then centrifuged at 15 000 × g for 10 min at 4°C. The supernatant was collected, and the total protein concentration was determined using the BCA protein assay kit. Next, each sample (50 μg total protein) was separated by 10% SDS polyacrylamide gels and electrophoretically transferred onto PVDF membranes. The membranes were blocked with 5% non‐fat dried milk proteins in 0.05% Tris‐buffered saline with Tween (TBST) for 2 h at room temperature. The membranes were then incubated with primary antibodies (1:1000 dilution) at 4°C overnight. After washing with TBST, incubation with a 1:5000 dilution of HRP‐conjugated secondary antibodies was performed for 1 h at room temperature. Specific bands were visualized with an ECL detection kit (ThermoFisher Scientific).
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean ± SEM. All data were analysed using GraphPad Prism 5 (GraphPad InStat Software, San Diego, CA, USA). Comparison between groups was made with the one‐way ANOVA followed by Dunnett's test. For all one‐way ANOVAs, post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. Statistical significance was set to P < 0.05.
Materials
Dexamethasone, SB, LPS, trichostatin A (TSA) and albumin‐fluorescein isothiocyanate‐conjugated albumin (FITC albumin) were all purchased from Sigma‐Aldrich (St. Louis, MO, USA). Histone HCK9ac antibody was purchased from Genetex (Irvine, CA, USA). Rabbit polyclonal antibodies against claudin‐1 and claudin‐3 were obtained from Invitrogen/Zymed Laboratories, San Francisco, CA, USA), mouse monoclonal antibodies against occludin (Invitrogen/Zymed Laboratories). Mouse TNF‐α, IL‐6 and IL‐1β elisa kits were obtained from eBioscience. All of the antibodies for Western blots were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). HRP‐conjugated goat anti‐rabbit and goat anti‐mouse antibodies were provided by GE Healthcare 5 (Little Chalfont, Bucks, UK). All other chemicals were of reagent grade.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Effects of SB on LPS‐induced histopathological impairment of mammary gland
Compared with the control group, histopathological examination of LPS‐challenged mammary glands revealed massive recruitment of neutrophils into the alveolar lumen and milk ducts. However, these LPS‐induced pathological changes were significantly attenuated by treatment with SB (50, 100 and 200 mg·kg−1) or dexamethasone (5 mg·kg−1) (Figure 2).
Figure 2.
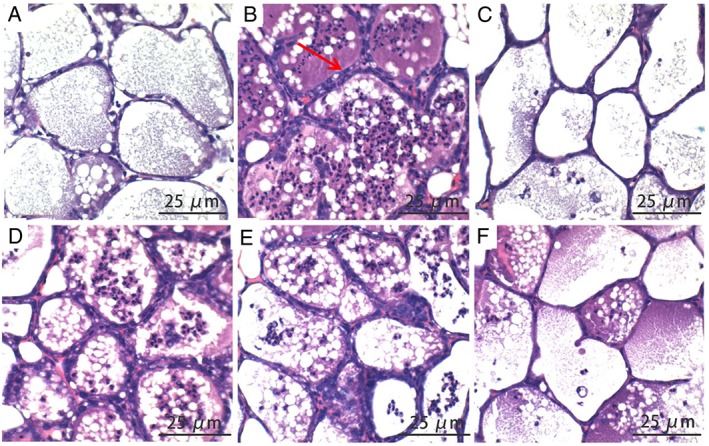
Effects of SB on LPS‐induced histopathological impairment of mammary gland. BALB/c mice were challenged by intramammary injection of 10 μg LPS. Mammary glands were collected 24 h after challenge with LPS. Haematoxylin–eosin staining of formalin‐fixed mammary gland. Histopathological sections of mammary gland. Scale bars: 25 μm. Normal mammary gland (A) and 24 h after LPS injection, showing infiltration of inflammatory cells (B), the LPS + DEX group (C), the LPS + SB (50 mg·kg−1) (D), the LPS + SB (100 mg·kg−1) (E) and the LPS + SB (200 mg·kg−1) (F). The histological morphology and pathology results showed that treatment with SB and dexamethasone alleviated LPS‐induced pathological changes.
Effects of SB on MPO activity
Activation of MPO, which directly reflects neutrophil accumulation, is a functional biomarker of neutrophils. The results of MPO immunohistochemistry and activity assay are shown in Figure 3. LPS challenge significantly increased the activity and distribution of MPO in the mammary gland, compared with the control group. However, treatment with SB (50, 100 and 200 mg·kg−1) or dexamethasone (5 mg·kg−1) significantly decreased the activity and distribution of MPO in mammary gland tissues, challenged with LPS.
Figure 3.
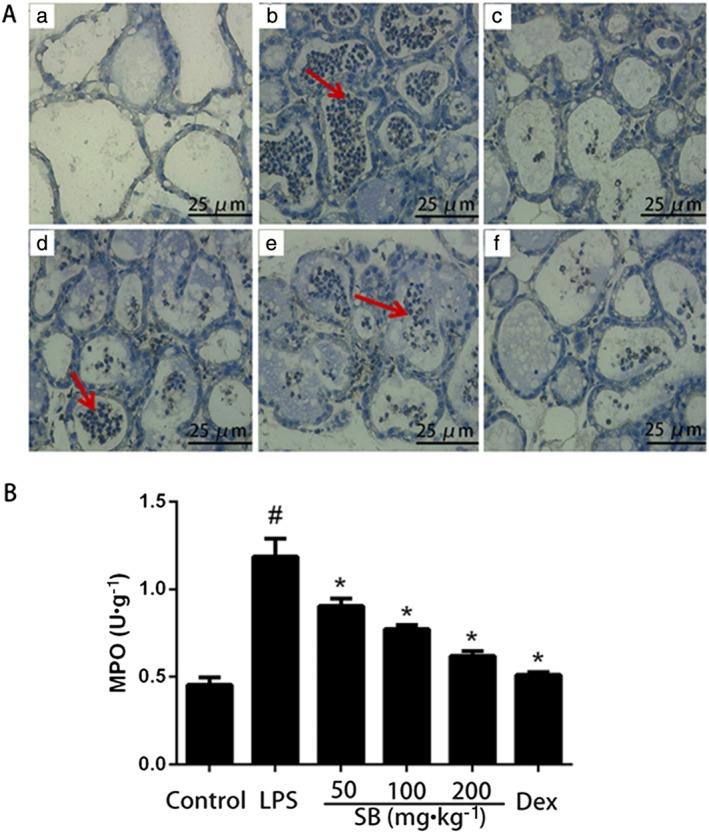
(A) Representative photomicrographs of mammary gland: MPO immunohistochemical staining (×400) in BALB/c mice with LPS treatment. Almost no MPO immunostaining was observed in mammary glands from the control group (a) and the LPS + Dex group (c). The intensity in the LPS group (b) was significantly higher, and there was a gradual decrease in intensity in the LPS + SB (50 mg·kg−1) (d), the LPS + SB (100 mg·kg−1) (e) and the LPS + SB (200 mg·kg−1) (f). Neutrophil accumulation in the mammary gland was assessed by the MPO assay (B). MPO activity in mammary gland BALB/c mice 24 h after intramammary challenge with 10 μg LPS, compared with control group. Data shown are means ± SEM (n = 12). #P<0.05, significantly different from control; *P<0.05, significantly different from LPS alone.
Effects of SB on inflammatory cytokines levels in mammary gland
The levels of pro‐inflammatory cytokines TNF‐α, IL‐1β and IL‐6 were measured by elisa. As shown in Figure 4, the levels of TNF‐α, IL‐6 and IL‐1β in the mammary tissues were significantly increased after LPS administration. SB (50, 100 and 200 mg·kg−1) or dexamethasone (5 mg·kg−1) inhibited the production of TNF‐α and IL‐6, induced by LPS, in a dose‐dependent manner.
Figure 4.
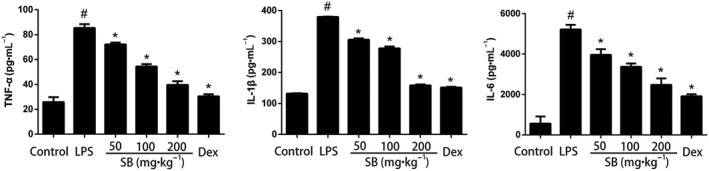
Effects of SB on levels of inflammatory cytokines in mammary gland. Levels of TNF‐α, IL‐1β and IL‐6 were measured by elisa. Data shown are means ± SEM (n = 12). #P<0.05, significantly different from control; *P<0.05, significantly different from LPS alone; one‐way ANOVA followed by Dunnett's test.
Effects of SB on the blood‐milk barrier structure and function in LPS‐induced mastitis model
To evaluate the effects of SB on the blood‐milk barrier, the leakage of FITC‐albumin from the interstitial side into the alveolar lumen was assessed. As shown in Figure 5, before LPS injection, FITC‐albumin was clearly localized on the interstitial side, including the intercellular regions of the neighbouring alveolar epithelial cells. However, 24 h after LPS injection FITC‐albumin was distributed in parts of the mammary alveolar lumen. Treatment of the mice with SB reduced the amount of FITC‐positive reactions in the alveolar lumen.
Figure 5.
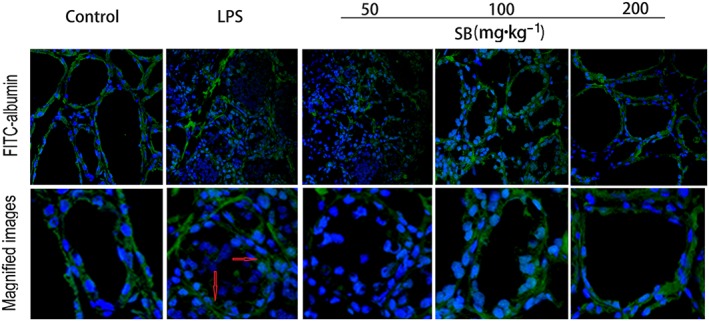
Effects of SB on the permeability of the blood‐milk barrier in LPS‐induced mastitis and the localization of FITC‐albumin was observed. Green and blue show FITC‐albumin and nuclei (DAPI) respectively. Control group, FITC‐albumin can be seen in interstitial side. LPS group, the mammary gland 24 h after LPS injection was treated with FITC‐albumin, FITC‐albumin can be seen in the lumen. Treatment with SB reduced the leakage of FITC‐albumin. Original magnification 400×.
To investigate the protective effects of SB on the LPS‐induced disruption of tight junctions, the levels of marker proteins for tight junctions, such as claudin‐1, claudin‐3 and occludin, were determined by Western blotting. As shown in Figure 6, claudin‐1 was barely detected in the mammary gland before LPS injection and LPS clearly induced expression of claudin‐1. However, SB pretreatment inhibited the expression of claudin‐1. Another tight junction marker, claudin‐3, but not occludin, was down‐regulated in mice treated with LPS alone, compared with the control group. SB pretreatment significantly increased the expression of both of these tight junction markers, suggesting the importance of SB for maintaining the integrity of the junction complex.
Figure 6.
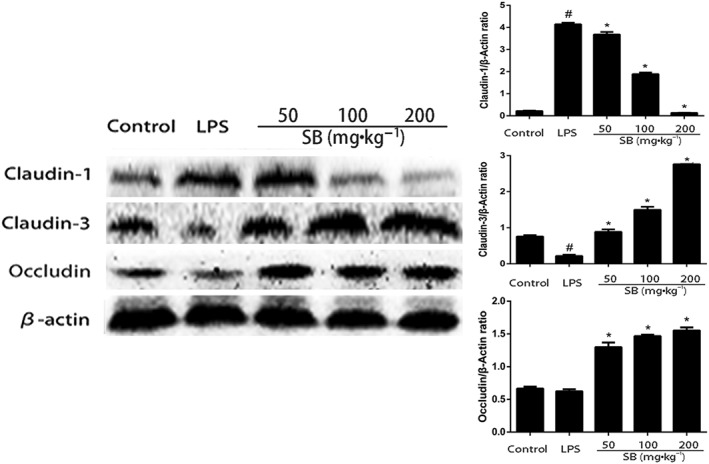
Expression of tight junction proteins was determined by Western blotting. Quantification of tight junction proteins was determined by densitometry and has been normalized to β‐actin. Data shown are means ± SEM (n = 12). #P<0.05, significantly different from control; *P<0.05, significantly different from LPS alone; one‐way ANOVA followed by Dunnett's test.
Effects of SB on cell viability
The potential cytotoxicity of SB on mMECs was analysed by MTT assay. As shown in Figure 7, viability of the mMECs was not affected by SB, over a wide concentration range (0.1‐ 2 mM).
Figure 7.
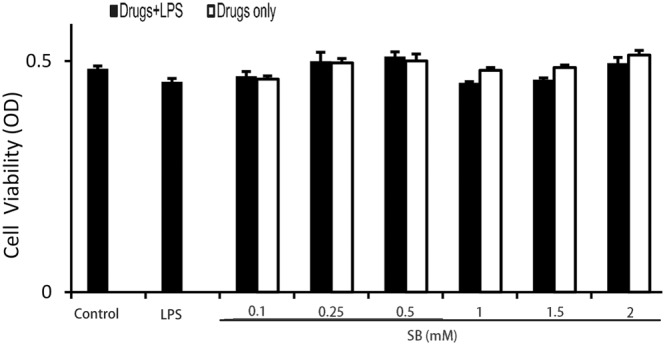
Effects of SB on cell viability. Cells were cultured with different concentrations of SB (0.1, 0.25, 0.5, 1, 1.5 or 2 mM) for 18 h. The cell viability was determined by MTT assay. Data shown are means ± SEM (n = 6).
Effects of SB on cell cytokine production in LPS‐induced mMECs
Previous studies have demonstrated that SB inhibits HDAC activity (Davie, 2003). To determine whether SB behaves as an HDAC inhibitor in mMECs, we treated mMECs with the well‐characterized HDAC inhibitor TSA, in combination with LPS (Figure 8A). The results showed that SB, as well as TSA, suppressed TNF‐α, IL‐6 and IL‐1β expression in LPS‐stimulated mMECs in a dose‐dependent manner (Figure 8B).
Figure 8.
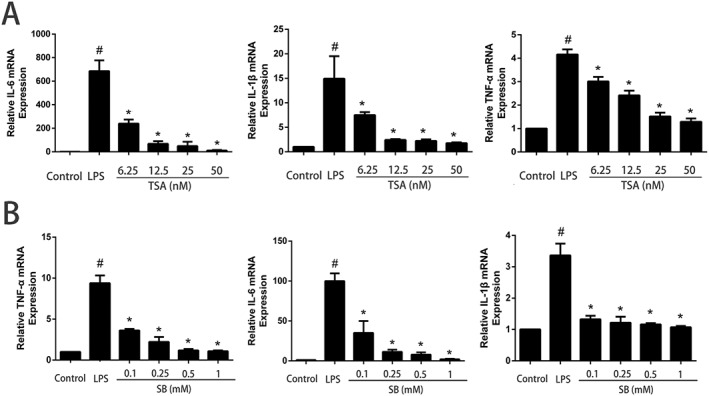
Effects of SB and TSA on cytokine production in LPS‐induced mMECs. Cells were pretreated with SB (0.1, 0.25, 0.5 or 1 mM) or TSA (6.25, 12.5, 25 or 50 nM) for 12 h followed by incubation with 1 μg·mL−1 LPS for 1 h. Total RNA was extracted from mMECs and analysed for mRNA of TNF‐α, IL‐1β, IL‐6 and GAPDH and was measured by qRT‐PCR using specific primers. Data shown are means ± SEM (n = 6). #P<0.05, significantly different from control; *P<0.05, significantly different from LPS alone; one‐way ANOVA followed by Dunnett's test.
Effects of SB on HDACs in LPS‐induced mMECs
To further demonstrate that SB acts as an HDAC inhibitor, we treated mMECs with varying amounts of SB and quantified acetylation of histone H3 by Western blot. As observed with the known HDAC inhibitor TSA, SB also increased histone acetylation in a dose‐dependent manner, suggesting that butyrate behaves as an HDAC inhibitor in mMECs (Figure 9).
Figure 9.
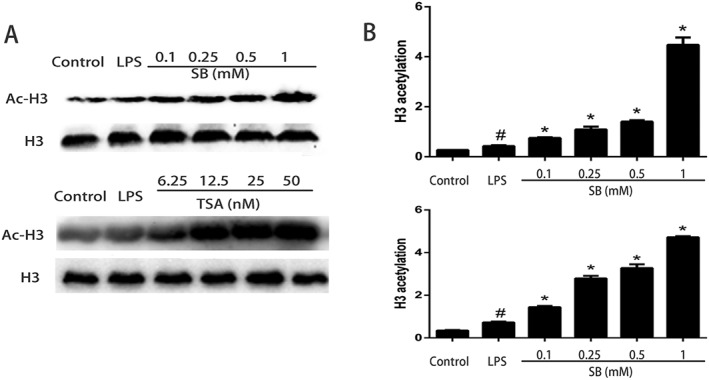
Effects of SB or TSA on histone acetylation in LPS‐induced mMECs. Cells were pretreated with SB (0.1, 0.25, 0.5 and 1 mM) or TSA (6.25, 12.5, 25 and 50 nM) for 12 h followed by incubation with 1 μg·mL−1 LPS for 1 h. (A) Protein samples were probed for acetylated histone H3 (Ac‐H3) by Western blotting with specific antibodies. (B) Quantification of H3 acetylation was determined by densitometry and is normalized to β‐actin. Data shown are means ± SEM (n = 6). #P<0.05, significantly different from control; *P<0.05, significantly different from LPS alone; one‐way ANOVA followed by Dunnett's test.
Effects of SB on the NF‐κB signalling pathway in LPS‐induced mMECs
LPS stimulate NF‐κB signalling pathway with phosphorylation of NF‐κB and IκB. To explore whether NF‐κB is involved in regulation of inflammatory responses by SB, we evaluated the phosphorylation level of NF‐κB and IκB. The results showed that the expression of phosphorylated IκB and p65 increased significantly in the LPS group compared with the control group. Also, pretreatment with various concentrations of SB significantly inhibited the phosphorylation of IκB and NF‐κB P65 in a dose‐dependent manner (Figure 10A). However, IκB is degraded after phosphorylation to facilitate nuclear translocation of NF‐κB. Therefore, we investigated the time‐dependent change of phosphorylated‐IκB in mMECs treated with LPS alone or with LPS with SB pretreatment (Figure 10B). Stimulation of mMECs with LPS primarily resulted in increased phosphorylation of IκB. Pretreatment of mMECs with SB abolished LPS‐induced phosphorylation of IκB. These results indicated that the activation of NF‐κB plays a crucial role in the process of SB moderating the inflammation in our model of mMECs exposed to LPS.
Figure 10.
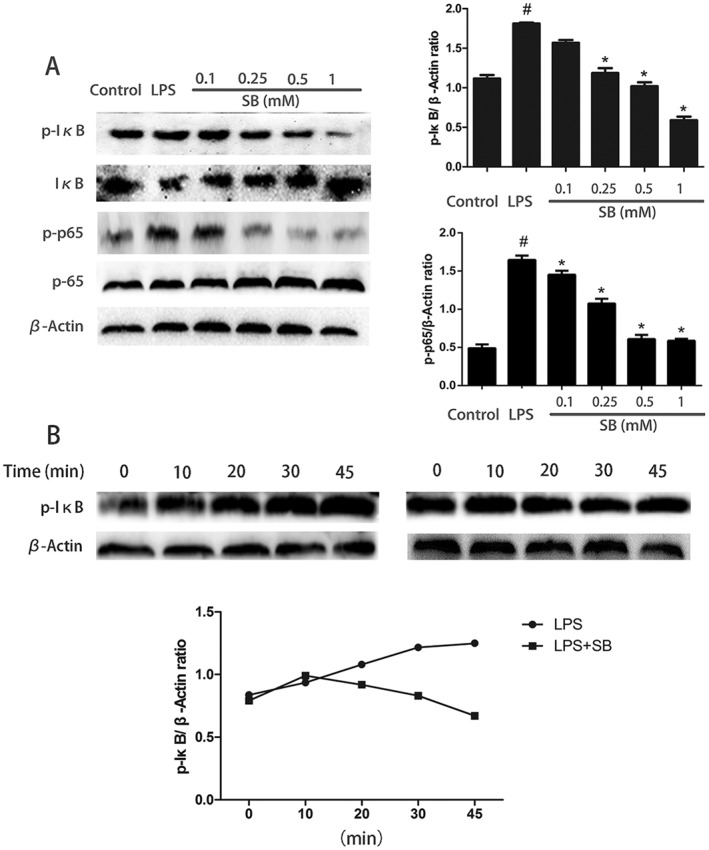
Effects of SB on the NF‐κB signalling pathway in LPS‐induced mMECs. Cells were pretreated with SB (0.1, 0.25, 0.5 or 1 mM) for 12 h followed by incubation with 1 μg·mL−1 LPS for 1 h. Protein samples were analysed by Western blotting with specific antibodies. Quantification of protein samples was determined by densitometry and is normalized to β‐actin (A). In (B), the time‐dependent change of phosphorylated‐IκB in mMECs treated with LPS alone or with LPS plus SB (B). Data shown are means ± SEM (n = 6). #P<0.05, significantly different from control; *P<0.05, significantly different from LPS alone; one‐way ANOVA followed by Dunnett's test.
Discussion
The therapeutic benefits of SB in the treatment of several inflammatory diseases have been previously recognized. Our studies have shown that SB treatment significantly ameliorated mammary tissue injury in LPS‐induced mastitis. To identify the mechanisms of these beneficial effects of SB on LPS‐stimulated model of mastitis, the anti‐inflammatory activity of SB was assessed. Tissue damage during mastitis can initially be caused by bacteria and their products. Mastitis is characterized by an infiltration of immune cells, such as polymorphonuclear neutrophils and macrophages, into the mammary gland (Zhao and Lacasse, 2008). Our finding showed that infiltration of immune cells was markedly reduced with SB treatment. MPO activity directly reflects neutrophils accumulation in the mammary gland and our results indicated that MPO activity was significantly reduced with SB treatment.
Mastitis is a common infectious condition of the mammary gland and LPS, a powerful bacterial virulence factor, is a common trigger of inflammation (Atabai and Matthay, 2002; Maybauer et al., 2006). LPS is widely used to induce mastitis models for the study of this disease (Yang et al., 2014). Mammary alveolar MECs form less permeable tight junctions that prevent the leakage of milk components and interstitial fluid during lactation (Stelwagen et al., 1997; Nguyen and Neville, 1998; Itoh and Bissell, 2003 ). However, the blood‐milk barrier formed by alveolar tight junctions becomes leaky in mastitis (Nguyen and Neville, 1998). In this study, the leakage of FITC‐albumin from the interstitial side into the alveolar lumen was observed, following treatment with LPS. Correspondingly, treatment of the mammary gland with SB caused reduced this LPS‐induced leakage of FITC‐albumin into the alveolar lumen. As recognised, the structure and functional integrity of tight junctions are maintained by the tight junction proteins, claudin and occludin (Furuse et al., 1994; Shen et al., 2006). Among these tight junction proteins, the claudin family is known to be the main determinant of barrier properties (Amasheh et al., 2011). Occludin, which was the first transmembrane protein identified, has critical effects in maintaining intercellular permeability and transepithelial resistance (Mitic and Anderson, 1998). In our experiments, we showed that SB up‐regulated the expression of these tight junction‐associated proteins. The protective effects of SB on the blood‐milk barrier could thus be mediated by maintaining the functional integrity of tight junctions, thereby reducing the severity of inflammation in the mammary gland.
Cytokines, such as TNF‐α, IL‐1β and IL‐6, are known to be involved in host defence against inflammatory diseases (Boudjellab et al., 2000). TNF‐α can amplify the inflammatory cascade via triggering the release of other pro‐inflammatory cytokines and enhancing the activation and accumulation of leukocytes (Hibi et al., 2003). The expression of these cytokines is significantly increased in mouse mammary glands exposed to LPS (Li et al., 2013). Our finding that pretreatment with SB reduced the levels of TNF‐α, IL‐1β and IL‐6 in the mammary gland suggests that SB ameliorated LPS‐induced mastitis via the suppression of pro‐inflammatory mediators. Increasing evidence suggests that pro‐inflammatory cytokines act as an important regulator in inflammation‐related disruption of tight junctions in the epidermis (Kirschner et al., 2009), intestinal epithelium (Schulzke et al., 2009), corneal epithelium (Yi et al., 2000) and blood–brain barrier (Stamatovic et al., 2008). A decrease in cytokine production may facilitate the recovery of epithelial barrier function (Maggini et al., 2007). Thus, the decrease in pro‐inflammatory cytokines, following treatment with SB in LPS‐induced mice, may play a role not only in anti‐inflammatory responses, but also in restoring epithelial barrier function.
The decrease in pro‐inflammatory cytokines gene expression prompted us to examine the effect of SB on NF‐κB activation. NF‐κB is a nuclear transcription factor that plays a major role in the regulation of gene expression involved with many cellular processes (Baeuerle and Baichwal, 1997). Once activated, the NF‐κB subunit p65 separates from IκB and translocates from the cytoplasm to the nucleus (Hoshino et al., 1999). Increasing evidence indicates that the NF‐κB pathway contributes to inflammatory diseases and to the overexpression of pro‐inflammatory cytokines, such as TNF‐α, IL‐6 and IL‐1β (Moynagh, 2005). Butyrate reduced production of pro‐inflammatory cytokines by modulating the activity of NF‐κB (Zapolska‐Downar et al., 2004) and it has been proposed that, through targeting NF‐κB, it may thus be possible to suppress inflammation in the mammary gland. Moreover, some studies (Aveleira et al., 2010; Kamekura et al., 2010; Tang et al., 2010; Choi et al., 2012) have indicated that the activation of the NF‐κB pathway increases tight junction permeability. Therefore, we investigated the mechanism by which SB affected NF‐κB activation in LPS‐exposed mMECs. Our results showed that LPS markedly induced the activation of NF‐κB p65 subunit and the degradation of IκBα, and pretreatment with SB inhibited the phosphorylation of these molecules in a dose‐dependent manner. These results suggest that the anti‐inflammatory effects of SB are related to the down‐regulation of NF‐κB in LPS‐induced mMECs.
It is well established that class I HDACs 1, 2 and 3 play vital roles in the regulation of pro‐inflammatory gene expression in immune cells (Ziesche et al., 2013; Dekker et al., 2014). Modulation of these enzymes using HDAC inhibitors has emerged as a promising treatment not only for cancer (Kelly et al., 2003) but also for asthma, inflammatory diseases, neurodegenerative diseases, rheumatoid arthritis and malaria (Bhavsar et al., 2008; Grabiec et al., 2008; Andrews et al., 2009; Dietz and Casaccia, 2010). Previous studies showed that HDAC inhibitors could inhibit NF‐κB activation. To further investigate the anti‐inflammatory mechanism of SB, we examined the possibility that SB acts as an HDAC inhibitor in LPS‐stimulated mMECs. We found that SB or the known HDAC inhibitor TSA, increased the acetylation of lysine 9 of histone 3 (H3K9). Moreover, both SB and TSA inhibited the amounts of TNF‐α, IL‐1β and IL‐6, suggesting that SB down‐regulated the inflammatory responses through HDAC inhibition.
In conclusion, our study has demonstrated that SB protected against LPS‐induced mastitis by ameliorating blood‐milk barrier disruption and inhibiting inflammatory responses. The anti‐inflammatory mechanism involved inhibition of HDACs, which subsequently inhibited LPS‐induced NF‐κB activation and the production of pro‐inflammatory cytokines. This study provides new insights into the protective actions of SB, which may serve as a potential prophylactic agent to protect the barrier function in mastitis.
Author contributions
J.‐J.W. performed experiments and analysed the data. Y.‐h.F. and Z.‐T.Y. conceived and designed the experiments. Y.‐h.F., Z.‐T.Y. and J.‐J.W. interpreted the data and wrote the paper. The authors sincerely thank Z.‐K.W., X.Z., Y.‐N.W. and Z.‐T.Y. for their support during the study.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (no. 31602122 and 31572583) and China Postdoctoral Science Foundation funded project (2016M600233).
Wang, J.‐J. , Wei, Z.‐K. , Zhang, X. , Wang, Y.‐N. , Fu, Y. , and Yang, Z.‐T. (2017) Butyrate protects against disruption of the blood‐milk barrier and moderates inflammatory responses in a model of mastitis induced by lipopolysaccharide. British Journal of Pharmacology, 174: 3811–3822. doi: 10.1111/bph.13976.
Contributor Information
Yun‐he Fu, Email: fuyunhesky@sina.com.
Zheng‐Tao Yang, Email: yangzhengtao01@sina.com.
References
- Akers RM, Nickerson SC (2011). Mastitis and its impact on structure and function in the ruminant mammary gland. J Mammary Gland Biol Neoplasia 16: 275–289. https://doi.org/10.1007/s10911‐011‐9231‐3. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. https://doi.org/10.1111/bph.13348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. https://doi.org/10.1111/bph.13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amasheh S, Fromm M, Gunzel D (2011). Claudins of intestine and nephron – a correlation of molecular tight junction structure and barrier function. Acta Physiol (Oxford, England) 201: 133–140. https://doi.org/10.1111/j.1748‐1716.2010.02148.x. [DOI] [PubMed] [Google Scholar]
- Andrews KT, Tran TN, Wheatley NC, Fairlie DP (2009). Targeting histone deacetylase inhibitors for anti‐malarial therapy. Curr Top Med Chem 9: 292–308. https://doi.org/10.2174/156802609788085313. [DOI] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P et al (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T‐cell generation. Nature 504: 451–455. https://doi.org/10.1038/nature12726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atabai K, Matthay MA (2002). The pulmonary physician in critical care. 5: acute lung injury and the acute respiratory distress syndrome: definitions and epidemiology. Thorax 57: 452–458. https://doi.org/10.1136/thorax.57.5.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aveleira CA, Lin CM, Abcouwer SF, Ambrosio AF, Antonetti DA (2010). TNF‐alpha signals through PKCzeta/NF‐kappaB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 59: 2872–2882. https://doi.org/10.2337/db09‐1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle PA, Baichwal VR (1997). NF‐kappa B as a frequent target for immunosuppressive and anti‐inflammatory molecules. Adv Immunol 65: 111–137. [PubMed] [Google Scholar]
- Bhavsar P, Ahmad T, Adcock IM (2008). The role of histone deacetylases in asthma and allergic diseases. J Allergy Clin Immunol 121: 580–584. https://doi.org/10.1016/j.jaci.2007.12.1156. [DOI] [PubMed] [Google Scholar]
- Boudjellab N, Chan‐Tang HS, Zhao X (2000). Bovine interleukin‐1 expression by cultured mammary epithelial cells (MAC‐T) and its involvement in the release of MAC‐T derived interleukin‐8. Comp Biochem Physiol A Mol Integr Physiol 127: 191–199. https://doi.org/10.1016/S1095‐6433(00)00257‐9. [DOI] [PubMed] [Google Scholar]
- Burton JL, Erskine RJ (2003). Immunity and mastitis. Some new ideas for an old disease. Vet Clin North Am Food Anim Pract 19: 1–45 v, https://doi.org/10.1016/S0749‐0720(02)00073‐7. [DOI] [PubMed] [Google Scholar]
- Chang PV, Hao L, Offermanns S, Medzhitov R (2014). The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A 111: 2247–2252. https://doi.org/10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HJ, Kim J, Park SH, Do KH, Yang H, Moon Y (2012). Pro‐inflammatory NF‐kappaB and early growth response gene 1 regulate epithelial barrier disruption by food additive carrageenan in human intestinal epithelial cells. Toxicol Lett 211: 289–295. https://doi.org/10.1016/j.toxlet.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. https://doi.org/10.1111/bph.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davie JR (2003). Inhibition of histone deacetylase activity by butyrate. J Nutr 133 (7 Suppl): 2485S–2493S. [DOI] [PubMed] [Google Scholar]
- Dekker FJ, van den Bosch T, Martin NI (2014). Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today 19: 654–660. https://doi.org/10.1016/j.drudis.2013.11.012. [DOI] [PubMed] [Google Scholar]
- Dietz KC, Casaccia P (2010). HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol Res 62: 11–17. https://doi.org/10.1016/j.phrs.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelblum KL, Turner JR (2009). The tight junction in inflammatory disease: communication breakdown. Curr Opin Pharmacol 9: 715–720. https://doi.org/10.1016/j.coph.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillon S, Robinson ZD, Colgan SP, Furuta GT (2009). Epithelial function in eosinophilic gastrointestinal diseases. Immunol Allergy Clin North Am 29: 171–178 xii‐xiii, https://doi.org/10.1016/j.iac.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Furuse M, Itoh M, Hirase T, Nagafuchi A, Yonemura S, Tsukita S et al (1994). Direct association of occludin with ZO‐1 and its possible involvement in the localization of occludin at tight junctions. J Cell Biol 127 (6 Pt 1): 1617–1626. https://doi.org/10.1083/jcb.127.6.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabiec AM, Tak PP, Reedquist KA (2008). Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther 10: 226 https://doi.org/10.1186/ar2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi T, Inoue N, Ogata H, Naganuma M (2003). Introduction and overview: recent advances in the immunotherapy of inflammatory bowel disease. J Gastroenterol 38 (Suppl 15): 36–42. [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y et al (1999). Cutting edge: Toll‐like receptor 4 (TLR4)‐deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol (Baltimore, Md. : 1950) 162: 3749–3752. [PubMed] [Google Scholar]
- Itoh M, Bissell MJ (2003). The organization of tight junctions in epithelia: implications for mammary gland biology and breast tumorigenesis. J Mammary Gland Biol Neoplasia 8: 449–462. https://doi.org/10.1023/B:JOMG.0000017431.45314.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamekura R, Kojima T, Takashima A, Koizumi J, Ogasawara N, Go M et al (2010). Thymic stromal lymphopoietin induces tight junction protein claudin‐7 via NF‐kappaB in dendritic cells. Histochem Cell Biol 133: 339–348. https://doi.org/10.1007/s00418‐009‐0674‐1. [DOI] [PubMed] [Google Scholar]
- Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor‐Curtelli B, Tong W et al (2003). Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res 9 (10 Pt 1): 3578–3588. [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. https://doi.org/10.1111/j.1476‐5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner N, Poetzl C, von den Driesch P, Wladykowski E, Moll I, Behne MJ et al (2009). Alteration of tight junction proteins is an early event in psoriasis: putative involvement of proinflammatory cytokines. Am J Pathol 175: 1095–1106. https://doi.org/10.2353/ajpath.2009.080973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Oyama S, Numata A, Rahman MM, Kumura H (2013). Lipopolysaccharide disrupts the milk‐blood barrier by modulating claudins in mammary alveolar tight junctions. PLoS One 8: e62187 https://doi.org/10.1371/journal.pone.0062187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Nusrat A (2009). Dynamic regulation of epithelial cell fate and barrier function by intercellular junctions. Ann N Y Acad Sci 1165: 220–227. [DOI] [PubMed] [Google Scholar]
- Li D, Zhang N, Cao Y, Zhang W, Su G, Sun Y et al (2013). Emodin ameliorates lipopolysaccharide‐induced mastitis in mice by inhibiting activation of NF‐kappaB and MAPKs signal pathways. Eur J Pharmacol 705: 79–85. https://doi.org/10.1016/j.ejphar.2013.02.021. [DOI] [PubMed] [Google Scholar]
- Machado RA, Constantino Lde S, Tomasi CD, Rojas HA, Vuolo FS, Vitto MF et al (2012). Sodium butyrate decreases the activation of NF‐kappaB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast‐induced nephropathy. Nephrol Dial Transplant 27: 3136–3140. https://doi.org/10.1093/ndt/gfr807. [DOI] [PubMed] [Google Scholar]
- Maggini S, Wintergerst ES, Beveridge S, Hornig DH (2007). Selected vitamins and trace elements support immune function by strengthening epithelial barriers and cellular and humoral immune responses. Br J Nutr 98 (Suppl 1): S29–S35. [DOI] [PubMed] [Google Scholar]
- Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D et al (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461: 1282–1286. https://doi.org/10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maybauer MO, Maybauer DM, Herndon DN (2006). Incidence and outcomes of acute lung injury. N Engl J Med 354: 416–417 author reply 416–417. [PubMed] [Google Scholar]
- Mazmanian SK, Round JL, Kasper DL (2008). A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453: 620–625. https://doi.org/10.1038/nature07008. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. https://doi.org/10.1111/bph.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitic LL, Anderson JM (1998). Molecular architecture of tight junctions. Annu Rev Physiol 60: 121–142. https://doi.org/10.1146/annurev.physiol.60.1.121. [DOI] [PubMed] [Google Scholar]
- Moynagh PN (2005). The NF‐κB pathway. J Cell Sci 118: 4589–4592. https://doi.org/10.1242/jcs.02579. [DOI] [PubMed] [Google Scholar]
- Nguyen DA, Neville MC (1998). Tight junction regulation in the mammary gland. J Mammary Gland Biol Neoplasia 3: 233–246. https://doi.org/10.1023/A:1018707309361. [DOI] [PubMed] [Google Scholar]
- Ramakrishna BS (2009). Probiotic‐induced changes in the intestinal epithelium: implications in gastrointestinal disease. Trop Gastroenterol 30: 76–85. [PubMed] [Google Scholar]
- Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H et al (2009). Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci 1165: 294–300. [DOI] [PubMed] [Google Scholar]
- Shen L, Black ED, Witkowski ED, Lencer WI, Guerriero V, Schneeberger EE et al (2006). Myosin light chain phosphorylation regulates barrier function by remodeling tight junction structure. J Cell Sci 119 (Pt 10): 2095–2106. https://doi.org/10.1242/jcs.02915. [DOI] [PubMed] [Google Scholar]
- Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H et al (2014). Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40: 128–139. https://doi.org/10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM et al (2013). The microbial metabolites, short‐chain fatty acids, regulate colonic Treg cell homeostasis. Science (New York, N.Y.) 341: 569–573. https://doi.org/10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44 (D1): D1054–D1068. https://doi.org/10.1093/nar/gkv1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Andjelkovic AV (2008). Brain endothelial cell–cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol 6: 179–192. https://doi.org/10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelwagen K, Farr VC, McFadden HA, Prosser CG, Davis SR (1997). Time course of milk accumulation‐induced opening of mammary tight junctions, and blood clearance of milk components. Am J Physiol 273 (1 Pt 2): R379–R386. [DOI] [PubMed] [Google Scholar]
- Tang Y, Clayburgh DR, Mittal N, Goretsky T, Dirisina R, Zhang Z et al (2010). Epithelial NF‐kappaB enhances transmucosal fluid movement by altering tight junction protein composition after T cell activation. Am J Pathol 176: 158–167. https://doi.org/10.2353/ajpath.2010.090548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Furuse M (2000). The structure and function of claudins, cell adhesion molecules at tight junctions. Ann N Y Acad Sci 915: 129–135. [DOI] [PubMed] [Google Scholar]
- Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC et al (2008). Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455: 1109–1113. https://doi.org/10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JM, de Souza R, Kendall CW, Emam A, Jenkins DJ (2006). Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40: 235–243. https://doi.org/10.1097/00004836‐200603000‐00015. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yin R, Cong Y, Yang Z, Zhou E, Wei Z et al (2014). Oxymatrine lightened the inflammatory response of LPS‐induced mastitis in mice through affecting NF‐kappaB and MAPKs signaling pathways. Inflammation 37: 2047–2055. https://doi.org/10.1007/s10753‐014‐9937‐7. [DOI] [PubMed] [Google Scholar]
- Yi X, Wang Y, Yu FS (2000). Corneal epithelial tight junctions and their response to lipopolysaccharide challenge. Invest Ophthalmol Vis Sci 41: 4093–4100. [PubMed] [Google Scholar]
- Yin L, Laevsky G, Giardina C (2001). Butyrate suppression of colonocyte NF‐kappa B activation and cellular proteasome activity. J Biol Chem 276: 44641–44646. https://doi.org/10.1074/jbc.M105170200. [DOI] [PubMed] [Google Scholar]
- Zapolska‐Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M (2004). Butyrate inhibits cytokine‐induced VCAM‐1 and ICAM‐1 expression in cultured endothelial cells: the role of NF‐kappaB and PPARalpha. J Nutr Biochem 15: 220–228. https://doi.org/10.1016/j.jnutbio.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Zhang C, Zhai S, Wu L, Bai Y, Jia J, Zhang Y et al (2015). Induction of size‐dependent breakdown of blood‐milk barrier in lactating mice by TiO2 nanoparticles. PLoS One 10: e0122591 https://doi.org/10.1371/journal.pone.0122591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Lacasse P (2008). Mammary tissue damage during bovine mastitis: causes and control. J Anim Sci 86 (13 Suppl): 57–65. https://doi.org/10.2527/jas.2007‐0302. [DOI] [PubMed] [Google Scholar]
- Ziesche E, Kettner‐Buhrow D, Weber A, Wittwer T, Jurida L, Soelch J et al (2013). The coactivator role of histone deacetylase 3 in IL‐1‐signaling involves deacetylation of p65 NF‐kappaB. Nucleic Acids Res 41: 90–109. [DOI] [PMC free article] [PubMed] [Google Scholar]