

Abstract
Background and Purpose
Effective treatments of nausea are limited. In this study we evaluated the ability of the peripherally restricted fatty acid amide hydrolase (FAAH) inhibitor, URB937, to suppress acute and anticipatory nausea in rats and examined the pharmacological mechanism of this effect.
Experimental Approach
We investigated the potential of URB937 (administered i.p.) to reduce the establishment of lithium chloride‐induced conditioned gaping (model of acute nausea) and to reduce the expression of contextually‐elicited conditioned gaping (model of anticipatory nausea) in rats. The role of CB1 receptors, CB2 receptors and PPARα in the anti‐nausea effect of URB937 was examined. The potential of URB937 to suppress FAAH activity in tissue collected from the area postrema (AP), prefrontal cortex (PFC), liver and duodenum and to elevate levels of FAAH substrates – anandamide (AEA), N‐oleoylethanolamide (OEO) and N‐palmitoylethanolamide (PEA) – in the AP was also evaluated.
Key Results
URB937 reduced acute nausea by a PPARα‐dependent mechanism and reduced anticipatory nausea by a CB1 receptor‐dependent mechanism. The PPARα agonist, GW7647, similarly attenuated acute nausea. URB937 reduced FAAH activity in the liver and the duodenum but not in the PFC. In addition, URB937 reduced FAAH activity and elevated levels of fatty‐acid ethanolamides in the AP, a brain region that is not protected by the blood–brain barrier.
Conclusions and Implications
The anti‐nausea action of URB937 may occur in the AP and may involve PPARα to suppress acute nausea and CB1 receptors to suppress anticipatory nausea.
Abbreviations
- 2‐AG
2‐arachidonoyl‐sn‐glycerol
- AEA
anandamide
- AP
area postrema
- CB1
cannabinoid 1
- CB2
cannabinoid 2
- CTA
conditioned taste aversion
- FAAH
fatty acid amide hydrolase
- FAEs
fatty‐acid ethanolamides
- IIC
interoceptive insular cortex
- LiCl
lithium chloride
- MAGL
monoacylglycerol lipase
- OEA
oleoylethanolamide
- PEA
palmitoylethanolamide
- PFC
prefrontal cortex
- URB937
cyclohexylcarbamic acid 3′‐carbamoyl‐6‐hydroxybiphenyl‐3‐yl ester
Introduction
Nausea and vomiting are distressing symptoms associated with several disorders such as side effects of chemotherapy in cancer treatment, chronic gastrointestinal disorders, gastroparesis and cyclic vomiting syndrome. Current anti‐emetic therapies are highly effective in reducing vomiting but only weak in reducing nausea (Roscoe et al., 2000; Hickok et al., 2003). Because nausea is so poorly understood, effective treatments are limited, highlighting the need to understand the mechanisms of nausea to develop new therapeutics.
Rats do not vomit, but they do display conditioned gaping reactions to a flavour that has been previously paired with ‘sickness’ (Grill and Norgren, 1978). Considerable behavioural evidence confirms that only manipulations that produce nausea and vomiting in other species promote conditioned gaping behaviours in rats, although even non‐emetic treatments produce taste avoidance in rats. Furthermore, treatments that reduce nausea and vomiting in other species consistently prevent conditioned gaping behaviours in rats but not taste avoidance. Therefore, conditioned gaping in rats is a much more selective measure of nausea than conditioned taste avoidance (Parker, 2014). Conditioned gaping in rats requires similar orofacial musculature as vomiting in emetic species (Travers and Norgren, 1986) and is topographically similar to the orofacial components of retching in the shrew (Parker, 2003). These conditioned gaping reactions are not only displayed to nausea‐paired flavours, but they are also displayed to nausea‐paired contextual cues, serving as a model of anticipatory nausea experienced by patients receiving chemotherapy treatment upon their return to the clinic (Limebeer et al., 2008; Rock et al., 2014).
Abundant evidence indicates that manipulations that enhance activity of the endocannabinoid system interfere with both acute and anticipatory nausea in these rat gaping models (Sticht et al., 2015). The endocannabinoid system consists of the cannabinoid receptors (CB1 and CB2), the natural ligands for those receptors, anandamide (AEA) (Devane et al., 1992) and 2‐arachidonoyl‐sn‐glycerol (2‐AG) (Mechoulam et al., 1995), and their degrading enzymes. AEA and other fatty‐acid ethanolamides (FAEs), including oleoylethanolamide (OEA) and palmitoylethanolamide (PEA), are rapidly degraded by fatty acid amide hydrolase (FAAH) (Deutsch and Chin, 1993); however, unlike AEA, OEA and PEA are agonists at the PPARα (Fu et al., 2003, 2012), not CB1 or CB2 receptors. 2‐AG is rapidly degraded by monoacylglycerol lipase (MAGL) (Dinh et al., 2002). Both FAAH and MAGL are distributed throughout the brain and periphery. The action of AEA, OEA and PEA can be prolonged by up to 24 h by pharmacological inhibition of their degradation by FAAH, and the action of 2‐AG can be prolonged by up to 24 h by MAGL inhibition (Cravatt et al., 1996), providing effective strategies for reducing acute and anticipatory nausea as assessed by the rat gaping models (Rock et al., 2014; Sticht et al., 2015).
Sticht et al. (2016) demonstrated that elevation of 2‐AG by MAGL inhibition in the interoceptive insular cortex (IIC), a cortical site responsible for the experience of nausea (Penfield and Faulk, 1955; Napadow et al., 2013), reduces nausea‐induced conditioned gaping in rats. However, the site of action of the anti‐nausea effects of FAAH inhibition remains unknown because FAAH inhibition in the IIC neither reduced nausea‐induced conditioned gaping nor elevated AEA (Sticht et al., 2016), but systemic administration of the FAAH inhibitor, PF3845 (Ahn et al., 2009), suppressed acute nausea (Rock et al., 2015), and both PF3845 and URB597 reduced anticipatory nausea (Rock et al., 2008, 2015). Therefore, it is possible that the anti‐nausea action of FAAH inhibition is peripherally mediated. We report new data here using the highly selective, peripherally restricted FAAH inhibitor, URB937 (Clapper et al., 2010).
URB937 suppresses FAAH and enhances AEA levels outside of the CNS and has the ability to suppress behavioural responses in rodent models of peripheral nerve injury and inflammation through a CB1 receptor mediated effect (Clapper et al., 2010). URB937 and the brain penetrant, URB597, have comparable potencies in membrane preparations of rat brain FAAH and are equally effective at blocking liver FAAH activity when administered systemically in mice (1 mg·kg−1, i.p.). The compounds markedly differ, however, in their ability to access the CNS, as URB937 suppresses FAAH activity in peripheral tissues but not in the brain (Clapper et al., 2010). Therefore, this compound provides a tool to determine the potential of peripheral FAAH inhibition on nausea‐induced behaviours. Here, we evaluate the ability of URB937 to interfere with acute and anticipatory nausea in the rat gaping models. Since FAAH inhibition elevates AEA, but also PEA and OEA, which act on PPARα receptors rather than CB1 or CB2 receptors, we assessed the role of these receptors in the anti‐nausea effects of URB937. The potential of the PPARα agonist, GW7647, to reduce acute nausea was also evaluated. Finally, the ability of URB937 to suppress FAAH activity in the liver, duodenum, prefrontal cortex (PFC) and area postrema (AP; an area of weak blood–brain barrier to allow toxins to produce the vomiting reflex in humans and other animals) and to elevate levels of FAEs in the AP was evaluated.
Methods
As conditioned gaping in rats is a selective measure of nausea (Parker, 2014), we used this as a preclinical model to evaluate whether compounds reduce lithium chloride (LiCl)‐induced conditioned gaping in Sprague Dawley rats.
Animals
All procedures with animals complied with the legislation of the Animals for Research Act of Ontario, as well as the guidelines of the Canadian Council on Animal Care (CCAC). All animal use protocols were approved by the Institutional Animal Care Committee at the University of Guelph, which is accredited by the CCAC. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Male Sprague Dawley rats (180), obtained from Charles River Laboratories (St Constant, QC, Canada), were used for assessment of acute nausea and anticipatory nausea and endocannabinoid/FAAH analysis. Their body weights ranged from 244 to 314 g on the day of conditioning for acute nausea and from 308 to 398 g on the day of testing for anticipatory nausea (approximately 8 weeks in age). Rats were housed in a specific pathogen‐free facility in opaque plastic shoebox cages (48 × 26 × 20 cm), containing bed‐o‐cob bedding from Harlan Laboratories, Inc. (Mississauga, ON, Canada), a brown paper towel and Crink‐l'Nest™ from The Andersons, Inc. (Maumee, OH, USA). Additionally, the rats were provided with a soft white paper container that was 14 cm long and 12 cm in diameter. Rats were pair‐housed for anticipatory nausea or individually housed for assessment of acute nausea (to prevent cage mates from chewing cannulae). The colony room was maintained at an ambient temperature of 21oC and a 12/12 h reverse light–dark schedule (lights off at 07 h) and maintained on ad libitum chow and water. All experimental manipulations occurred during the dark phase cycle.
Experimental procedures
In vivo procedures
Experiment 1: effects of URB937 on acute nausea and its mechanism of action
All rats were surgically implanted with an intraoral cannula under isofluorane anaesthesia according to the procedure described by Limebeer et al. (2010). Animals were anaesthetized with isoflurane gas and administered the antibiotic depocillin (0.33 mL·kg−1, s.c.) and the non‐steroidal anti‐inflammatory/analgesic drug carprofen (5 mg·kg−1, i.p.), as well as a topical anaesthetic (50/50% marcaine/lidocaine; Hospira, Montreal, QC, Canada) at the surgical site (0.1 mL, s.c.). A 15‐G stainless steel needle was inserted at the mid‐area on the back of the neck, guided s.c. below the ear and across the cheek until it exited into the oral cavity behind the first molar. A 10 cm long section of polyethylene tubing (PE 90, I.D. 0.86 mm, O.D. 1.27 mm) was inserted into the needle. Once inserted, the needle was removed, leaving the tubing in place. A circular section of surgical mesh along with three elastic squares (8 × 8 mm) was threaded onto the tubing and drawn down to the neck to secure the cannula. Within the mouth, the cannula was held in place by a flanged end of the tubing over a circular section of surgical mesh that rested flush against the inside of the mouth. Twenty‐four hours after surgery, rats were administered a second dose of carprofen (5 mg·kg−1) and monitored for 3 days following surgery. During this time, rats were weighed and the health of the animal was assessed; a visual check for urine/faeces in the home cage, activity, vocalization, dehydration, rigidity and presence of porphyrin staining around the eye was performed, as well as adjustment of the elastics and visual inspection of the surgical site. The cannulae were flushed daily, for 3 days, with chlorhexidine antiseptic.
Following recovery from surgery, the rats received an adaptation trial in which they were placed in the taste reactivity chamber with their cannula attached to an infusion pump (Model KDS100, KD Scientific, Hollliston, MA, USA) for fluid delivery. Water was infused into their intraoral cannuale for 2 min at a rate of 1 mL·min−1.
On the day following the adaptation trial, the rats received a conditioning trial in which they were administered a pretreatment injection of vehicle (VEH) or URB937 (0.3, 1 or 3 mg·kg−1) 2 h prior to placement in the chamber. To assess the CB1 receptor, CB2 receptor or PPARα mediation of the effects of URB937, additional groups also received a pretreatment injection of rimonabant (1 mg·kg−1), AM‐630 (1 mg·kg−1) or MK‐886 (1 mg·kg−1) 30 min prior to placement in the chamber. On the conditioning trial, the rats were randomly assigned to one of 10 pretreatment groups (n = 8 per group): VEH‐VEH; 0.3 mg·kg−1 URB937‐VEH; 1 mg·kg−1 URB937‐VEH; 3.0 mg·kg−1 URB937‐VEH; VEH–1 mg·kg−1 rimonabant; 3 mg·kg−1 URB937–1 mg·kg−1 rimonabant; VEH–1 mg·kg−1 AM‐630; 3 mg·kg−1 URB937–1 mg·kg−1 AM‐630; VEH–1 mg·kg−1 MK‐886; 3 mg·kg−1 URB937–1 mg·kg−1 MK‐886. One rat in group VEH–1 mg·kg−1 AM‐630 was removed from the study (preventing behavioural testing from occurring) and was killed by carbon dioxide due to loss of the intra‐oral cannula. Four additional rats were added to group 3 mg·kg−1 URB937–1 mg·kg−1 AM‐630 because preliminary LSD post hoc comparisons suggested a trend towards significance. Additional animals were added to this group to explore the effect (however, the additional animals did not modify the results of the statistical analysis).
During conditioning, the rats were individually placed in the chamber and intraorally infused with 0.1% saccharin solution for 2 min at a rate of 1 mL·min−1 while the orofacial responses were video recorded from the mirror beneath the chamber with the feed sent to a computer via fire‐wire connection. Immediately after the saccharin infusion, all rats were injected with 20 mL·kg−1 of 0.15 M LiCl and returned to their home cage.
Seventy‐two hours later, the rats were tested drug‐free. They were again intraorally infused with 0.1% saccharin solution for 2 min at the rate of 1 mL·min−1 while the orofacial reactions were video recorded. The videotapes were later scored by an observer blind to the experimental conditions using The Observer for the behaviour of gaping (large openings of the mouth and jaw, with lower incisors exposed).
To determine if the pretreatment interfered with learning per se, conditioned taste aversion (CTA) was assessed in a single bottle test. Rats were water restricted at 15:00 h following their test session. The next morning, a bottle containing 0.1% saccharin solution was placed on the cage at 08:00 h. Measures of saccharin consumption were taken for the next 6 h.
Experiment 2: effect of PPARα agonist, GW7647, on acute nausea
The rats were adapted and conditioned as in Experiment 1, with the exception of the pretreatment condition. In Experiment 2, the rats were injected with VEH or GW7647 (3 mg·kg−1) 30 min prior to the infusion of 0.1 % saccharin solution. Immediately following the infusion, the rats were injected with equivolume saline (SAL) or LiCl. The rats received a conditioned taste avoidance test on the next day. The groups were (n = 8 per group) as follows: VEH‐SAL; VEH‐LiCl; GW7647‐SAL; GW7647‐LiCl.
Experiment 3: effect of URB937 on anticipatory nausea, its mechanism of action and effect on locomotor activity
All rats received four conditioning trials, with 72 h between trials. On each conditioning trial, each rat was injected with LiCl and immediately placed in the distinctive conditioning context for 30 min. The test trial occurred 72 h after the final conditioning trial. On the test trial, the rats received a pretreatment injection of VEH or URB937 2 h prior to placement in the conditioning chamber. To assess the involvement of CB1, CB2 receptors or PPARα in the effects, additional groups also received a pretreatment injection of rimonabant (1 mg·kg−1), AM‐630 (1 mg·kg−1) or MK‐866 (1 mg·kg−1) 30 min prior to placement in the chamber. The rats all then received an i.p. injection of saline (20 mL·kg−1) and were placed in the conditioning chamber for 5 min while their orofacial reactions were videotaped from the mirror beneath the chamber; the videotapes were later scored for the number of gapes. Immediately following the test trial, the rats were placed in the novel activity chamber for 15 min and their locomotor activity was automatically videotracked using EthoVision. Prior to the test trial, the rats were randomly assigned to one of the five pretreatment groups, with n = 8 per group: VEH‐VEH; 3 mg·kg−1 URB937‐VEH; 3 mg·kg−1 URB937–1 mg·kg−1 Rim; 3 mg·kg−1 URB937–1 mg·kg−1 AM‐630; 3 mg·kg−1 URB937–1 mg·kg−1 MK‐886.
Apparatus
The taste reactivity chambers used for acute nausea experiments were made of clear Plexiglas (22.5 × 26 × 20 cm) that sat on a table with a clear glass top. A mirror beneath the chamber at a 45o angle facilitated viewing of the ventral surface of the rat to observe orofacial responses. A Sony videocamera (Handycam, Henry's Camera Waterloo, ON, Canada) was used to videotape the rats from the mirror beneath the chamber. The videotapes were later scored using ‘The Observer’ event recording software (Noldus Information Technology Inc, Leesburg, VA, USA).
The anticipatory nausea context was made of black Plexiglas sides (22.5 × 26 × 20 cm) with an opaque lid. The chamber was placed on a table with a clear Plexiglas top and a mirror on a 45o angle below. The room was dark with two 50 W lights on either side of the conditioning chamber. The behaviour of the rat during the anticipatory nausea test was recorded from the mirror beneath the chamber with a Sony Handycam videocamera.
Immediately following the test for anticipatory nausea, the rats received a test for locomotor activity. The activity chamber was constructed of white Plexiglas with the dimensions of 60 × 25 × 25 cm and located in a different room than the anticipatory nausea chamber.
The room was illuminated with a red light. A video camera mounted on an extension pole captured the activity of the rat, which was sent to a computer for analysis of distance (cm) travelled using the Ethovision software programme (Noldus Information technology Inc, Leesburg, VA, USA).
Ex vivo procedures
Drug‐naïve rats were killed 120 min after receiving an injection of either vehicle or URB937 (3 mg·kg−1, i.p.). Rats were killed by rapid decapitation (restrained in a decapicone; Braintree Scientific, MA, USA), and their brains and peripheral tissue (liver and duodenum) were immediately extracted. To obtain the rat tissue used for neurochemical analysis, it was necessary to kill these rats by decapitation (rather than by inhalation of carbon dioxide or cervical dislocation), as inhalation of carbon dioxide leads to altered neurotransmitters in the brain (EFSA, 2005), and cervical dislocation may affect neuropeptide levels and brain histology (EFSA, 2005). Indeed, Hawkins et al. (2016) state that the Canadian Council on Animal Care agrees that decapitation is acceptable when no other method of killing the animals is possible. The prefrontal cortex (PFC) and area postrema (AP) were subsequently dissected on ice, rapidly frozen over dry ice and were stored at −80°C until the time of processing, which was performed at the University of California, Irvine.
Quantification of FAEs and 2‐AG by LC/MS
Frozen AP samples were weighed (~20 mg) and homogenized in methanol (1 mL) containing AEA‐d4 (10 pmol), OEA‐d4 (200 pmol), PEA‐d4 (200 pmol) and 2‐AG‐d5 (500 pmol) as internal standards. Homogenates were extracted with chloroform (2 vol) and washed with water (1 vol). Organic phases were collected, dried under nitrogen and fractionated by open‐bed silica gel column chromatography. Briefly, the extract was dissolved in chloroform and loaded onto small glass columns packed with Silica Gel G (60A 230–400 Mesh ASTM; Whatman, Clifton, NJ, USA). AEA and PEA were eluted with chloroform/methanol (9:1 w.v‐1). Organic phases were evaporated under nitrogen and reconstituted in 100 μL of methanol. LC/MS analyses were performed on an Agilent 1200 LC system coupled to an Agilent G6410A triple quadrupole (QQQ) MS detector (Agilent Technologies, Inc., Santa Clara, CA, USA) equipped with an electrospray ionization interface. FAEs were separated using a XDB Eclipse C18 column (2.1 × 50 mm i.d., 1.8 μm), eluted with an isocratic method of methanol in water (A: 20% water +0.25% acetic acid +5 mM ammonium acetate; and B: 80% methanol +0.25% acetic acid +5 mM ammonium acetate in 8 min) at a flow rate of 0.4 mL·min−1. Column temperature was kept at 40°C. MS detection was in the positive mode, capillary voltage is set at 4 kV, fragmentor voltage is varied from 120 to 140 V and collision energy was 20 eV. Helium was used as collision gas while nitrogen was used as drying gas at a flow rate of 12 L·min−1 at 350°C. Nebulizer pressure was set at 50 PSI (82.7 kPa). We quantified FAEs with an isotope‐dilution method monitoring proton adducts of the molecular ions [M + H]+ in multiple reaction‐monitoring (MRM) mode in the positive ion mode. The MRM transitions monitored for FAEs detection and quantification are the following: PEA 300.3➔62.1; OEA 326.3➔62.1; AEA 348.3➔62.1; 2AG 379.3➔287.2; PEA‐d4 304.3➔66.1; OEA‐d4 330.3➔66.1; AEA‐d4 352.3➔66.1; 2AG‐d5 384.3➔287.2. We prepared standard calibration curves by adding a constant amount of deuterium‐labelled standards to increasing amounts of the corresponding unlabelled FAEs, followed by MS analysis as described above. The relative concentrations of unlabelled versus labelled ions were plotted against their relative response (i.e. peak area), and the calibration curves were constructed using linear regression. R 2 was 0.998 for all analytes, indicating a linear response. The concentrations of the internal deuterated standards were as follows: PEA‐d4 was kept at 200 nM, OEA‐d4 was 200 nM, AEA‐d4 was 10 nM and 2AG‐d5 was 500 nM. The non‐deuterated reference standards were at concentrations ranging from 1 to 2 μM, for a total of 11 points with a serial dilution of 1:2:2:2.5:2:2:2.5:2:2:2.5:2.
Determination of FAAH activity
Tissues were weighed, homogenized in ice‐cold Tris–HCl buffer (50 mM, 5–9 vol, pH 7.5) containing 0.32 M sucrose and centrifuged at 1000× g for 10 min at 4°C. Supernatants were collected and protein concentrations determined using a bicinchoninic acid assay kit (Pierce, Rockford, IL, USA). FAAH activity was measured at 37°C for 30 min in 0.5 mL of Tris–HCl buffer (50 mM, pH 7.5) containing fatty acid‐free BSA (0.05%, w.v‐1), tissue homogenates (50 mg protein from PFC, AP and duodenum and 10 mg from liver), 10 mM AEA and AEA‐(ethanolamine‐3H) (10 000 cpm, specific activity 60 Ci·mmol−1). Reactions were stopped with chloroform/methanol (1:1, 1 mL), and radioactivity was measured in 600 μL of the aqueous layers by liquid scintillation counting.
Experimental design and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). In vivo data were analysed using SPSS Statistics (IBM, Version 23). Statistical significance was set at P < 0.05. Power analyses using G*Power (3.1.9.2 for Windows; Faul et al., 2007) indicated that for Experiments 1 and 2 (four groups each), to achieve power = 0.96, a total of 32 rats (n = 8 per group) is necessary (with an effect size f = 0.8, α err prob = 0.05). For Experiment 3 (five groups), to achieve power = 0.96, a total of 35 rats (n = 7 per group) is necessary (with an effect size f = 0.8, α err prob = 0.05). In addition, power analyses using Sigma Plot (version 11.0, Systat Software, San Jose, CA, USA) indicated that for ex vivo experiments (two groups), to achieve power = 0.96, a total of 24 rats (n = 6 per group per analysis) is necessary (with an effect size f = 0.8, α err prob = 0.05) based on our previous results (Moreno‐Sanz et al., 2012).
In Experiment 1 of the in vivo studies, to evaluate the effect of various doses of URB937 on acute nausea, the number of gapes among each dose group (VEH, 0.3, 1 and 3 mg·kg−1 URB937; n = 8 per group) were entered into a single factor ANOVA. To evaluate the effect of the potential of rimonabant, AM‐630 or MK‐886 to reverse the effect of URB937 (3 mg·kg−1) on acute nausea, the number of gapes displayed by the VEH‐VEH and 3 mg·kg−1 URB937‐VEH were entered into each of three between‐groups ANOVAs with the inclusion of the followng: (i) VEH–1 mg·kg−1 rimonabant (n = 8), 3 mg·kg−1 URB937–1 mg·kg−1 rimonabant (n = 8); (ii) VEH–1 mg·kg−1 AM‐630 (n = 7), 3 mg·kg−1 URB937–1 mg·kg−1 AM‐630 (n = 12); and (ii) VEH–1 mg·kg−1 MK‐886 (n = 8), 3 mg·kg−1 URB937–1 mg·kg−1 MK‐886 (n = 8). Each of these analyses was also performed for the amount of saccharin solution consumed on the subsequent day in the 6 h conditioned taste avoidance test.
In Experiment 2, to evaluate the potential of PPARα agonism to reduce acute nausea, the number of gapes and amount of saccharin solution consumed in the subsequent 6 h taste avoidance test by each pretreatment group (VEH‐SAL, VEH‐LiCl, GW7647‐SAL, GW7647‐LiCl; n = 8 per group) were entered into a between‐groups ANOVA.
In Experiment 3, to evaluate the potential of URB937 (3 mg·kg−1) to reduce anticipatory nausea and to determine the mechanism of action, the number of gapes elicited by the LiCl‐paired chamber was entered into a between‐groups ANOVA with the groups of (n = 8 per group): VEH‐VEH, URB937‐VEH, URB937‐rimonabant, URB937‐AM‐630, URB937‐MK‐886. Bonferroni post hoc comparison tests were used for all analyses.
Differences in levels of AEA, OEA, PEA and 2‐AG in the AP of rats treated with either vehicle (PEG/Tween‐80/SAL, 1:1:18; n = 6) or URB937 (3 mg·kg−1, i.p.; n = 6) were analysed by independent t‐tests. FAAH activity in each tissue was expressed as % of activity in URB937‐treated animals (n = 6) compared to VEH‐treated animals (n = 6) and analysed by a t‐test. Results are expressed as mean ± SEM, and the significance of differences was determined using one‐way ANOVA followed by Dunnett's test as post hoc and Student's t‐test. Differences were considered significant if P < 0.05. Statistical analyses were conducted using GraphPad Prism Version 4.0 (San Diego, CA, USA).
Drugs
All drugs were administered by i.p. injection. LiCl (Sigma‐Aldrich, Oakville, Ontario, Canada) was prepared in a 0.15 M solution with sterile water and was administered at a volume of 20 mL·kg−1 (127.2 mg·kg−1). URB937 was prepared in a vehicle solution consisting of 1:1:18 [PEG 400 (Sigma, St Louis, MO, USA): Tween 80 (Sigma): saline] and was mixed at a concentration of 0.3, 1 and 3 mg·mL−1 and administered at 1 mL·kg−1 (0.3, 1 and 3 mg·kg−1). Rimonabant (Rim; Sequoia Research Products, Pangbourne, United Kingdrom), AM‐630 (Tocris, Minneapolis, MN, USA) and MK‐886 (Cayman Chemicals, Ann Arbor, Michigan, USA) were prepared in a vehicle consisting of 1:1:18 (EtOH: Tween 80: saline). The ethanol/drug solution was measured into the graduated cylinder, and the Tween 80 was then added and the mixture vortexed. The ethanol was evaporated using a nitrogen stream after which 9 mL saline was added [final vehicle, 1:9 (Tween 80: saline)], mixed at a concentration of 1 mg·mL−1 and administered in a volume of 1 mL·kg−1 (1 mg·kg−1). GW7647 (Cayman Chemicals) was prepared in a vehicle of 1:9 (Tween 80: saline) and mixed at a concentration of 3 mg.mL−1 and administered in a volume of 1 mL·kg−1 (3 mg·kg−1) for rats.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
In vivo experiments
Experiment 1: effects of URB937 on acute nausea and its mechanism of action
The peripherally restricted FAAH inhibitor, URB937, reduced LiCl‐induced acute nausea in a dose‐dependent manner, at doses of 1 and 3 mg·kg−1, but not 0.3 mg·kg−1, i.p. Figure 1 presents the mean number of gapes displayed by each pretreatment group. The ANOVA revealed a significant effect of pretreatment condition, F(3, 28) = 8.2; P < 0.05. Bonferroni post hoc comparison tests revealed that relative to VEH, pretreatment with 1 mg·kg−1 (P < 0.05) or 3 mg·kg−1 (P < 0.05) URB937 suppressed LiCl‐induced conditioned gaping reactions; the dose of 0.3 mg·kg−1 approached statistical significance (P = 0.055). No treatment modified the strength of the LiCl‐induced taste avoidance (data not shown).
Figure 1.
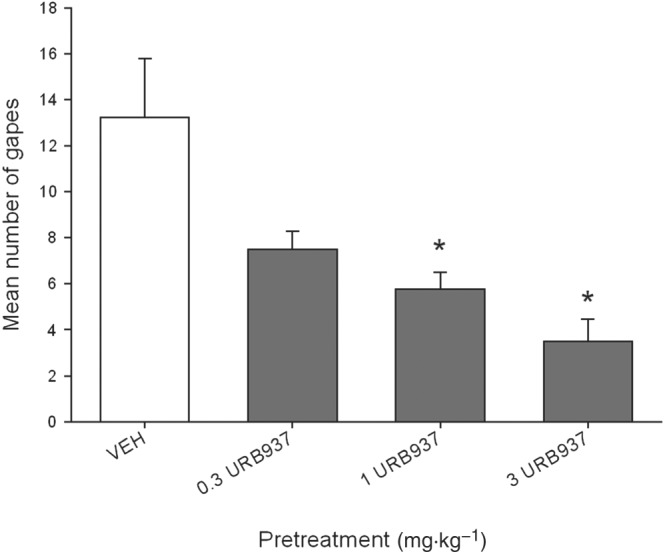
Effect of URB937 (0.3, 1, 3 mg·kg−1, i.p.) or VEH administered 120 min prior to a saccharin‐LiCl pairing on conditioned gaping reactions in a subsequent drug‐free taste reactivity test (a rat model of acute nausea). Each bar represents the mean ± SEM (n = 8) number of gapes. The asterisks indicate a significant difference from the VEH‐treated control animals (*P < 0.05).
The effect of URB937 on acute nausea was reversed by the PPARα antagonist, MK‐866, but not by the CB1 receptor antagonist, rimonabant, or the CB2 receptor antagonist, AM‐630. Figure 2 presents the mean number of gapes displayed by the VEH‐VEH and the URB937‐VEH groups compared with each of the antagonists to test for the mechanism of action (Section A: CB1 receptor mediated mechanism; Section B: CB2 receptor‐mediated mechanism; Section C: PPARα mediated mechanism). To evaluate the potential of CB1 receptor antagonism to prevent the anti‐nausea effect of URB937, the ANOVA for the data in Figure 2A revealed a significant effect of pretreatment, F(3, 28) = 11.6; P < 0.05; subsequent Bonferroni comparison tests revealed that both groups URB937‐VEH and URB‐937‐rimonabant displayed suppressed gaping (P < 0.05) at test compared with both VEH‐VEH and VEH‐rimonabant. The ANOVA for the data in Figure 2B (CB2 receptor mechanism) revealed a significant effect of pretreatment, F(3, 31) = 6.3; P < 0.05. Bonferroni post hoc tests revealed that both URB937‐VEH and URB937‐AM‐630 displayed suppressed gaping (P < 0.05) at test compared with VEH‐VEH; group VEH‐AM‐630 did not significantly differ from any group. Finally, the ANOVA for the data in Figure 2C (PPARα mechanism) revealed a significant effect of pretreatment, F(3, 28) = 5.9; P < 0.05. By Bonferroni tests, group URB937‐VEH displayed significantly less conditioned gaping than group VEH‐VEH (P < 0.05) and URB937‐MK‐886 (P < 0.05). No pretreatment condition modified the strength of the LiCl‐induced CTA in any analysis (data not depicted).
Figure 2.
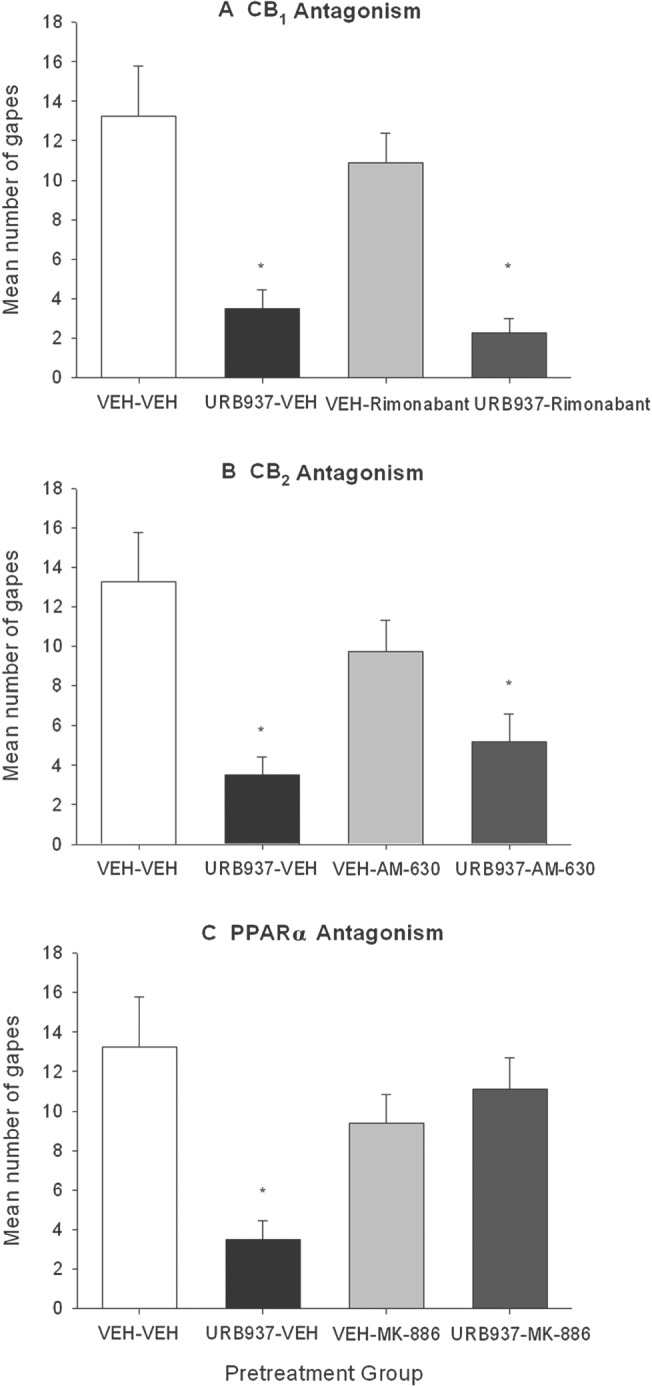
Effect of administration of rimonabant (1 mg·kg−1, i.p.) (A), AM‐630 (1 mg·kg−1, i.p.) (B) or MK‐886 (1 mg·kg−1, i.p.) (C) on URB937 (3 mg·kg−1, i.p.) or VEH administered 120 min prior to a saccharin‐LiCl pairing on conditioned gaping reactions in a subsequent drug‐free taste reactivity test (a rat model of acute nausea). Groups were as follows: VEH‐VEH (n = 8); 0.3 mg·kg−1 URB937‐VEH (n = 8); 1 mg·kg−1 URB937‐VEH (n = 8); 3.0 mg·kg−1 URB937‐VEH (n = 8); VEH‐1 mg·kg−1 rimonabant (n = 8); 3 mg·kg−1 URB937–1 mg·kg−1 rimonabant (n = 8); VEH–1 mg·kg−1 AM‐630 (n = 7); 3 mg·kg−1 URB937–1 mg·kg−1 AM‐630 (n = 12); VEH–1 mg·kg−1 MK‐886 (n = 8); 3 mg·kg−1 URB937–1 mg·kg−1 MK‐886 (n = 8). Each bar represents the mean ± SEM number of gapes. The asterisks indicate a significant difference from the VEH‐treated control animals (*P < 0.05).
Experiment 2: effect of PPARα agonist, GW7647, on acute nausea
Consistent with the results of Experiment 1, pretreatment with the PPARα agonist, GW7647, suppressed LiCl‐induced conditioned gaping reactions. Figure 3 presents the mean number of gapes at test following the various treatments. The between‐groups ANOVA revealed a significant main effect, F(3, 28) = 21.6; P < 0.05. Subsequent Bonferroni post hoc comparison tests revealed that group VEH‐LiCl differed significantly (P < 0.05) from all groups, including GW7647‐LiCl, which did not differ from any other group. No pretreatment modified the strength of LiCl‐induced taste avoidance (data not shown).
Figure 3.
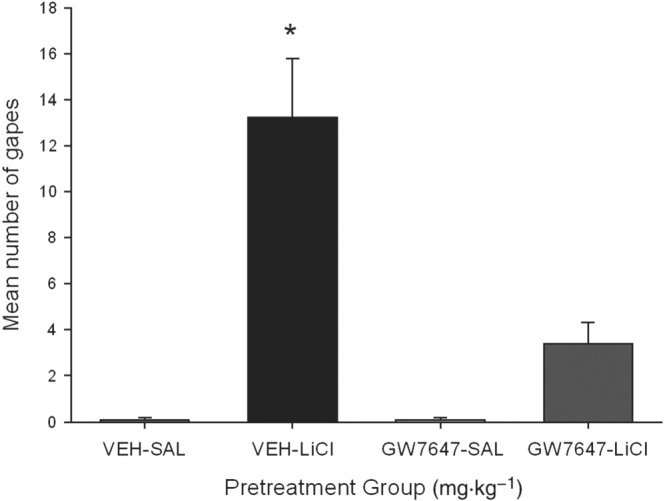
Effect of the PPARα agonist GW7647 (3 mg·kg−1, i.p.) or VEH administered 30 min prior to a saccharin infusion (paired with either LiCl or saline) on conditioned gaping reactions in a subsequent drug‐free taste reactivity test (a rat model of acute nausea). Each bar represents the mean ± SEM (n = 8) number of gapes. The asterisks indicate a significant difference from all other groups (*P < 0.05).
Experiment 3: effect of URB937 on anticipatory nausea, its mechanism of action and effect on locomotor activity
URB937 interfered with the expression of anticipatory nausea; however, this effect was likely mediated by the action of AEA on CB1 receptors. Rock et al. (2015) also found that the suppression of anticipatory nausea by the globally active FAAH inhibitors, URB597 and PF3845, was CB1 receptor mediated, not PPARα mediated. Figure 4 presents the mean number of gapes elicited by re‐exposure to the context previously paired with LiCl‐induced nausea by rats pretreated at test with each of the various drugs. The between‐groups ANOVA revealed a significant effect of treatment, F(4, 35) = 15.3; P < 0.05). Subsequent Bonferroni tests revealed that group VEH‐VEH displayed significantly more gaping than any group (P < 0.05) other than group URB937‐rimonabant. No other groups differed from one another.
Figure 4.
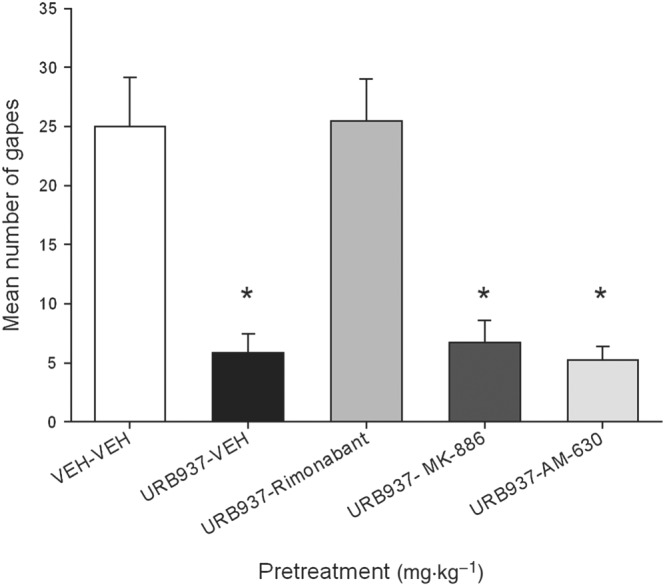
Effect of URB937 (3 mg·kg−1, i.p.) or VEH (n = 8 per group) administered 120 min prior to the anticipatory nausea test. Additional groups (n = 8 per group) were also administered rimonabant (1 mg·kg−1, i.p.), AM‐630 (1 mg·kg−1, i.p.) or MK‐886 (1 mg·kg−1, i.p.) 30 min prior to the test. The mean number of conditioned gaping responses was measured during the anticipatory nausea test trial. Each bar represents the mean ± SEM. The asterisks indicate a significant difference from the VEH‐VEH group (*P < 0.05).
None of the pretreatments modified overall activity in the subsequent test for locomotor activity. Figure 5 presents the mean distance (cm) moved during the 15 min locomotor test. The ANOVA was non‐significant.
Figure 5.
The mean distance (cm) travelled was measured in a 15 min activity test that occurred after the anticipatory nausea test in Experiment 3. Each bar represents the mean ± SEM.
Ex vivo experiments
As previously reported (Clapper et al., 2010), treatment with URB937 (3 mg·kg−1, i.p.) completely blocked FAAH activity in peripheral tissues such as liver and duodenum, while leaving brain tissue unaltered (PFC; Figure 6A). Interestingly, FAAH activity was partially but significantly inhibited in the AP (51.7 ± 9.7% of activity left, P < 0.05), a region that is not fully protected by the blood–brain barrier, which is coherent with the molecular mechanism responsible for the peripheral distribution of URB937. To further examine the effect of this partial inhibition, we analysed the local concentrations of FAAH substrates in the AP. As shown in Figure 6B–D, levels of AEA, OEA and PEA were significantly elevated by treatment with URB937 while levels of 2‐AG (Figure 6E) remained unchanged.
Figure 6.
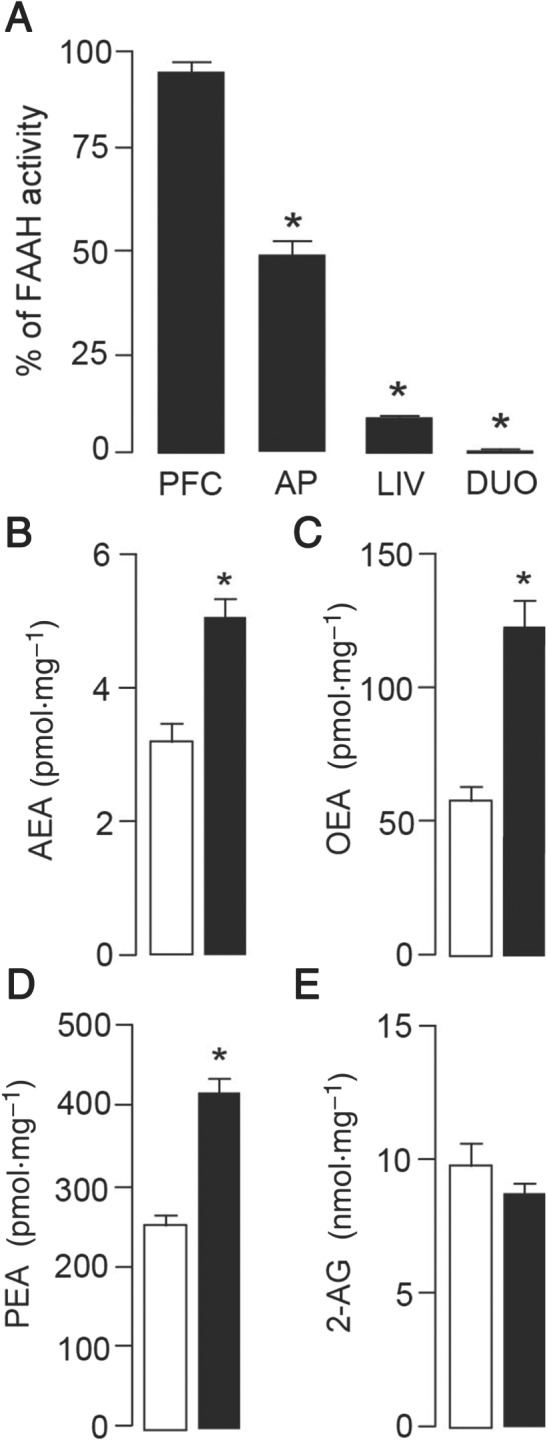
Effects of URB937 on FAAH activity and levels of FAEs in rats. (A) A single injection of URB937 (3 mg·kg−1, i.p., n = 6) blocked FAAH activity in peripheral tissues [liver (LIV) and duodenum (DUO)] 2 h after administration. Cortical (PFC) FAAH activity remained unaltered, and a partial inhibition was observed in the AP compared to animals treated with VEH (n = 6). Analysis of local concentrations of FAEs, which are substrates for FAAH, revealed significantly increased levels of AEA (B), OEA (C) and PEA (D) but not 2‐AG (E) in the AP of rats treated with URB937 (3 mg·kg−1, i.p., n = 6, solid bars) compared to VEH‐treated animals (n = 6, open bars). Each bar represents the mean ± SEM. The asterisks indicate a significant difference from the VEH control animals (*P < 0.05).
Discussion
The peripherally restricted FAAH inhibitor, URB937, at doses of 1 and 3 mg·kg−1, reduced acute nausea as reflected by the prevention of LiCl‐induced conditioned gaping elicited at test but did not modify learning per se as indicated by the conditioned avoidance of LiCl‐paired saccharin (see Parker, 2014). Interestingly, the suppression of LiCl‐induced conditioned gaping by URB937 was prevented by pretreatment with the PPARα antagonist, MK‐886, but not by the CB1 receptor antagonist, rimonabant, or the CB2 receptor antagonist, AM‐630. These findings are consistent with those of Rock et al. (2015) showing that when systemically administered, the global FAAH inhibitor, PF3845 (Ahn et al., 2009), suppressed acute nausea and this suppression was reversed by the PPARα antagonist, MK‐886, but not by the CB1 receptor antagonist, rimonabant. Since FAAH inhibition not only elevates AEA but also OEA and PEA, these results and of those of Rock et al. (2015) suggest that FAAH inhibition (either global or peripheral) produces its acute anti‐nausea effect by the action OEA or PEA on PPARα. Indeed, Venkatesan and colleagues (2016) found increased levels of OEA and PEA (but not AEA or 2‐AG) in patients with cyclic vomiting syndrome when they reported feeling ill (as compared to a self‐reported ‘well’ phase).
Since the PPARα antagonist, MK‐886, reversed the suppressive effect of URB937 on acute nausea, we also evaluated the potential of the PPARα agonist, GW7647, to act as an anti‐nausea agent in the gaping model. Indeed, like URB937, GW7647 also suppressed LiCl‐induced acute nausea in the gaping model, verifying the anti‐nausea effects of systemic PPARα agonism. Given that URB937 suppressed FAAH and elevated OEA and PEA (as well as AEA) in the AP, an area with a weakened blood–brain barrier, which has been shown to prevent LiCl‐induced conditioned gaping reactions when lesioned (Eckel and Ossenkopp, 1996), it is possible that agonism of PPARα in the AP is responsible for the anti‐nausea effects of both URB937 and PF3845. It is interesting to note that OEA (10 mg·kg−1, i.p.) dramatically elevated c‐Fos mRNA expression in the AP (Romano et al., 2014), verifying the activity of PPARα in this region of the brain. Consistent with previous findings, URB937 suppressed FAAH levels in the liver and duodenum, but not the in the PFC, confirming its peripheral site of action (Clapper et al., 2010).
Sticht et al. (2016) demonstrated that endocannabinoid regulation of nausea in the IIC, a cortical site responsible for the experience of nausea (Penfield and Faulk, 1955; Napadow et al., 2013), is mediated by the action of 2‐AG, not AEA, on CB1 receptors. Intra‐IIC administration of the MAGL inhibitor MJN110 (which elevated 2‐AG) suppressed acute LiCl‐induced nausea as expressed by conditioned gaping reactions, but the FAAH inhibitors, PF8345 and URB597, did not. In fact, when PF3845 was administered systemically or intra‐IIC, there was no accompanying increase in levels of AEA in the IIC, but levels of OEA and PEA were elevated. Therefore, the site of action of FAAH inhibition in the regulation of acute nausea was previously unknown. Here, we report that this site of action may be at the level of the AP, potentially representing the initial central relay station for nausea‐inducing stimulation to eventually activate the IIC by some as of yet unknown mechanism.
The peripherally restricted FAAH inhibitor, URB937, also suppressed the expression of contextually elicited conditioned gaping reactions, as a model of anticipatory nausea expressed by chemotherapy patients when they return to the treatment environment (Limebeer et al., 2008). Also in a similar manner as the global FAAH inhibitors, PF3845 and URB597, the suppression of anticipatory gaping with URB937 was reversed by rimonabant but not by the PPARα antagonist, MK‐886, or the CB2 receptor antagonist, AM‐630. As intracranial administration of PF3845 or URB597 into the IIC does not suppress anticipatory nausea (Limebeer et al., 2016), the action of AEA on other brain region(s) may be responsible for the suppression of this conditioned response. Here, we suggest that the action of AEA on CB1 receptors located in the AP (Van Sickle et al., 2003) may be the initial central site of action producing the suppression of anticipatory nausea in this model.
The AP has been shown to be a critical site in LiCl‐induced CTA (suppressed consumption) and also LiCl‐induced conditioned gaping. Indeed, when systemically administered, lithium penetrates and accumulates at high concentrations in the AP (Sandner et al., 1994). AP‐lesioned rats do not display LiCl‐induced conditioned gaping reactions, despite having intact unconditioned reactions to palatable (sucrose) and unpalatable (quinine) taste stimuli (Ossenkopp and Eckel, 1995; Eckel and Ossenkopp, 1996). Furthermore, increases in c‐Fos‐like immunoreactivity are observed in the AP 2 h after LiCl (i.p.) (Swank et al., 1995; Thiele et al., 1996), further suggesting that this site is critical in LiCl‐induced conditioned gaping. Together, these results indicate that direct (or indirect) activation of the AP is necessary for the establishment of conditioned gaping.
In conclusion, we report new data showing that inhibition of peripheral FAAH activity attenuates acute nausea through a mechanism mediated by activation of the PPARα and reduces anticipatory nausea through a mechanism mediated by activation of CB1 receptors, probably through the elevation of local FAEs levels in the AP. These results suggest that peripheral FAAH inhibition may be a potential therapeutic target for the alleviation of acute nausea (via action of PEA and OEA at PPARα) and/or anticipatory nausea (via action of AEA at CB1 receptors), highlighting the need for clinical trials to examine this possible anti‐nausea mechanism.
Author contributions
E.R., C.L. and G.P. performed the in vivo behavioural testing at the University of Guelph. E.R. and C.L. collected the tissue for analysis. G.M.‐S. and R.A. performed the in ex vivo procedures at UC, Irvine. L.P. and E.R. wrote the paper, with suggestions and revisions from G.M.‐S. and D.P.; E.R., G.M.‐S., D.P. and L.P. conceived and coordinated the work. All the authors approved the final version of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was supported by research grants to L.A.P. from the Government of Canada: Natural Sciences and Engineering Research Council of Canada (NSERC: 920157) and from the Government of Canada: Canadian Institute of Health Research (CIHR: 1371220) and to D.P. from the National Institute on Drug Abuse (DA DA012413).
Rock, E. M. , Moreno‐Sanz, G. , Limebeer, C. L. , Petrie, G. N. , Angelini, R. , Piomelli, D. , and Parker, L. A. (2017) Suppression of acute and anticipatory nausea by peripherally restricted fatty acid amide hydrolase inhibitor in animal models: role of PPARα and CB1 receptors. British Journal of Pharmacology, 174: 3837–3847. doi: 10.1111/bph.13980.
References
- Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK et al (2009). Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol 16: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapper JR, Moreno‐Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A et al (2010). Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nat Neurosci 13: 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996). Molecular characterization of an enzyme that degrades neuromodulatory fatty‐acid amides. Nature 384: 83–87. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch DG, Chin SA (1993). Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem Pharmacol 46: 791–796. [DOI] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G et al (1992). Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946–1949. [DOI] [PubMed] [Google Scholar]
- Dinh TP, Freund TF, Piomelli D (2002). A role for monoglyceride lipase in 2‐arachidonoylglycerol inactivation. Chem Phys Lipids 121: 149–158. [DOI] [PubMed] [Google Scholar]
- Eckel LA, Ossenkopp KP (1996). Area postrema mediates the formation of rapid, conditioned palatability shifts in lithium‐treated rats. Behav Neurosci 110: 202–212. [PubMed] [Google Scholar]
- European Food Safety Authority Panel on Animal Health and Welfare – EFSA (2005). Opinion of the Scientific Panel on Animal Health and Welfare on a request from the Commission related to “Aspects of the biology and welfare of animals used for experimental and other scientific purposes”. EFSA J 3: 1–183. [Google Scholar]
- Faul F, Erdfelder E, Lang A‐G, Buchner A (2007). G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods 39: 175–191. [DOI] [PubMed] [Google Scholar]
- Fu J, Bottegoni G, Sasso O, Bertorelli R, Rocchia W, Masetti M et al (2012). A catalytically silent FAAH‐1 variant drives anandamide transport in neurons. Nat Neurosci 15: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodriguez De Fonseca F et al (2003). Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR‐alpha. Nature 425: 90–93. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Norgren R (1978). Chronically decerebrate rats demonstrate satiation but not bait shyness. Science 201: 267–269. [DOI] [PubMed] [Google Scholar]
- Hawkins P, Prescott MJ, Carbone L, Dennison N, Johnson C, Makowska IJ et al (2016). A good death? Report of the second Newcastle meeting on laboratory animal euthanasia. Animals (Basel) 6: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickok JT, Roscoe JA, Morrow GR, King DK, Atkins JN, Fitch TR (2003). Nausea and emesis remain significant problems of chemotherapy despite prophylaxis with 5‐hydroxytryptamine‐3 antiemetics: a University of Rochester James P. Wilmot Cancer Center Community Clinical Oncology Program Study of 360 cancer patients treated in the community. Cancer 97: 2880–2886. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limebeer CL, Krohn JP, Cross‐Mellor S, Litt DE, Ossenkopp KP, Parker LA (2008). Exposure to a context previously associated with nausea elicits conditioned gaping in rats: a model of anticipatory nausea. Behav Brain Res 187: 33–40. [DOI] [PubMed] [Google Scholar]
- Limebeer CL, Vemuri VK, Bedard H, Lang ST, Ossenkopp KP, Makriyannis A et al (2010). Inverse agonism of cannabinoid CB1 receptors potentiates LiCl‐induced nausea in the conditioned gaping model in rats. Br J Pharmacol 161: 336–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limebeer CL, Rock EM, Puvanenthirarajah N, Niphakis MJ, Cravatt BF, Parker LA (2016). Elevation of 2‐AG by monoacylglycerol lipase inhibition in the visceral insular cortex interferes with anticipatory nausea in a rat model. Behav Neurosci 130: 261–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Ben‐Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR et al (1995). Identification of an endogenous 2‐monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol 50: 83–90. [DOI] [PubMed] [Google Scholar]
- Moreno‐Sanz G, Sasso O, Guijarro A, Oluyemi O, Bertorelli R, Reggiani A et al (2012). Pharmacological characterization of the peripheral FAAH inhibitor URB937 in female rodents: interaction with the Abcg2 transporter in the blood‐placenta barrier. Br J Pharmacol 167: 1620–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napadow V, Sheehan JD, Kim J, Lacount LT, Park K, Kaptchuk TJ et al (2013). The brain circuitry underlying the temporal evolution of nausea in humans. Cereb Cortex 23: 806–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkopp KP, Eckel LA (1995). Toxin‐induced conditioned changes in taste reactivity and the role of the chemosensitive area postrema. Neurosci Biobehav Rev 19: 99–108. [DOI] [PubMed] [Google Scholar]
- Parker LA (2003). Taste avoidance and taste aversion: evidence for two different processes. Learn Behav 31: 165–172. [DOI] [PubMed] [Google Scholar]
- Parker LA (2014). Conditioned flavor avoidance and conditioned gaping: rat models of conditioned nausea. Eur J Pharmacol 722: 122–133. [DOI] [PubMed] [Google Scholar]
- Penfield W, Faulk ME (1955). The insula; further observations on its function. Brain 78: 445–470. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Mechoulam R, Piomelli D, Parker LA (2008). The effect of cannabidiol and URB597 on conditioned gaping (a model of nausea) elicited by a lithium‐paired context in the rat. Psychopharmacology (Berl) 196: 389–395. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Parker LA (2014). Anticipatory nausea in animal models: a review of potential novel therapeutic treatments. Exp Brain Res 232: 2511–2534. [DOI] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Ward JM, Cohen A, Grove K, Niphakis MJ et al (2015). Interference with acute nausea and anticipatory nausea in rats by fatty acid amide hydrolase (FAAH) inhibition through a PPARalpha and CB1 receptor mechanism, respectively: a double dissociation. Psychopharmacology (Berl) 232: 3841–3848. [DOI] [PubMed] [Google Scholar]
- Romano A, Karimian Azari E, Tempesta B, Mansouri A, Micioni Di Bonaventura MV, Ramachandran D et al (2014). High dietary fat intake influences the activation of specific hindbrain and hypothalamic nuclei by the satiety factor oleoylethanolamide. Physiol Behav 136: 55–62. [DOI] [PubMed] [Google Scholar]
- Roscoe JA, Morrow GR, Hickok JT, Stern RM (2000). Nausea and vomiting remain a significant clinical problem: trends over time in controlling chemotherapy‐induced nausea and vomiting in 1413 patients treated in community clinical practices. J Pain Symptom Manage 20: 113–121. [DOI] [PubMed] [Google Scholar]
- Sandner G, Di Scala G, Oberling P, Abbe JC, Stampfler A, Sens JC (1994). Distribution of lithium in the rat brain after a single administration known to elicit aversive effects. Neurosci Lett 166: 1–4. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht MA, Limebeer CL, Rafla BR, Abdullah RA, Poklis JL, Ho W et al (2016). Endocannabinoid regulation of nausea is mediated by 2‐arachidonoylglycerol (2‐AG) in the rat visceral insular cortex. Neuropharmacology 102: 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht MA, Rock EM, Limebeer CL, Parker LA (2015). Endocannabinoid mechanisms influencing nausea. Int Rev Neurobiol 125: 127–162. [DOI] [PubMed] [Google Scholar]
- Swank MW, Schafe GE, Bernstein IL (1995). c‐Fos induction in response to taste stimuli previously paired with amphetamine or LiCl during taste aversion learning. Brain Res 673: 251–261. [DOI] [PubMed] [Google Scholar]
- Thiele TE, Roitman MF, Bernstein IL (1996). c‐Fos induction in rat brainstem in response to ethanol‐ and lithium chloride‐induced conditioned taste aversions. Alcohol Clin Exp Res 20: 1023–1028. [DOI] [PubMed] [Google Scholar]
- Travers JB, Norgren R (1986). Electromyographic analysis of the ingestion and rejection of sapid stimuli in the rat. Behav Neurosci 100: 544–555. [DOI] [PubMed] [Google Scholar]
- Van Sickle MD, Oland LD, Mackie K, Davison JS, Sharkey KA (2003). Delta9‐tetrahydrocannabinol selectively acts on CB1 receptors in specific regions of dorsal vagal complex to inhibit emesis in ferrets. Am J Physiol Gastrointest Liver Physiol 285: G566–G576. [DOI] [PubMed] [Google Scholar]
- Venkatesan T, Zadvornova Y, Raff H, Hillard CJ (2016). Endocannabinoid‐related lipids are increased during an episode of cyclic vomiting syndrome. Neurogastroenterol Motil 28: 1409–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]