

Abstract
Background and Purpose
Doxorubicin is a highly effective anticancer drug, but its clinical application is hampered by cardiotoxicity. Asymptomatic diastolic dysfunction can be the earliest manifestation of doxorubicin cardiotoxicity. Therefore, a search for therapeutic intervention that can interfere with early manifestations and possibly prevent later development of cardiotoxicity is warranted. Increased doxorubicin‐dependent ROS may explain, in part, Ca2+ and Na+ overload that contributes to diastolic dysfunction and development of heart failure. Therefore, we tested whether the administration of ranolazine, a selective blocker of late Na+ current, immediately after completing doxorubicin therapy, could affect diastolic dysfunction and interfere with the progression of functional decline.
Experimental Approach
Fischer 344 rats received a cumulative dose of doxorubicin of 15 mg·kg−1 over a period of 2 weeks. After the assessment of diastolic dysfunction, the animals were treated with ranolazine (80 mg·kg−1, daily) for the following 4 weeks.
Key Results
While diastolic and systolic function progressively deteriorated in doxorubicin‐treated animals, treatment with ranolazine relieved diastolic dysfunction and prevented worsening of systolic function, decreasing mortality. Ranolazine lowered myocardial NADPH oxidase 2 expression and oxidative/nitrative stress. Expression of the Na+/Ca2+ exchanger 1 and Nav 1.5 channels was reduced and of the sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 2 protein was increased. In addition, ranolazine lowered doxorubicin‐induced hyper‐phosphorylation and oxidation of Ca2+/calmodulin‐dependent protein kinase II, and decreased myocardial fibrosis.
Conclusions and Implications
Ranolazine, by the increased Na+ influx, induced by doxorubicin, altered cardiac Ca2+ and Na+ handling and attenuated diastolic dysfunction induced by doxorubicin, thus preventing the progression of cardiomyopathy.
Linked Articles
This article is part of a themed section on New Insights into Cardiotoxicity Caused by Chemotherapeutic Agents. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.21/issuetoc
Abbreviations
- BNP
brain natriuretic peptide
- CaMKII
Ca2+/calmodulin‐dependent protein kinase II
- DHE
dihydroethidium
- E Dec t
deceleration time of E wave
- EF
ejection fraction
- FS
fractional shortening
- HF
heart failure
- INa
Na+ current
- IVRT
isovolumetric relaxation time
- LV
left ventricle
- NCX1
Na+/Ca2+ exchanger 1
- NOX2 and NOX4
NADPH oxidase isoforms 2 and 4
- ox‐CaMKII
oxidized CaMKII
- p‐CaMKII
phosphorylated CaMKII
- PLB
phospholamban
- RNS
reactive nitrogen species
- SERCA2
sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 2
Tables of Links
| TARGETS | ||
|---|---|---|
| Enzymes a | Voltage‐gated ion channels b | Transporters c |
| Ca2+/calmodulin‐dependent kinase II http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989 | Nav1.5 channels | Na+/Ca2+ exchanger 1 |
| SERCA2 |
| LIGANDS |
|---|
| Doxorubicin http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=989 |
| Ranolazine |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a,b,c).
Introduction
Doxorubicin is among the most effective antitumor agents successfully used for the management of several neoplastic diseases, but the risk of dose‐dependent cardiotoxicity limits its clinical applications. Furthermore, although the reduction of the cumulative dose below 450 mg·m−2 diminishes the incidence of cardiac toxicity, functional abnormalities occur also at significantly lower doses (Lipshultz et al., 2005; Vejpongsa and Yeh, 2014). Besides the adjustment of doxorubicin dose, none of the several additional strategies for primary prevention of cardiotoxicity (i.e. slow infusion, liposomal formulations, anthracycline analogues and dexrazoxane) is routinely used (Raj et al., 2014).
On the other hand, pharmacological interventions with angiotensin‐converting enzyme inhibitors, angiotensin II receptor blockers and/or β‐blockers are used only after patients have developed symptoms of cardiac damage or are given prophylactically in patients at high risk of cardiotoxicity. Therefore, they do not constitute a line of therapy that prevents cardiotoxic manifestations (Menna et al., 2012). As for secondary prevention, the definition of cardiotoxicity needs to be extended, taking into account symptoms other than the reduction in ejection fraction (EF). Recently, it has been observed that diastolic dysfunction can be present early during the course or after ending chemotherapy, in the absence of clear clinical signs that may prognosticate heart failure (HF) (Prestor et al., 2000; Elli et al., 2013; Minotti, 2013). In both adult and paediatric patients, diastolic dysfunction, as the earliest manifestation of doxorubicin cardiotoxicity, is associated with impaired active relaxation and elevated passive stiffness leading to left ventricle (LV) dysfunction that first appears during diastole and subsequently involves systole (Carver et al., 2007; Salvatorelli et al., 2015; Serrano et al., 2015).
Although a complete agreement about the molecular mechanisms underlying anthracycline cardiotoxicity has not been achieved, many studies support the hypothesis that oxidative stress plays a central role (Schimmel et al., 2004; Raj et al., 2014). In cardiomyocytes exposed to doxorubicin, ROS mediate the down‐regulation of sarcoplasmic/endoplasmic reticulum Ca2+‐ATPase 2 (SERCA2) (Arai et al., 2000). Similarly, in human and animal models of diastolic dysfunction, increased levels of cardiac ROS account for changes in Ca2+ handling proteins (Kass et al., 2004). Ca2+ overload that contributes to diastolic dysfunction is aggravated by alterations in intracellular Na+ homeostasis. Cytosolic Na+ overload occurs as a result an increase of the late Na+ current (I Na), carried by voltage‐gated Nav1.5 channels that can be activated by ROS directly, or indirectly with the involvement of Ca2+/calmodulin‐dependent protein kinase II (CaMKII) (Erickson et al., 2011; Wagner et al., 2011; Coppini et al., 2013a). At the same time, disturbed Na+ and Ca2+ handling in diseased myocytes may increase ROS production (Kohlhaas et al., 2010), generating a vicious cycle that sustains oxidative stress, as well as intracellular Na+ and Ca2+ overload. A major determinant of the levels of myocardial ROS is NADPH oxidase, whose involvement has been demonstrated in HF, including that following anthracycline cardiomyopathy (Grieve et al., 2006; Deng et al., 2007; Zhao et al., 2010).
Ranolazine, a potent and selective inhibitor of I Na, improved diastolic function in both animal models and humans (Sossalla et al., 2008, 2010; Hwang et al., 2009; Figueredo et al., 2011). An explanation for its cardioprotective effect includes the reduction of pathological increases in cytosolic Na+ and Ca2+ accumulation (Sossalla et al., 2011; Coppini et al., 2013a; Williams et al., 2014; De Angelis et al., 2016). Furthermore, ranolazine protected cardiomyocytes from doxorubicin‐induced oxidative stress (Tocchetti et al., 2014). With this background, the use of ranolazine to mitigate the lifetime risk of cardiotoxicity or to alleviate doxorubicin‐induced LV diastolic dysfunction in cancer patients is gaining interest (Minotti, 2013; Corradi et al., 2014), but several compelling arguments are still not validated by experimental evidence.
The INTERACT study (www.clinicaltrialsregister.eu: EudraCT n. 2009‐016930‐29) is a Phase IIB clinical trial conducted on patients who have completed standard dose chemotherapy for the treatment of non‐Hodgkin lymphoma or the adjuvant treatment of breast cancer or colorectal cancer. It was designed to test the efficacy of ranolazine in relieving early‐detected diastolic dysfunction. Therefore, in line with INTERACT, we tested the hypothesis that the administration of ranolazine immediately after ending doxorubicin therapy may interfere with diastolic abnormalities and attenuate the deterioration of heart function that progresses with time towards overt HF, in a rat model of anthracycline cardiomyopathy.
Methods
Animal protocols
All animal care and experimental procedures in this study conformed to the National ethical guidelines (Italian Ministry of Health; D.L.vo 26, March 4, 2014) and was approved by the Ministry of Health (protocol n. 275/2013‐B) and the local ethics committee. All the animal procedures are in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Three‐month‐old female Fischer 344 rats (Envigo Laboratories, Udine, Italy) were housed under controlled temperature, humidity and 12 h light/dark cycle, and with food and water provided ad libitum. Sixty‐six animals were used for the experiments described here. Rats (n = 45) received six i.p. injections of 2.5 mg·kg−1 of doxorubicin, every other day, over a period of 2 weeks to reach a cumulative dose of 15 mg·kg−1 (De Angelis et al., 2015). Control rats (n = 15) were injected with saline. Before starting ranolazine treatment (i.e. 2 weeks after the first injection of doxorubicin), rats were monitored by echocardiography for the assessment of diastolic dysfunction and a small portion of them (n = 5 per group) was killed for preliminary analysis. The remaining animals were randomly divided into two experimental groups: DOXO + RAN group (n = 20) received ranolazine daily (80 mg·kg−1 by gavage) for the following 4 weeks; the DOXO group (n = 20) was given with equal volumes of saline. Animals were killed 6 weeks after the first injection of doxorubicin. As ranolazine is a weak inhibitor of drug transporter P‐glycoprotein and CYP3A enzymes, which are responsible for doxorubicin metabolism (Minotti, 2013), in the view of a potential clinical application, a sequential administration (ranolazine treatment after ending chemotherapy) has been preferred to a concomitant treatment. The temporal plan of in vivo procedures are shown in Figure 1. The dose of ranolazine was comparable with those used clinically in humans (Reagan‐Shaw et al., 2008). The effects of ranolazine treatment on heart function of saline‐injected rats (RAN group; n = 6) were also evaluated.
Figure 1.
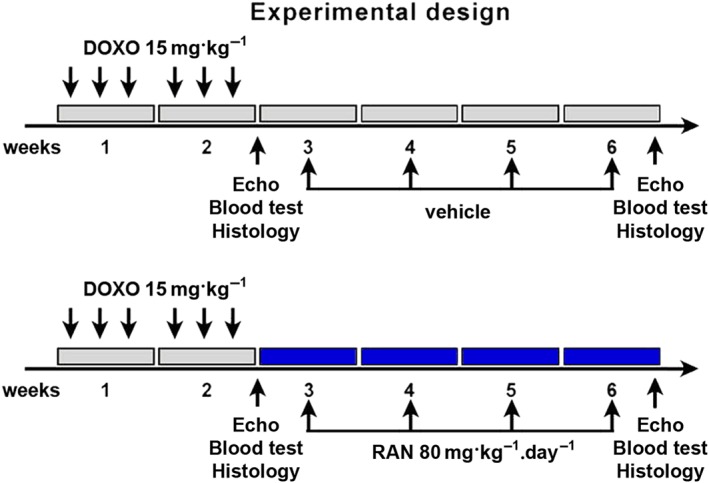
Experimental Design. Temporal scheme of in vivo procedures.
Echocardiography
Rats were anaesthetised with ketamine (100 mg·kg−1) and medetomidine (0.25 mg·kg−1) and echocardiographic parameters were collected with Vevo 770 (VisualSonics, Toronto, Ontario, Canada) equipped with a 25 MHz linear transducer. Body temperature was maintained at ~37°C with a heating pad. Diastolic function was assessed by Doppler echocardiography. Specifically, mitral blood flow velocities were evaluated from a four‐chamber apical view by using pulsed‐wave Doppler with the sample volume placed at the mitral leaflet tips. Similarly, tissue Doppler recording was conducted placing the sample volume at the septal insertion site of the mitral leaflet. Functional measurements included peak velocities of the E wave and A wave, the E/A ratio, isovolumetric relaxation time (IVRT) and deceleration time of E wave (E Dec t). Tissue Doppler measurements of mitral E and A wave velocities (E′, A′ and E′/A′ ratio) were also made. From the short‐axis view, M‐mode tracing of the LV at the level of the papillary muscles was obtained. The measurement of internal LV diameter and posterior wall thickness, during diastole and systole, served to calculate systolic indices EF and fractional shortening (FS) (Di Meglio et al., 2012).
Circulating brain natriuretic peptide analysis
At the end of the experiment, blood was collected from the abdominal aorta. Plasma was obtained by centrifugation at 4000 × g (15 min, 4°C) and stored at −80°C for further analysis. Brain natriuretic peptide (BNP) levels were measured by using an enzyme immunoassay according to the manufacturer's instructions. All standards and samples were run in duplicate.
Rat cardiomyocyte isolation and culture
Adult rat left ventricular myocytes (ARVM) were obtained from 3‐month‐old rats (Pacini et al., 2013; Ferrantini et al., 2014). Animal studies were performed according to the University of Florence laboratory animal guidelines. Isoflurane was used for anaesthesia. The heart was rapidly removed and retrogradely perfused with perfusion buffer [120 mM NaCl, 15 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4, 5 mM NaHCO3, 10 mM Na‐HEPES, 30 mM taurine, 10 mM 2,3‐butanedione monoxime (BDM), 5.5 mM glucose, pH 7.2] for 10 min at 37°C. The heart was digested with 0.33 mg·mL−1 Liberase TH for 8 min. The heart was then cut into small pieces, and the slurry was filtered through a sterile 150 nm mesh. The filtrate was centrifuged at 400 g for 4 min to separate myocytes from non‐myocytes, and the myocyte pellet was resuspended in 10 mL of cultured medium. The myocyte suspension was layered over 60 μg·mL−1 of BSA and allowed to settle for 15 min to further separate myocytes from fibroblasts. The myocyte pellet was resuspended in culture medium. Myocyte concentration was determined using a cyclohexameter and plated on 100 mm laminin coated plastic culture dishes at a density of 100 to 150 cells mm−2. The ARVM culture was maintained in serum free culture medium supplemented with 2 mg·mL−1 albumin, 2 mM carnitine, 5 mM creatine, 5 mM taurine, 1 mg·mL−1 BDM and 100 μg·mL−1 penicillin–streptomycin. Doxorubicin was added to half of the culture wells (randomly selected) at a final concentration of 5 μM. Cells were maintained in culture for 24 h and then used for patch‐clamp experiments as detailed below.
Patch‐clamp measurements
Patch‐clamp studies were performed as previously described (Coppini et al., 2013a). For late I Na measurements, isolated ventricular cardiomyocytes were superfused with an extracellular solution containing 135 mM NaCl, 5 mM CsCl, 1 mM MgSO4, 10 mM glucose, 10 mM HEPES and 0.01 mM nitrendipine, pH 7.4, at 25°C. Pipette solution contained 120 mM l‐aspartic acid, 20 mM CsCl, 1 mM MgSO4, 4 mM Na2ATP, 0.1 mM Na3GTP and 10 mM HEPES, pH 7.3 (with CsOH). To measure late I Na and the extent of block by 10 μM ranolazine, 500 ms depolarizing steps to −10 mV from a holding potential of −120 mV were applied to cells at a rate of 0.2 Hz. In a subset of experiments, 150 ms depolarizing steps were applied at a rate of 5 Hz. The magnitude of late I Na was determined by integration of the area of the current between 25 and 225 ms after onset of the −10 mV clamp pulse (for 0.2 Hz recordings; 25–125 ms interval was used for 5 Hz recordings), using the integration (area) feature of the pCLAMP program (Molecular Devices, Sunnyvale, CA, USA).
Tissue preparation
For the preparation of frozen sections, isolated hearts were weighted, dissected and then placed onto a tissue mould and covered with OCT cryo‐embedding medium. The cryo mould containing the tissue block was placed in a metal beaker filled with isopentane already placed in liquid nitrogen. After ensuring tissue was completely frozen, the tissue block was stored at −80°C, ready for sectioning. Tissue sections (10 μm thick) were generated by a Leica CM3050 S cryostat (Leica Microsystems, Wetzlar, Germany). For molecular biology analysis, heart samples were frozen in liquid nitrogen and then stored at −80°C.
Histochemistry and immunocytochemistry
To evaluate generation of ROS, tissue sections were incubated with the oxidative fluorescent dye, dihydroethidium (DHE) (De Angelis et al., 2004). Peroxynitrite formation was assessed by using an anti‐3‐nitrotyrosine antibody. FITC conjugated was used as secondary antibody. Samples were analysed with a Leica DM5000B (Leica Microsystems) microscope and a Zeiss LSM700 confocal microscope (Zeiss, Oberkochen, Germany).
Quantitative reverse‐transcription polymerase chain reaction
Total RNA was extracted from heart tissue using the TRIzol reagent according to the manufacturer's instructions. Both cDNA synthesis and PCR were performed simultaneously by using the SuperScript III Platinum SYBR Green One‐Step qRT‐PCR Kit. Quantitative RT‐PCR was carried out using the iCycler iQ System (Bio‐Rad Laboratories, Hercules, CA, USA). The transcript levels of SERCA2, Na+/Ca2+ exchanger 1 (NCX1), Nav 1.5 channels, collagen I and collagen III were measured and the housekeeping gene encoding hypoxanthine phosphoribosyltransferase was used as internal control. Relative expression was calculated using the comparative cycle threshold (Ct) method (2−ΔΔCt).
Western blotting
Tissue samples were lysed in buffer containing 0.1% Triton X100 and a cocktail of protease and phosphatase inhibitors. Protein extracts were then separated on 8–12% SDS‐PAGE and transferred onto PVDF membrane. Membranes were probed with primary antibodies against CaMKII, p‐CaMKII‐Thr287, oxidized CaMKII (ox‐CaMKII), NADPH oxidase isoforms 2 and 4 (NOX2 and NOX4), Nav 1.5 channel, NCX1, SERCA2, phospholamban (PLB), MMP2 and collagen I. Loading conditions were determined with GAPDH. Peroxidase‐conjugated secondary antibodies were employed to detect primary antibodies. Antibody binding was visualized by ECL and the optical density of the bands was analysed with a Molecular Analysis software (Bio‐Rad Laboratories).
Gel zymography
Frozen tissue was mechanically homogenized on ice in lysis buffer (50 mM Tri‐HCl pH 7.4, 150 mM NaCl, 5 mM CaCl2, 1% Nonidet P40, 0.1% SDS) including protease inhibitors. Equal amounts of un‐denatured total cellular proteins were separated by electrophoresis. Afterwards, gels were incubated in Renaturation and Development buffers, stained with a solution containing 0.5% Coomassie Blue R‐250 in 30% methanol and 10% acetic acid and successively destained in 30% methanol and 10% acetic acid solution. The gelatinolytic activity of the MMP2 was shown as transparent bands against a dark blue background.
Data and statistical analysis
Data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The number of replicates was at least n = 5 per group for each data set and results were presented as mean ± SD unless stated otherwise. Data were analysed by using GraphPad Prism (GraphPad software, San Diego, CA, USA). Significance between two comparisons was determined by Student's t‐test and, for multiple comparisons, by one‐way ANOVA and Bonferroni's post test. Mortality curves were analysed by the standard Kaplan–Meier method with log‐rank test. All P values are two‐sided and P < 0.05 was considered statistically significant.
Materials
Doxorubicin was from Teva Pharmaceuticals Industries (Assago, Italy). BNP immunoassay kit, primary antibodies for CaMKII, MMP2, NOX2 and NOX4, NCX1 and SERCA2 were from Abcam (Cambridge, UK). Collagen I and Nav 1.5 channel were from Novus Biologicals (Littleton, CO, USA). Nitrotyrosine, PLB, p‐CaMKII‐Thr287, ox‐CaMKII antibodies and ECL were from Merck Millipore (Milan, Italy). Ranolazine, NaCl, KCl, KH2PO4, Na2HPO4, MgSO4, NaHCO3, Na‐HEPES, taurine, BDM, glucose, BSA, carnitine, creatine, CsCl, HEPES, nitrendipine, l‐aspartic acid, Na2ATP, Na3GTP, isopentane, protease and phosphatase inhibitor cocktails, Tri‐HCl, CaCl2, Nonidet P40, SDS, acetic acid, DHE and GAPDH antibody were from Sigma‐Aldrich (St. Louis, MO, USA). PVDF membrane was from GE Healthcare Life Sciences (Marlborough, MA, USA). Penicillin–streptomycin was from Euroclone (Pero, Italy). Renaturation and Development buffers and Coomassie Blue R‐250 were from Bio‐Rad Laboratories (Hercules, CA, USA). OCT was from Bio‐Optica (Milan, Italy). Laminin was from Thermo Fisher Scientific (Waltham, MA, USA). Liberase TH was from Roche Italia (Monza Italy). TRIzol and SYBR Green were from Life Technologies Italia (Milan, Italy). FITC conjugated secondary antibody was from Jackson ImmunoResearch (Suffolk, UK). Peroxidase‐conjugated secondary antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA).
Results
Doxorubicin and early diastolic dysfunction
After completion of doxorubicin treatment and just before starting treatment with ranolazine, rats were monitored by Doppler and standard echocardiography for the assessment of diastolic and systolic function. While systolic function was unchanged (Figure 2A), alterations in diastolic parameters by doxorubicin treatment demonstrated an impaired ventricular relaxation. Indeed, higher mitral inflow A wave velocity and lower E/A ratio together with longer IVRT and E Dec t were observed in the DOXO group in comparison to the CTRL rats (Figure 2B). No differences were found in other parameters (E, E′ and A′ waves and E′/A′ ratio) between the groups (Figure 2B,C). Because changes in cardiovascular biomarkers, such as natriuretic peptides, can have a prognostic value for the development of HF in asymptomatic patients with normal EF (Minotti, 2013; Lenneman and Sawyer, 2016), the levels of circulating BNP were evaluated. As a confirmation of diastolic dysfunction at this stage, increased plasma levels of BNP were found after doxorubicin treatment (Figure 2D).
Figure 2.
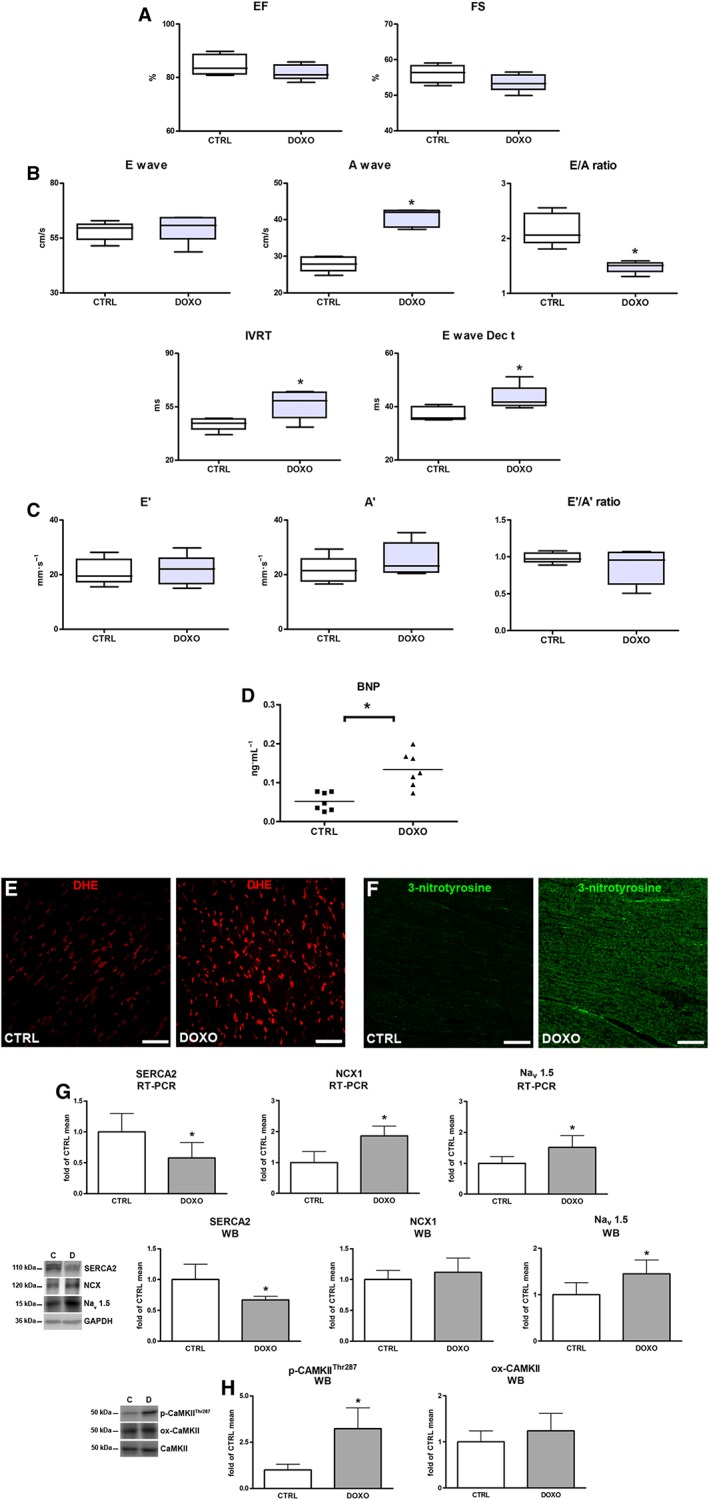
Early effects of doxorubicin (DOXO). (A) Echocardiographic systolic indices EF and FS. (B) Pulsed‐wave Doppler showing diastolic function: peak velocity of the E wave and A wave, E/A ratio, IVRT and E Dec t. (C) Tissue Doppler measurements of mitral E′, A′ and E′/A′ ratio. (D) BNP plasma levels. (E,F) Myocardial content of DHE (red) and 3‐nitrotyrosine (green). (G) Myocardial mRNA and protein expression of SERCA2, NCX1 and voltage‐gated Nav 1.5 channels. (H) Myocardial levels of phosphorylated and oxidized Ca2+/calmodulin‐dependent protein kinase II (p‐CaMKII‐Thr287; ox‐CaMKII). Scale bars: (E,F) 50 μm. *P < 0.05, significantly different from CTRL.
Oxidative/nitrative stress and early changes in cardiac Ca2+ and Na+ handling proteins
Cardiac excitation‐contraction coupling is affected by oxidative stress, leading to contractile dysfunction in HF (Köhler et al., 2014). In our model, oxidative/nitrative stress analysis showed an increased formation of ROS and reactive nitrogen species (RNS), as documented by DHE and 3‐nitrotyrosine staining in the DOXO group, compared with the levels in control rats (Figure 2E,F). Furthermore, altered mRNA and protein expression levels of SERCA2, NCX1 and Nav 1.5 channels in the hearts of doxorubicin‐treated animals were observed. Particularly, after the completion of doxorubicin treatment, SERCA2 expression significantly decreased, while that of Nav 1.5 channels increased (Figure 2G). The involvement of CaMKII, which triggers the activation of Nav 1.5 channels (Sossalla et al., 2011; Köhler et al., 2014), was demonstrated by its elevated phosphorylation levels observed in the DOXO group. The ox‐CaMKII level did not change (Figure 2H).
Proof of concept of doxorubicin effects on late I Na
To validate the hypothesis that increased late I Na following CaMKII‐dependent, Nav1.5 channel activation occurs upon doxorubicin exposure, we measured Na+ influx in isolated cardiomyocytes. For this purpose, ARVM were isolated and cultured for 24 h in the presence or absence of 5 μM doxorubicin and were then used for patch‐clamp measurement of late I Na (Figure 3A,B). Late I Na was significantly increased in doxorubicin‐treated cardiomyocytes as compared with cells exposed to vehicle (Figure 3C). Peak I Na was, however, not affected by doxorubicin treatment (Table 1). Interestingly, the application of 10 μM ranolazine in DOXO‐treated cardiomyocytes reduced late I Na to the level of control cells, while the drug did not significantly change the current recorded in vehicle‐treated cardiomyocytes (Figure 3C, Table 1). Because the effects of ranolazine are rate‐dependent, the inhibitory effects of this conmpound drug were more pronounced when the current was elicited at 5 Hz as compared with 0.2 Hz (Table 1).
Figure 3.
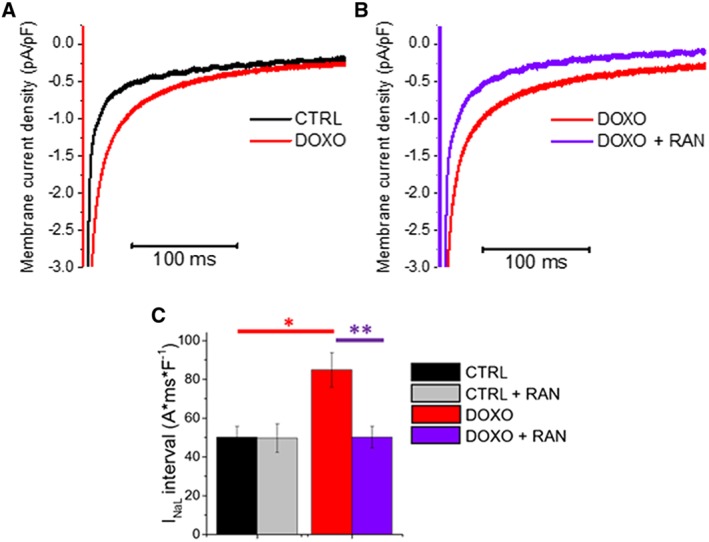
Effects of doxorubicin (DOXO) treatment on late I Na and inhibition by ranolazine (RAN). (A) Representative superimposed traces of late I Na from control (CTRL) and doxorubicin‐treated rat cardiomyocytes. (B) Representative superimposed traces of late I Na from a doxorubicin‐treated cardiomyocyte in the absence (DOXO, red) and presence of 10 μM ranolazine (DOXO + RAN, purple). (C) Average late I Na density integrals, calculated from 25 to 225 ms after the onset of a 500 ms depolarization step from −120 to −10 mV. Means ± SEM from 17 CTRL (four rats) and 18 DOXO (four rats) cardiomyocytes. *P < 0.05, significantly different from CTRL (unpaired t‐test ); **P < 0.05, significantly different from DOXO (paired t‐test ).
Table 1.
Additional patch‐clamp data, obtained from rat cardiomyocytes.
| Parameter | CTRL | DOXO | CTRL + RAN | DOXO + RAN | Statistics (P) | |
|---|---|---|---|---|---|---|
| DOXO versus CTRL | DOXO + RAN versus DOXO | |||||
| Cell capacitance (pF) | 243 ± 19 | 225 ± 19 | – | – | > 0.05 | – |
| Peak Na‐current density (pA/pF) at 0.2 Hz | 25.9 ± 2.9 | 24.1 ± 2.3 | 25.4 ± 2.7 | 20.4 ± 2.9 | > 0.05 | > 0.05 |
| Peak Na‐current density (pA/pF) at 5 Hz | 24.6 ± 3.1 | 23.5 ± 2.7 | 23.3 ± 3.2 | 19.3 ± 3.3 | > 0.05 | > 0.05 |
| Late Na‐current density integral at 5 Hz (A*ms*F−1) | 14.5 ± 2.9 | 31.1 ± 2.1 | 13.4 ± 2.6 | 14.8 ± 3.3 | <0.05 | <0.05 |
| % change of late Na‐current with ranolazine at 0.2 Hz | −8.3 ± 1.2 | −40.0 ± 3.5 | – | – | <0.05 | – |
| % change of late Na‐current with ranolazine at 5 Hz | −12.3 ± 3.2 | −55.0 ± 7.5 | – | – | <0.05 | – |
Data in the Table are means ± SEM from 17 control (CTRL) and 18 doxorubicin (DOXO)‐treated rat cardiomyocytes. Significant differences between group means (P<0.05) are shown in bold; unpaired t‐test (DOXO versus CTRL) or paired t‐test (DOXO + RAN versus. DOXO). There were no significant differences between the values from the CTRL and the CTRL + RAN groups.
Effects of ranolazine on animal survival and body weight
While doxorubicin treatment had detrimental consequences for the general condition of the animals, 4 weeks of ranolazine treatment had a beneficial effect on animal health and partly counteracted the progressive weight‐loss observed in the DOXO group of rats (Figure 4A). Moreover, the Kaplan–Meier analysis showed a reduction in mortality within the DOXO + RAN group when compared to the DOXO group (Figure 4B). There were no deaths in the control group.
Figure 4.
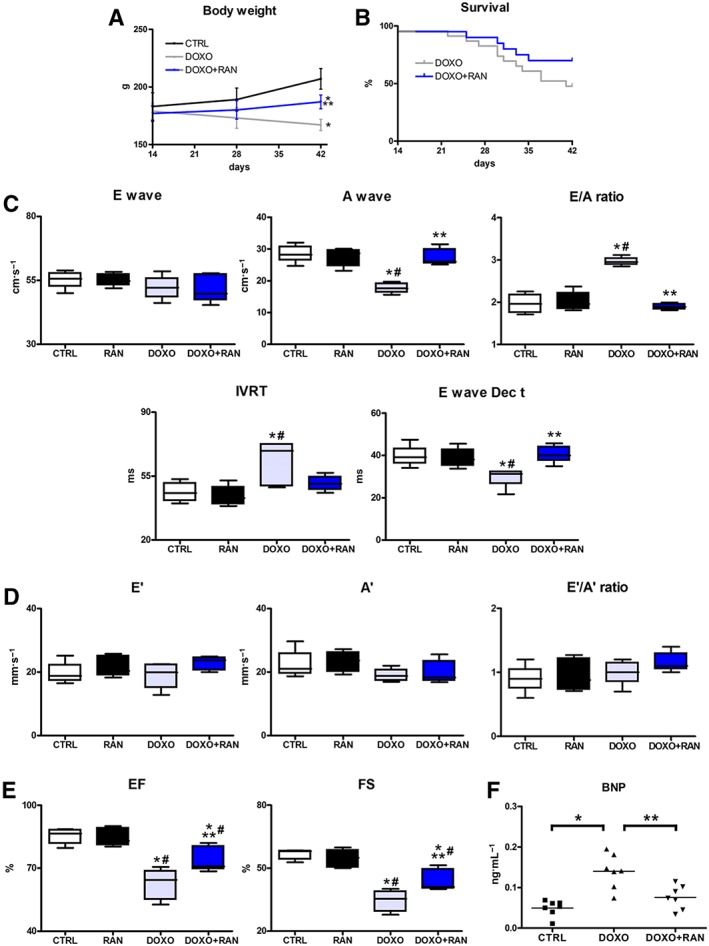
Ranolazine (RAN) and late effects of doxorubicin (DOXO). (A,B) Body weight trend and Kaplan–Meier survival analysis. (C) Pulsed‐wave Doppler showing diastolic function: peak velocity of the E wave and A wave, E/A ratio, IVRT and E Dec t. (D) Tissue Doppler measurements of mitral E′, A′ and E′/A′ ratio. (E) Echocardiographic systolic indices EF and FS. (F) BNP plasma levels. *P < 0.05, significantly different from CTRL; #P < 0.05, significantly different from RAN; **P < 0.05, significantly different from DOXO.
Effects of ranolazine on cardiac function
Four weeks after the last injection of doxorubicin, cardiac function was examined. Our data demonstrated a progressive deterioration of diastolic and systolic function in the DOXO group, compared with the intermediate time‐point, immediately after the end of doxorubicin treatment. On the contrary, rats receiving ranolazine showed a minor worsening of systolic function (not seen after doxorubicin treatment) and a relief of diastolic dysfunction. Specifically, the DOXO group of rats showed a progression of diastolic dysfunction as demonstrated by Doppler echocardiography. Pulsed‐wave Doppler displayed a restrictive filling pattern of the mitral valve inflow, characterized by elevation in the E/A ratio and shortening of the E Dec t. In DOXO + RAN rats, however, 4 week treatment with ranolazine was sufficient to preserve diastolic performance and to reverse the functional abnormalities caused by doxorubicin. Indeed, A wave velocity, E/A ratio, IVRT and E Dec t were comparable to parameters recorded in CTRL animals (Figure 4C). Tissue Doppler parameters wereunchanged and comparable among experimental groups (Figure 4D). On the other hand, as measured by M‐mode echocardiography, doxorubicin induced a significant reduction in EF and FS, with respect to control rats, and ranolazine was able to attenuate the deterioration of systolic function (Figure 4E). No effect of ranolazine on heart function was observed when this compound was administered to healthy animals. The diastolic and systolic parameters were comparable to that recorded in CTRL group (Figure 4C–E) and ranolazine did not affect heart rate (not shown). As ranolazine treatment had no effect on cardiac performance, no further investigations on the RAN group were pursued. Finally, the levels of circulating BNP, higher in the DOXO group, were significantly diminished after treatment with ranolazine (Figure 4F).
Ranolazine and oxidative stress
Myocardial NOX2 NADPH oxidase expression significantly increased in the DOXO group, compared with the CTRL group. The involvement of NOX4 isoform seemed to be less marked, given the slight modulation occurring in our experimental conditions. Additionally, the content of DHE and peroxynitrite (3‐nitrotyrosine) were higher in the myocardium of the DOXO group than in the CTRL group. Conversely, treatment with ranolazine decreased oxidative and nitrative stress, supporting the concept that such a reduction mediates the beneficial effects of ranolazine (Figure 5A–C).
Figure 5.
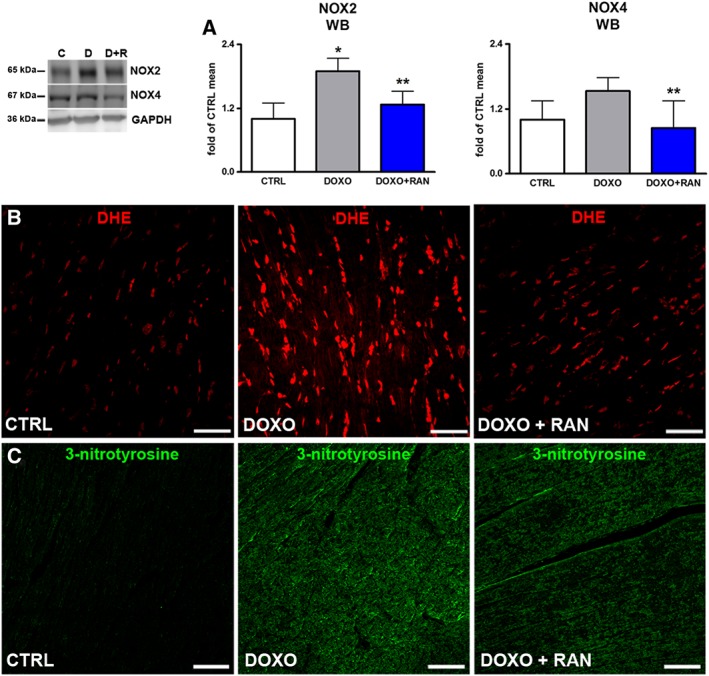
Oxidative stress. (A) Protein expression of NOX2 and NOX4 in rat hearts. (B) Myocardial content of DHE (red) and 3‐nitrotyrosine (green). Scale bars: (B,C) 50 μm. *P < 0.05, significantly different from CTRL; **P < 0.05, significantly different from DOXO.
Effects of ranolazine on cardiac Ca2+ and Na+ handling proteins
With respect to control, the DOXO group showed a decrease in SERCA2 and an increase in NCX1 mRNA and protein levels. Treatment with ranolazine reduced NCX1 expression and, most importantly, increased that of SERCA2 (Figure 6A). The beneficial effect of ranolazine on ameliorating Ca2+ re‐uptake was reflected by the increase in the SERCA2/PLB ratio (Figure 6B). These findings confirm the link between Na+ and Ca2+ handling in doxorubicin cardiotoxicity. Late I Na hyperactivation, due to ROS and RNS overproduction, might directly cause a cytosolic Ca2+ overload. Expression of mRNA and protein for Nav 1.5 channels were increased in the DOXO group. Long‐term ranolazine treatment significantly attenuated Nav 1.5 channel overexpression, probably as a consequence of the reduced Ca2+ overload (Figure 6C). Moreover, the increased phosphorylation and oxidation of CaMKII, a key regulator of Na+ and Ca2+ homeostasis, following doxorubicin exposure, were decreased by ranolazine (Figure 6D).
Figure 6.
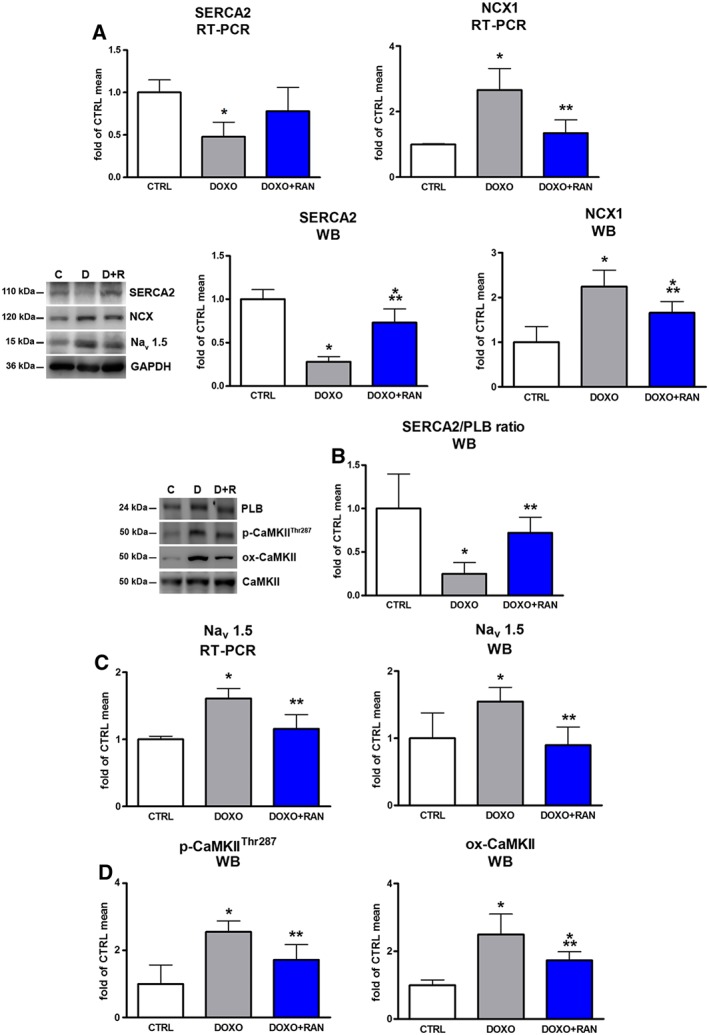
Cardiac excitation‐contraction coupling. (A) mRNA and protein expression of SERCA2, NCX1 in animal myocardium. (B) Myocardial SERCA2/PLB ratio. (C) RT‐PCR and Western blot analysis of Nav 1.5 channels in rat hearts. (D) Myocardial levels of phosphorylated and oxidized Ca2+/calmodulin‐dependent protein kinase II (p‐CaMKII‐Thr287; ox‐CaMKII). *P < 0.05, significantly different from CTRL; **P < 0.05, significantly different from DOXO.
Effects of ranolazine on myocardial remodelling
Extracellular matrix remodelling and interstitial fibrosis are constantly present in the development and progression of doxorubicin cardiotoxicity (De Angelis et al., 2015; Cappetta et al., 2016). Therefore, the effects of ranolazine on doxorubicin‐induced extracellular matrix remodelling and fibrosis was evaluated. When compared with control animals, MMP2 expression increased in the DOXO group. Treatment with ranolazine prevented this increase (Figure 7A). Moreover, MMP2 gelatinolytic activity in myocardial tissue from ranolazine‐treated rats was significantly reduced (Figure 7B). Consistent with these results, ranolazine also attenuated the increase in collagen type I along with the higher collagen type I‐to‐type III ratio observed in the DOXO group (Figure 7C).
Figure 7.
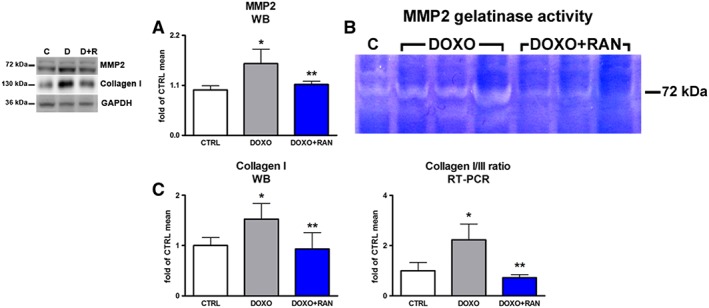
Myocardial remodelling and extracellular matrix turnover. (A) Myocardial expression of MMP2. (B) MMP2 gelatinase activity shown by gel zymography assay. (C) Collagen I expression and collagen I/III ratio. *P < 0.05, significantly different from CTRL; **P < 0.05, significantly different from DOXO.
Discussion
The late recognition of doxorubicin cardiotoxicity is associated with a poorer prognosis, due to potentially irreversible LV dysfunction, and the lack of clinical response to pharmacological therapy (Cardinale et al., 2010; Plana et al., 2014). In this view, the early detection of cardiac abnormalities, even when asymptomatic, may have important clinical implications. As diastolic dysfunction, as an early sign of cardiotoxicity, can predict and contribute to the onset of HF, there is a possibility that the intervention within this particular window may prevent the development of overt HF. In our experimental setting, at the completion of doxorubicin treatment, systolic indices were normal while altered diastolic parameters defined an impaired ventricular relaxation. The evidence of an early functional disorder was supported by the rise in plasma BNP, immediately after doxorubicin treatment. BNP has been used not only as a biomarker for the diagnosis of HF, but also to detect asymptomatic LV dysfunction and to predict the prognosis (Minotti, 2013; Ishigaki et al., 2015; Lenneman and Sawyer, 2016).
In doxorubicin‐induced cardiotoxicity and diastolic dysfunction related to other pathologies, increased oxidative stress and intracellular Ca2+ dysregulation are commonly present and closely interconnected (Octavia et al., 2012; Paulus and Tschöpe, 2013). In diastolic dysfunction, characterized by the presence of ROS, RNS and hypoxia, the hyperactivation of late I Na is constantly reported as well. A similar scenario can occur in the heart exposed to doxorubicin, where the continuous formation of ROS/RNS together with increased cytoplasmic Ca2+ may lead to activation of late I Na (Minotti, 2013; Corradi et al., 2014). In our study, high levels of oxidative stress together with significant alterations in Ca2+ handling proteins (SERCA2 and NCX1) were detected at the end of doxorubicin treatment. This is consistent with previous studies showing the association of LV diastolic dysfunction and doxorubicin, with SERCA2a‐dependent, abnormal active relaxation in cardiomyocytes (Neilan et al., 2007; Octavia et al., 2012). Doxorubicin exposure increased myocardial Nav 1.5 channel expression, which may have contributed to the enhancement of late I Na observed in electrophysiological studies conducted on isolated ventricular cardiomyocytes. Doxorubicin‐induced hyperactivation/overexpression of Nav 1.5 channels may cause an increase of intracellular [Na+], forcing NCX to work in the reverse mode thus amplifying the retention of intracellular Ca2+, along with ROS‐induced inhibition of SERCA2.
Regulation of Na+ and Ca2+ handling in several cardiac diseases including doxorubicin cardiotoxicity involves increased activation of myocardial CaMKII, a central mediator of the changes in cardiomyocyte expression‐profile (Erickson et al., 2011; Sag et al., 2011; Coppini et al., 2013b; Kreusser and Backs, 2014). The increase in CaMKII phosphorylation detected in our study can be both Ca2+ and ROS‐dependent, given that elevated oxidative stress and Ca2+ handling dysregulation are typical of doxorubicin cardiomyopathy. Interestingly, CaMKII also phosphorylates cardiac Nav 1.5 channels, driving late I Na augmentation, which, in turn, determines cytosolic Na+ overload. Na+ overload leads to decreased Ca2+ extrusion and elevated diastolic Ca2+, that slow down myocardial relaxation and increase diastolic tension, ultimately inducing diastolic dysfunction (Wagner et al., 2006; Erickson et al., 2008; Wagner et al., 2011; Coppini et al., 2013a).
Therefore, the use of ranolazine may be beneficial in diastolic dysfunction and chemotherapeutic cardiotoxicity, due to its capacity to reduce late I Na and cytosolic Na+, and consequently to counteract intracellular Ca2+ accumulation (Minotti, 2013; Corradi et al., 2014; Mihos et al., 2016; Banerjee et al., 2017). In a previous study in mice, ranolazine was shown to act as an antioxidant when co‐administered with doxorubicin (Tocchetti et al., 2014). However, the effect of ranolazine on diastolic dysfunction and Na+ and Ca2+ handling was not addressed. Moreover, pharmacokinetic profiles indicating a possible interaction between ranolazine and doxorubicin at the level of CYP3A and P‐glycoprotein (Jerling, 2006; Wilde et al., 2007; Minotti, 2013) prompted us to avoid the concomitant administration of these drugs, in line with the INTERACT study. With this background, the use of ranolazine to target the vicious cycle induced by anthracycline, fuelled by ROS and late I Na appears a reasonable strategy (Figure 8). Ranolazine treatment of animals exposed to doxorubicin translated into significant functional improvements. In contrast to a progressive deterioration of diastolic and systolic function observed in the DOXO group , 4 week treatment with ranolazine led to a relief in diastolic dysfunction and a minor worsening of systolic function with time. The prevention of the progression towards the failing phenotype was reflected by better general conditions and increase in survival rate. Afterwards, molecular studies were performed to explore the possible underlying mechanisms.
Figure 8.
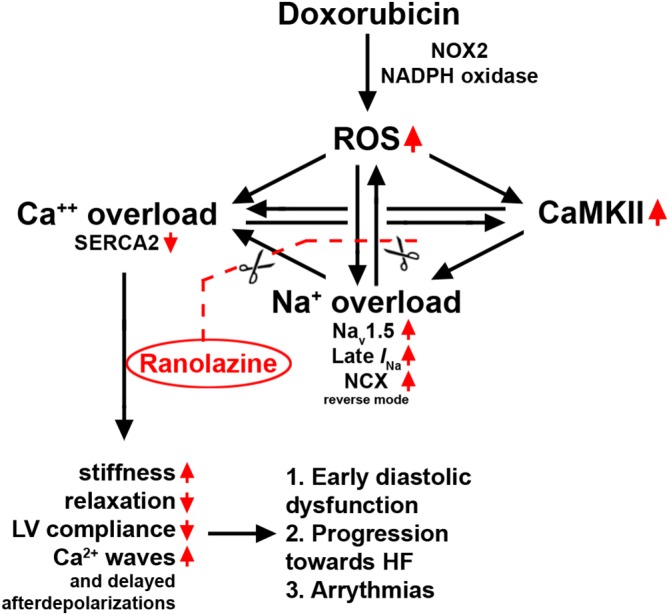
Schematic illustration of the positive feedback loops leading to to Ca++ overload, via increased late I Na, and the consequent cardiotoxicity, induced by doxorubicin. The possible sites of action of ranolazine, on Nav1.5 channels, to break this vicious cycle are also shown.
Due to the multifactorial nature of the pathogenesis of anthracycline cardiotoxicity, with a wide spectrum of molecular mechanisms involved, doxorubicin cardiotoxicity is still an unsolved riddle, where the cardiac phenotype is a result of processes that are reciprocally linked, often in a self‐propelling manner. The beneficial effects of ranolazine discussed above can be explained, at least in part, by our observations. Treatment with ranolazine reduced NCX1 and increased SERCA2 expression and SERCA2/PLB ratio. Moreover, expression of myocardial Nav 1.5 channels was restored to normal levels. In parallel, acute application of ranolazine to doxorubicin‐treated cardiomyocytes reduced late I Na to the level of control cells. These data suggest that ranolazine treatment, by counteracting doxorubicin‐induced increase in late I Na, was able to reverse the functional consequences of doxorubicin on cardiomyocytes, both directly (by normalizing Ca2+ fluxes) and indirectly (by normalizing the expression level of channels and transporters).
Changes in the expression of ion channels and Ca2+‐transporters by ranolazine treatment were likely to be the consequence of the reduced phosphorylation of CaMKII. Conversely, elevated p‐CaMKII and ox‐CaMKII levels in the DOXO group indicated a continuing CaMKII activation that may be driven by the persistent oxidative/nitrative stress, elevated long after the clearance of doxorubicin and its metabolites. A challenging hypothesis is that doxorubicin can be considered as a pharmacological stressor that leaves its ROS‐dependent “molecular signature” and triggers a chronic oxidative vicious cycle. A long‐lasting augmented ROS production may be implemented by NADPH oxidases, which are implicated in the development of chronic anthracycline cardiotoxicity (Deng et al., 2007; Octavia et al., 2012; Stěrba et al., 2013). In our experiments, four weeks after the last administration of doxorubicin, increased NOX2 and NOX4 were still evident, confirming preclinical and clinical studies of the importance of NADPH oxidases in doxorubicin‐induced cardiotoxicity (Toko et al., 2002; Pacher et al., 2003; Mizutani et al., 2005; Spallarossa et al., 2006; De Falco et al., 2016). Genetic disruption of NOX2 is known to protect against doxorubicin cardiotoxicity, while doxorubicin‐treated wild‐type mice display NOX2 and NOX4 up‐regulation and increased oxidase activity (Deng et al., 2007; Zhao et al., 2010). Notably, the association between cardiotoxicity and gene variants in the subunits of human NADPH oxidases reinforce the relevance of these enzymes in anthracycline cardiomyopathy (Wojnowski et al., 2005; Reichwagen et al., 2015). By counteracting doxorubicin‐induced increases of NOX2 and NOX4 expression, ranolazine positively affected late consequences of doxorubicin, supporting the early initiation of ranolazine treatment to prevent the development of late anthracycline cardiomyopathy.
Dysregulation of ion homeostasis and myocardial oxidative stress by doxorubicin have structural outcomes and influence the remodelling process. Treatment with ranolazine lowered MPP2 expression and activity along with a reduction in collagen content. The anti‐fibrotic effects of ranolazine may be the consequence of the reduced CaMKII activation, as CaMKII is among the main drivers of fibrotic remodelling in cardiac diseases (Kreusser and Backs, 2014).
Our observation that administration of ranolazine in healthy animals did not alter systolic and diastolic markers is compatible with the notion that, in the absence of disease, the amplitude of late I Na is small and its inhibition is not expected to change the function of the normal heart. Additionally, ranolazine did not modify late I Na in vehicle‐treated cardiomyocytes, while it markedly reduced the enhanced current measured in cells treated with doxorubicin. These data are supported by several studies showing the neutral effect of ranolazine on healthy animals or cardiomyocytes from normal hearts (Sabbah et al., 2002; Undrovinas et al., 2010; Coppini et al., 2013a; Tocchetti et al., 2014).
Limitations
We are aware that our study has several limitations. First, the direct link between doxorubicin and late I Na in vivo is missing, although the electrophysiological experiments performed in vitro showed that doxorubicin is able to raise late I Na in cardiomyocytes, thus demonstrating a mechanism by which ranolazine, by restoring a normal amplitude of late I Na, counteracts doxorubicin toxicity. These in vitro findings represent an indirect link, but they support the mechanistic hypothesis and strengthen the rationale of our study.
Second, the use of DHE for targeting superoxide anion generation has several technical limitations. DHE can undergo non‐specific oxidation by more than one oxidant, such as peroxynitrite or hydroxyl radical, which reduces its specificity as an index of superoxide anion. Despite this, the DHE assay is considered suitable for qualitative assessment and thus provides relevant information about ROS production. Because our study aimed at testing the effects of ranolazine on diastolic dysfunction in the early stages of doxorubicin‐induced HF, we did not identify the contribution of each specific oxidant to in the mediation of doxorubicin effects in the heart. DHE was used as one of the approaches that, together with NADPH oxidase and nitrotyrosine, have shown the already well‐documented involvement of ROS and RNS in doxorubicin cardiotoxicity.
Finally, given the recognized complexity of known (and unknown) aspects of doxorubicin‐induced HF, where a dissection of a single contributing event has been challenging for decades, the identification of a particular ‘master mechanism’ although ambitious, was not pursued. We tested a clinically relevant intervention that may interrupt one of the arms of the vicious cycle triggered by doxorubicin and explored the mechanistic aspects that can explain our findings. As with other individual studies on doxorubicin cardiotoxicity, our work represents a fragmentary view of a far more complex general picture and by necessity have to be viewed as “an attempt” to approach a definitive answer.
Conclusions
In the search for early interventions that may change an unfavourable course of doxorubicin cardiomyopathy, the importance of early diastolic dysfunction should not be underestimated, as it could serve not only as a tool for the prompt detection of cardiac damage in cancer patients but may also represent a therapeutic target. The use of ranolazine as a cardioprotective agent to reverse diastolic dysfunction may be viewed as a potential secondary prevention for late anthracycline cardiotoxicity. An interesting aspect of the potential use of ranolazine in oncological settings could also emerge from the current research showing that drugs targeting voltage‐gated Na+ channels may have anti‐invasion and anti‐metastatic effects (Djamgoz and Onkal, 2013; Driffort et al., 2014; Koltai, 2015).
Author contributions
D.C., A.D.A. and K.U. conceived the study, designed the experiments, wrote the paper, performed in vivo experiments and analysed data. G.E. designed the experiments, performed immunofluorescence and molecular biology experiments, and analysed data. R.C. designed and performed patch‐clamp experiments, analysed data and revised the manuscript. E.P., R.R., L.P.C., A.R., C.R. and C.S. performed histochemistry and molecular biology experiments and analysed data. L.S. isolated rat cardiomyocytes, performed patch‐clamp experiments, and analysed data. F.R. and L.B. revised the manuscript and gave final approval to the publication. All authors approved the final draft of the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
This work was partly supported by ‘Giovani Ricercatori’ grant entitled ‘Valutazione degli effetti cardioprotettivi della ranolazina in un modello animale di disfunzione diastolica indotta da doxorubicina’ from the Department of Experimental Medicine, University of Campania ‘Luigi Vanvitelli’; ‘Cardiotossicità da Doxorubicina: meccanismi fisiopatologici, indici precoci di danno miocardiaco e terapie innovative’ from Regione Campania, L.R. N.5/2002.
Cappetta, D. , Esposito, G. , Coppini, R. , Piegari, E. , Russo, R. , Ciuffreda, L. P. , Rivellino, A. , Santini, L. , Rafaniello, C. , Scavone, C. , Rossi, F. , Berrino, L. , Urbanek, K. , and De Angelis, A. (2017) Effects of ranolazine in a model of doxorubicin‐induced left ventricle diastolic dysfunction. British Journal of Pharmacology, 174: 3696–3712. doi: 10.1111/bph.13791.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The concise guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Yoguchi A, Takizawa T, Yokoyama T, Kanda T, Kurabayashi M et al. (2000). Mechanism of doxorubicin‐induced inhibition of sarcoplasmic reticulum Ca(2+)‐ATPase gene transcription. Circ Res 86: 8–14. https://doi.org/10.1161/01.RES.86.1.8. [DOI] [PubMed] [Google Scholar]
- Banerjee K, Ghosh RK, Kamatam S, Banerjee A, Gupta A (2017). Role of ranolazine in cardiovascular disease and diabetes: exploring beyond angina. Int J Cardiol 227: 556–564. https://doi.org/10.1016/j.ijcard.2016.10.102. [DOI] [PubMed] [Google Scholar]
- Cappetta D, Esposito G, Piegari E, Russo R, Ciuffreda LP, Rivellino A et al. (2016). SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol 205: 99–110. https://doi.org/10.1016/j.ijcard.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Cardinale D, Colombo A, Lamantia G, Colombo N, Civelli M, De Giacomi G et al. (2010). Anthracycline‐induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. J Am Coll Cardiol 55: 213–220. https://doi.org/10.1016/j.jacc.2009.03.095. [DOI] [PubMed] [Google Scholar]
- Carver JR, Shapiro CL, Ng A, Jacobs L, Schwartz C, Virgo KS et al. (2007). American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J Clin Oncol 25: 3991–4008. https://doi.org/10.1200/JCO.2007.10.9777. [DOI] [PubMed] [Google Scholar]
- Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F et al. (2013a). Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127: 575–584. https://doi.org/10.1161/CIRCULATIONAHA.112.134932. [DOI] [PubMed] [Google Scholar]
- Coppini R, Ferrantini C, Mazzoni L, Sartiani L, Olivotto I, Poggesi C et al. (2013b). Regulation of intracellular Na(+) in health and disease: pathophysiological mechanisms and implications for treatment. Glob Cardiol Sci Pract 2013: 222–242. https://doi.org/10.5339/gcsp.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi F, Paolini L, De Caterina R (2014). Ranolazine in the prevention of anthracycline cardiotoxicity. Pharmacol Res 79: 88–102. https://doi.org/10.1016/j.phrs.2013.11.001. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. https://doi.org/10.1111/bph.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis A, Cappetta D, Piegari E, Rinaldi B, Ciuffreda LP, Esposito G et al. (2016). Long‐term administration of ranolazine attenuates diastolic dysfunction and adverse myocardial remodeling in a model of heart failure with preserved ejection fraction. Int J Cardiol 217: 69–79. https://doi.org/10.1016/j.ijcard.2016.04.168. [DOI] [PubMed] [Google Scholar]
- De Angelis A, Piegari E, Cappetta D, Russo R, Esposito G, Ciuffreda LP et al. (2015). SIRT1 activation rescues doxorubicin‐induced loss of functional competence of human cardiac progenitor cells. Int J Cardiol 189: 30–44. https://doi.org/10.1016/j.ijcard.2015.03.438. [DOI] [PubMed] [Google Scholar]
- De Angelis A, Rinaldi B, Capuano A, Rossi F, Filippelli A (2004). Indomethacin potentiates acetylcholine‐induced vasodilation by increasing free radical production. Br J Pharmacol 142: 1233–1240. https://doi.org/10.1038/sj.bjp.0705877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Falco E, Carnevale R, Pagano F, Chimenti I, Fianchini L, Bordin A et al. (2016). Role of NOX2 in mediating doxorubicin‐induced senescence in human endothelial progenitor cells. Mech Ageing Dev 159: 37–43. https://doi.org/10.1016/j.mad.2016.05.004. [DOI] [PubMed] [Google Scholar]
- Deng S, Kruger A, Kleschyov AL, Kalinowski L, Daiber A, Wojnowski L (2007). Gp91phox‐containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic Biol Med 42: 466–473. https://doi.org/10.1016/j.freeradbiomed.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Di Meglio F, Nurzynska D, Castaldo C, Miraglia R, Romano V, De Angelis A et al. (2012). Cardiac shock wave therapy: assessment of safety and new insights into mechanisms of tissue regeneration. J Cell Mol Med 16: 936–942. https://doi.org/10.1111/j.1582‐4934.2011.01393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djamgoz MB, Onkal R (2013). Persistent current blockers of voltage‐gated sodium channels: a clinical opportunity for controlling metastatic disease. Recent Pat Anticancer Drug Discov 8: 66–84. https://doi.org/10.2174/1574892811308010066. [DOI] [PubMed] [Google Scholar]
- Driffort V, Gillet L, Bon E, Marionneau‐Lambot S, Oullier T, Joulin V, Collin C et al. (2014) Ranolazine inhibits NaV1.5‐mediated breast cancer cell invasiveness and lung colonization. Mol Cancer 13:264. https://doi.org/10.1186/1476‐4598‐13‐264 [DOI] [PMC free article] [PubMed]
- Elli M, Sungur M, Genç G, Ayyildiz P, Dagdemir A, Pinarli FG et al. (2013). The late effects of anticancer therapy after childhood Wilm's tumor: the role of diastolic function and ambulatory blood pressure monitoring. Jpn J Clin Oncol 43: 1004–1011. https://doi.org/10.1093/jjco/hyt105. [DOI] [PubMed] [Google Scholar]
- Erickson JR, He BJ, Grumbach IM, Anderson ME (2011). CaMKII in the cardiovascular system: sensing redox states. Physiol Rev 91: 889–915. https://doi.org/10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV et al. (2008). A dynamic pathway for calcium‐independent activation of CaMKII by methionine oxidation. Cell 133: 462–474. https://doi.org/10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrantini C, Coppini R, Sacconi L, Tosi B, Zhang ML, Wang GL, de Vries E et al. (2014) Impact of detubulation on force and kinetics of cardiac muscle contraction. J Gen Physiol143:783–797. https://doi.org/10.1085/jgp.201311125 [DOI] [PMC free article] [PubMed]
- Figueredo VM, Pressman GS, Romero‐Corral A, Murdock E, Holderbach P, Morris DL (2011). Improvement in left ventricular systolic and diastolic performance during ranolazine treatment in patients with stable angina. J Cardiovasc Pharmacol Ther 16: 168–172. https://doi.org/10.1177/1074248410382105. [DOI] [PubMed] [Google Scholar]
- Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC et al. (2006). Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol 47: 817–826. https://doi.org/10.1016/j.jacc.2005.09.051. [DOI] [PubMed] [Google Scholar]
- Hwang H, Arcidi JM Jr, Hale SL, Simkhovich BZ, Belardinelli L, Dhalla AK et al. (2009). Ranolazine as a cardioplegia additive improves recovery of diastolic function in isolated rat hearts. Circulation 120: S16–S21. https://doi.org/10.1161/CIRCULATIONAHA.108.844167. [DOI] [PubMed] [Google Scholar]
- Ishigaki T, Yoshida T, Izumi H, Fujisawa Y, Shimizu S, Masuda K et al. (2015). Different implication of elevated B‐type natriuretic peptide level in patients with heart failure with preserved ejection fraction and in those with reduced ejection fraction. Echocardiography 32: 623–629. https://doi.org/10.1111/echo.12707. [DOI] [PubMed] [Google Scholar]
- Jerling M (2006). Clinical pharmacokinetics of ranolazine. Clin Pharmacokinet 45: 469–491. https://doi.org/10.2165/00003088‐200645050‐00003. [DOI] [PubMed] [Google Scholar]
- Kass DA, Bronzwaer JG, Paulus WJ (2004). What mechanisms underlie diastolic dysfunction in heart failure? Circ Res 94: 1533–1542. https://doi.org/10.1161/01.RES.0000129254.25507.d6. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. https://doi.org/10.1111/j.1476‐5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler AC, Sag CM, Maier LS (2014). Reactive oxygen species and excitation‐contraction coupling in the context of cardiac pathology. J Mol Cell Cardiol 73: 92–102. https://doi.org/10.1016/j.yjmcc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M et al. (2010). Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 121: 1606–1613. https://doi.org/10.1161/CIRCULATIONAHA.109.914911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltai T (2015) Voltage‐gated sodium channel as a target for metastatic risk reduction with re‐purposed drugs. F1000Res 4:297. https://doi.org/10.12688/f1000research.6789.1 [DOI] [PMC free article] [PubMed]
- Kreusser MM, Backs J (2014) Integrated mechanisms of CaMKII‐dependent ventricular remodeling. Front Pharmacol5:36. https://doi.org/10.3389/fphar.2014.00036 [DOI] [PMC free article] [PubMed]
- Lenneman CG, Sawyer DB (2016). Cardio‐oncology: an update on cardiotoxicity of cancer‐related treatment. Circ Res 118: 1008–1020. https://doi.org/10.1161/CIRCRESAHA.115.303633. [DOI] [PubMed] [Google Scholar]
- Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD et al. (2005). Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol 23: 2629–2636. https://doi.org/10.1200/JCO.2005.12.121. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. https://doi.org/10.1111/bph.12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menna P, Paz OG, Chello M, Covino E, Salvatorelli E, Minotti G (2012). Anthracycline cardiotoxicity. Expert Opin Drug Saf 1: S21–S36. https://doi.org/10.1517/14740338.2011.589834. [DOI] [PubMed] [Google Scholar]
- Mihos CG, Krishna RK, Kherada N, Larrauri‐Reyes M, Tolentino A, Santana O (2016). The use of ranolazine in non‐anginal cardiovascular disorders: a review of current data and ongoing randomized clinical trials. Pharmacol Res 103: 49–55. https://doi.org/10.1016/j.phrs.2015.10.018. [DOI] [PubMed] [Google Scholar]
- Minotti G (2013). Pharmacology at work for cardio‐oncology: ranolazine to treat early cardiotoxicity induced by antitumor drugs. J Pharmacol Exp Ther 346: 343–349. https://doi.org/10.1124/jpet.113.204057. [DOI] [PubMed] [Google Scholar]
- Mizutani H, Tada‐Oikawa S, Hiraku Y, Kojima M, Kawanishi S (2005). Mechanism of apoptosis induced by doxorubicin through the generation of hydrogen peroxide. Life Sci 76: 1439–1453. https://doi.org/10.1016/j.lfs.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Neilan TG, Blake SL, Ichinose F, Raher MJ, Buys ES, Jassal DS et al. (2007). Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 116: 506–514. https://doi.org/10.1161/CIRCULATIONAHA.106.652339. [DOI] [PubMed] [Google Scholar]
- Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL (2012). Doxorubicin‐induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol 52: 1213–1225. https://doi.org/10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Pacher P, Liaudet L, Bai P, Mabley JG, Kaminski PM, Virág L et al. (2003). Potent metalloporphyrin peroxynitrite decomposition catalyst protects against the development of doxorubicin‐induced cardiac dysfunction. Circulation 107: 896–904. https://doi.org/10.1161/01.CIR.0000048192.52098. [DOI] [PubMed] [Google Scholar]
- Pacini L, Suffredini S, Ponti D, Coppini R, Frati G, Ragona G, Cerbai E, Calogero A (2013) Altered calcium regulation in isolated cardiomyocytes from Egr‐1 knock‐out mice. Can J Physiol Pharmacol91:1135–1142. https://doi.org/10.1139/cjpp‐2012‐0419 [DOI] [PubMed]
- Paulus WJ, Tschöpe C (2013). A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62: 263–271. https://doi.org/10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer‐Crosbie M et al. (2014). Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 27: 911–939. https://doi.org/10.1016/j.echo.2014.07.012. [DOI] [PubMed] [Google Scholar]
- Prestor VV, Rakovec P, Kozelj M, Jereb B (2000). Late cardiac damage of anthracycline therapy for acute lymphoblastic leukemia in childhood. Pediatr Hematol Oncol 17: 527–540. https://doi.org/10.1080/08880010050122799. [DOI] [PubMed] [Google Scholar]
- Raj S, Franco VI, Lipshultz SE (2014). Anthracycline‐induced cardiotoxicity: a review of pathophysiology, diagnosis, and treatment. Curr Treat Options Cardiovasc Med 16: 315 https://doi.org/10.1007/s11936‐014‐0315‐4. [DOI] [PubMed] [Google Scholar]
- Reagan‐Shaw S, Nihal M, Ahmad N (2008). Dose translation from animal to human studies revisited. FASEB J 22: 659–661. https://doi.org/10.1096/fj.07‐9574LSF. [DOI] [PubMed] [Google Scholar]
- Reichwagen A, Ziepert M, Kreuz M, Gödtel‐Armbrust U, Rixecker T, Poeschel V et al. (2015). Association of NADPH oxidase polymorphisms with anthracycline‐induced cardiotoxicity in the RICOVER‐60 trial of patients with aggressive CD20(+) B‐cell lymphoma. Pharmacogenomics 16: 361–372. https://doi.org/10.2217/pgs.14.179. [DOI] [PubMed] [Google Scholar]
- Sabbah HN, Chandler MP, Mishima T, Suzuki G, Chaudhry P, Nass O et al. (2002). Ranolazine, a partial fatty acid oxidation (pFOX) inhibitor, improves left ventricular function in dogs with chronic heart failure. J Card Fail 8: 416–422. https://doi.org/10.1054/jcaf.2002.129232. [DOI] [PubMed] [Google Scholar]
- Sag CM, Köhler AC, Anderson ME, Backs J, Maier LS (2011). CaMKII‐dependent SR Ca leak contributes to doxorubicin‐induced impaired Ca handling in isolated cardiac myocytes. J Mol Cell Cardiol 51: 749–759. https://doi.org/10.1016/j.yjmcc.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatorelli E, Menna P, Cantalupo E, Chello M, Covino E, Wolf FI et al. (2015). The concomitant management of cancer therapy and cardiac therapy. Biochim Biophys Acta 1848: 2727–2737. https://doi.org/10.1016/j.bbamem.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Schimmel KJ, Richel DJ, van den Brink RB, Guchelaar HJ (2004). Cardiotoxicity of cytotoxic drugs. Cancer Treat Rev 30: 181–191. https://doi.org/10.1016/j.ctrv.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Serrano JM, González I, Del Castillo S, Muñiz J, Morales LJ, Moreno F et al. (2015). Diastolic dysfunction following anthracycline‐based chemotherapy in breast cancer patients: incidence and predictors. Oncologist 20: 864–872. https://doi.org/10.1634/theoncologist.2014‐0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S, Maurer U, Schotola H, Hartmann N, Didié M, Zimmermann WH et al. (2011). Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIdelta(C) can be reversed by inhibition of late Na(+) current. Basic Res Cardiol 106: 263–272. https://doi.org/10.1007/s00395‐010‐0136‐x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K et al. (2010). Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J Am Coll Cardiol 55: 2330–2342. https://doi.org/10.1016/j.jacc.2009.12.055. [DOI] [PubMed] [Google Scholar]
- Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schöndube FA et al. (2008). Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol 45: 32–43. https://doi.org/10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spallarossa P, Altieri P, Garibaldi S, Ghigliotti G, Barisione C, Manca V et al. (2006). Matrix metalloproteinase‐2 and ‐9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc Res 69: 736–745. https://doi.org/10.1016/j.cardiores.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Stěrba M, Popelová O, Vávrová A, Jirkovský E, Kovaříková P, Geršl V et al. (2013). Oxidative stress, redox signaling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 18: 899–929. https://doi.org/10.1089/ars.2012.4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tocchetti CG, Carpi A, Coppola C, Quintavalle C, Rea D, Campesan M et al. (2014). Ranolazine protects from doxorubicin‐induced oxidative stress and cardiac dysfunction. Eur J Heart Fail 16: 358–366. https://doi.org/10.1002/ejhf.50. [DOI] [PubMed] [Google Scholar]
- Toko H, Oka T, Zou Y, Sakamoto M, Mizukami M, Sano M et al. (2002). Angiotensin II type 1a receptor mediates doxorubicin‐induced cardiomyopathy. Hypertens Res 25: 597–603 https://doi.org/10.1291/hypres.25.597. [DOI] [PubMed] [Google Scholar]
- Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN, Undrovinas A (2010). Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci 60: 245–257. https://doi.org/10.1007/s12576‐010‐0092‐0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vejpongsa P, Yeh ET (2014). Prevention of anthracycline‐induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol 64: 938–945. https://doi.org/10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T et al. (2011). Reactive oxygen species‐activated Ca/calmodulin kinase IIδ is required for late I (Na) augmentation leading to cellular Na and Ca overload. Circ Res 108: 555–565. https://doi.org/10.1161/CIRCRESAHA.110.221911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P et al. (2006). Ca2+/calmodulin‐dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest 116: 3127–3138. https://doi.org/10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilde S, Jetter A, Rietbrock S, Kasel D, Engert A, Josting A et al. (2007). Population pharmacokinetics of the BEACOPP polychemotherapy regimen in Hodgkin's lymphoma and its effect on myelotoxicity. Clin Pharmacokinet 46: 319–333. https://doi.org/10.2165/00003088‐200746040‐00005. [DOI] [PubMed] [Google Scholar]
- Williams S, Pourrier M, McAfee D, Lin S, Fedida D (2014). Ranolazine improves diastolic function in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 306: H867–H881. https://doi.org/10.1152/ajpheart.00704.2013. [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Kulle B, Schirmer M, Schlüter G, Schmidt A, Rosenberger A et al. (2005). NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin‐induced cardiotoxicity. Circulation 112: 3754–3762. https://doi.org/10.1161/CIRCULATIONAHA.105.576850. [DOI] [PubMed] [Google Scholar]
- Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM et al. (2010). Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res 70: 9287–9297. https://doi.org/10.1158/0008‐5472.CAN‐10‐2664. [DOI] [PMC free article] [PubMed] [Google Scholar]