

Abstract
Background and Purpose
Idiopathic pulmonary fibrosis (IPF) is a fatal respiratory disease characterized by excessive fibroblast activation ultimately leading to scarring of the lungs. Although, the activation of β2‐adrenoceptors (β2‐AR) has been shown to inhibit pro‐fibrotic events primarily in cell lines, the role of β2‐adrenoceptor agonists has not yet been fully characterized. The aim of our study was to explore the anti‐fibrotic activity of the long‐acting β2‐adrenoceptor agonist olodaterol in primary human lung fibroblasts (HLF) and in murine models of pulmonary fibrosis.
Experimental Approach
We assessed the activity of olodaterol to inhibit various pro‐fibrotic mechanisms, induced by different pro‐fibrotic mediators, in primary HLF from control donors and patients with IPF (IPF‐LF). The in vivo anti‐fibrotic activity of olodaterol, given once daily by inhalation in either a preventive or therapeutic treatment regimen, was explored in murine models of lung fibrosis induced by either bleomycin or the overexpression of TGF‐β1.
Key Results
In both HLF and IPF‐LF, olodaterol attenuated TGF‐β‐induced expression of α‐smooth muscle actin, fibronectin and endothelin‐1 (ET‐1), FGF‐ and PDGF‐induced motility and proliferation and TGF‐β/ET‐1‐induced contraction. In vivo olodaterol significantly attenuated the bleomycin‐induced increase in lung weight, reduced bronchoalveolar lavage cell counts and inhibited release of pro‐fibrotic mediators (TGF‐ß, MMP‐9 and tissue inhibitor of metalloproteinase‐1). Forced vital capacity was increased only with the preventive treatment regimen. In the TGF‐β‐overexpressing model, olodaterol additionally reduced the Col3A1 mRNA expression.
Conclusion and Implications
Olodaterol showed anti‐fibrotic properties in primary HLF from control and IPF patients and in murine models of lung fibrosis.
Abbreviations
- BALF
bronchoalveolar lavage fluid
- COPD
chronic obstructive pulmonary disease
- ECM
extracellular matrix
- ET‐1
endothelin‐1
- FCS
fetal calf serum
- FVC
forced vital capacity
- HLF
human lung fibroblast
- IPF(‐LF)
idiopathic pulmonary fibrosis (lung fibroblasts)
- α‐SMA
α‐smooth muscle actin
Introduction
Idiopathic pulmonary fibrosis (IPF) is characterized by an irreversible decrease in oxygen diffusion capacity that is caused by a replacement of normal lung parenchyma by fibrotic scar tissue (Raghu et al., 2011). Although the exact pathogenic processes and pathways underlying the initiation and progression of IPF are not yet fully understood, it is well known that the major player in the process of lung stiffening is the myofibroblast. Under fibrotic conditions, myofibroblasts differentiate from resident fibroblasts, which in turn exhibit higher proliferation rates. Myofibroblasts are characterized by the production of excess extracellular matrix (ECM) and by the expression of α‐smooth muscle actin (α‐SMA), which confers contractile properties on the cells. The recruitment of circulating mesenchymal precursor cells, namely, fibrocytes (Kuwana et al., 2003), as well as epithelial cell trans‐differentiation (Todd et al., 2012) may also provide a source of myofibroblasts. One of the main pro‐fibrotic mediators, activating these cells and stimulating their differentiation towards ECM protein producing cells, is TGF‐β (Desmouliere et al., 1993; Swigris and Brown, 2010). Other prominent growth factors including PDGF‐BB, FGF‐2 and EGF stimulate the proliferation and recruitment of fibroblasts (Allen and Spiteri, 2002). Further, the vasoconstrictor peptide endothelin‐1 (ET‐1) participates in the pathogenesis of pulmonary fibrosis (Shi‐Wen et al., 2006; Swigris and Brown, 2010).
Recently, the tyrosine kinase inhibitor nintedanib (Ofev®, Boehringer Ingelheim), targeting the receptors for PDGF (PDGFR), FGF (FGFR) and VEGF (VEGFR) (Hilberg et al., 2008; Richeldi et al., 2014), and pirfenidone (Esbriet®, Roche), with known anti‐fibrotic and anti‐inflammatory properties (Noble et al., 2011), were shown to slow disease progression in patients with IPF, resulting in market authorization in many countries including the US and EU. While both drugs are able to significantly slow the pace of disease progression, for many patients, symptoms, like dyspnoea and cough, are still debilitating. Therefore, the need for additional treatment options is still high (Borie et al., 2016).
Agonists at β2‐adrenoceptors are well‐established therapeutic options for the maintenance treatment of chronic obstructive pulmonary disease (COPD). Upon agonist binding to the receptor, intracellular cAMP is synthesized. Besides their well‐proven bronchodilatory properties, β2‐adrenoceptor agonists, as well as other agents that increase cAMP levels, were shown to attenuate various pro‐fibrotic mechanisms, such as proliferation or collagen and α‐SMA expression, in cultured fibroblasts (Liu et al., 2004; Schiller et al., 2010; Lamyel et al., 2011). Inhibition of PDEs was also shown to inhibit TGF‐β‐induced fibrotic events in fibroblasts (Dunkern et al., 2007), and the PDE4 inhibitor roflumilast was shown to attenuate bleomycin‐induced lung fibrosis in mice (Cortijo et al., 2009). This indicates that cAMP elevating agents might also have therapeutic potential for the treatment of pulmonary fibrosis.
The primary aim of our study was to assess the ability of olodaterol (Striverdi®, Boehringer Ingelheim), an inhaled long‐acting β2‐adrenoceptor agonist approved for the once‐daily maintenance treatment of COPD (Bouyssou et al., 2010a; van Noord et al., 2011), to inhibit pathogenic mechanisms involved in lung fibrosis. For this purpose, we performed different in vitro assays, including fibroblast proliferation and migration, myofibroblast differentiation and pro‐fibrotic mediator release using primary human lung cells from patients with IPF and control donors. To further confirm our data in the in vivo situation, we explored the activity of olodaterol in murine models of lung fibrosis.
Methods
All assays performed comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Culture conditions and assay set up
Normal human lung fibroblasts (HLF) (Lonza, Walkersville, MD, USA) from control donors and fibroblasts from donors with IPF (IPF‐LF) (Asterand, Detroit, MI, USA) were grown in fibroblast basal medium (FBM) (CC‐3131, Lonza Walkersville, Inc., Walkersville, MD, USA) supplemented with FGM‐2 SingleQuot Kit Supplements & Growth Factors (CC‐4126, Lonza). Cells were grown in a humidified incubator at 37°C and 5% CO2 and passaged by trypsinization (ReagentPack Subculture Reagents, CC‐5034, Lonza) at approximately 80% confluence. All assays were performed at passage 7 or 8.
For assay set‐up, cells were seeded in fibroblast growth medium plus supplements in assay relevant densities. For all TGF‐β‐stimulated assays and the cAMP assay, cells were seeded at 4500 cells per well in 96‐well plates. Proliferation assays were performed in 96‐well plates at an initial seeding cell density on day 0 of 2000 cells per well. Cell motility assays were performed with 750 cells per well in 12‐well plates. After 24 h, the cell culture medium was changed to starvation medium (FBM without supplements).
Compound treatment
After a 24 h starvation period, cells were pre‐incubated for 30 min with different concentrations of olodaterol and subsequently stimulated with the assay relevant stimulus for the indicated time in the presence of the compound.
cAMP assay
Cells were stimulated with ICI‐118,551 (30 nM), CGP‐20712A (100 nM) or stimulation buffer (containing IBMX) at 37°C for 30 min. After antagonist pre‐incubation, various olodaterol concentrations (1 pM to 1 μM in stimulation buffer) were added for another 30 min. cAMP release was determined with the cAMP‐Glo™ assay kit (Promega, Madison, WI, USA) according to the instruction manual.
α‐SMA Western blot replacement assay
For determination of α‐SMA expression, cells were stimulated with TGF‐β (4 ng·mL−1) for 48 h. Lysates were assayed in a Western blot replacement assay [Meso Scale Discovery (MSD), Rockville, MD, USA]. In short, lysates were pipetted onto high binding plates (MSD) and were sequentially incubated with a mouse anti‐human α‐SMA‐antibody (Sigma Aldrich, Deisenhofen, Germany) and an anti‐mouse sulfotag‐antibody (MSD). For analysis, a MSD sector imager (MSD) was used.
Immunocytochemistry
For determination of collagen I assembly, the ‘Scar‐in‐a‐Jar’ method was used as described elsewhere (Chen et al., 2009). Briefly, fibroblasts were grown on tissue culture (TC)‐treated glass culture slides (Becton Dickinson, Heidelberg, Germany) at 50 000 cells per well. After a starving period of 24 h, cells were cultured under a mixture of 70 and 400 kDa Ficoll™ in DMEM, supplemented with 100 μM L‐ascorbic acid 2‐phosphate in presence of 10 nM olodaterol and 5 ng·mL−1 TGF‐β for 72 h. Cell layers were fixed with 70% ethanol and blocked with 1% FCS in PBS. The slides were incubated for 3 h at 37°C with a mouse anti‐human collagen type I antibody (Sigma) followed by the incubation with the secondary antibody (AlexaFluor 488 goat anti‐mouse, Life Technologies) for 1 h at 37°C. The slides were mounted with ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher, Waltham, MA, USA) and dried overnight before pictures were taken.
elisas
Cells were stimulated with TGF‐β (4 ng·mL−1) for 48 h. Supernatants were used to determine fibrotic cytokines using appropriate elisas. Quantikine human ET‐1 elisa was from R&D Systems, Inc. (Minneapolis, MN, USA). Human fibronectin platinum elisa was from eBioscience (San Diego, CA, USA). Human Pro‐collagen I Peptide elisa was from Takara Bio Inc (Kusatsu Shiga, Japan).
Unstimulated, starved cells, lysed in RIPA buffer (Sigma) plus protease‐inhibitor (Thermo Fisher, Waltham, MA, USA), were used for the human β2‐adrenoceptor elisa kit (Cusabio biotech CO, Wuhan, China). All elisas were performed according to the instruction manual.
Proliferation assay
Cells were stimulated with PDGF‐BB (50 ng·mL−1), FGF‐basic (20 ng·mL−1), EGF (3 ng·mL−1) or FCS (1%). After 72 h of stimulation, 10‐times concentrated BrdU was added to the stimulus medium and the cells were incubated for another 18 h. Cell proliferation was assessed by BrdU assay (Roche, Mannheim, Germany) according to the instruction manual.
Motility assay
Cells were stimulated with 20 ng·mL−1 PDGF‐BB or FGF‐basic. The motility of the cells was determined by time‐lapse microscopy using a Cell‐IQ® imaging system (Cenibra life science solution, Bramsche, Germany) and manual cell tracking performed with the Cell‐IQ® analyser software. Cells were tracked continuously for approximately 72 h. Fifteen cells were analysed in each well, and the mean values of the cell movement velocity (pixel·h−1) were calculated for each well. In each experiment, three wells were sampled per condition.
Contraction assay
Collagen matrices in 48‐well plates were prepared using 500 μL of a mixture containing 1.7 × 105 fibroblasts and 1 mg·mL−1 bovine type I collagen per well (PureCol®, Advanced BioMatrix, Carlsbad, CA, USA) in DMEM medium without serum. Polymerization of collagen matrices lasted for 60 min at 37°C. To initiate lattice contraction, FBM medium was added and freshly polymerized matrices were released from the underlying culture dish by using a spatula. The gels were then incubated at 37°C in a 5% CO2 atmosphere and photographed after 24 h (starting area). Treatment with different concentrations of olodaterol and subsequent stimulation with TGF‐β (5 ng·mL−1) + ET‐1 (100 ng·mL−1) was performed for 72 h. The area of each gel was calculated using imaging software analysis. Data are expressed as the percentage of area compared with the original gel size and normalized to maximum contraction.
Western blot analysis
Cells were seeded at 4 × 105 cells per well in a 12‐well plate. After the starvation period, cells were pre‐incubated with different concentrations of olodaterol and subsequently stimulated with FGF (20 ng·mL−1) for 5 min. For gel electrophoresis, the XCell SureLock™ Mini‐Cell System (Invitrogen) and NuPAGE 4–12% Bis‐Tris Gels (Invitrogen) were used. Proteins were blotted in the Mini Trans‐Blot Cell system (Bio‐Rad Laboratories). Nitrocellulose membranes were blocked with Western blocking buffer for 30–60 min and subsequently incubated overnight at 4°C with the respective primary antibody. The next day, membranes were incubated for 90 min at room temperature with the respective secondary antibody. All primary antibodies were diluted 1:1000 in 5% BSA in TBS. The secondary antibody was diluted 1:5000 in TBS. Membranes were developed using SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher, Waltham, MA, USA) and analysed with the Stella 3200 analyser (Raytest, Straubenhardt, Germany). Bands were quantified by intensity/area‐background (%) with the AIDA 422 software (Raytest). Bands were normalized to GAPDH after stripping the membranes.
Bleomycin‐ and AAV6.2m‐TGF‐β‐induced fibrosis model
Animals
All animal care and experimental procedures complied with German national guidelines and legal regulations and were approved by the ethical committee of the Regierungspräsidium Tübingen (Germany) (Permit No. 35/9185.81‐8/12‐012 and /12‐030). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Ten‐ to twelve‐week‐old male C57BL/6 mice were obtained from Charles River Laboratories (Sulzfeld, Germany). Mice were fed standard chow diet and tap water ad libitum and housed under controlled temperature (22°C), under a 12 h light–dark cycle. The group size was calculated by a statistician and defined in the respective permit. A total of 110 mice were used in the experiments described here.
Bleomycin or AAV6.2m‐TGF‐β administration and olodaterol treatment
The models chosen, bleomycin challenge and TGF‐ß overexpression in mice, have been used for several years to study lung fibrosis (Moore et al., 2013). On day 0, mice were anesthetized with isoflurane (3–5%); either bleomycin (Calbiochem, Darmstadt, Germany), at a dose of 0.5 mg·kg−1 (1.5–2 U·mg−1) in sterile isotonic saline (2 mL·kg−1), or adeno‐associated virus (AAV) 6.2‐TGF‐β1, at a dose of 2.5 × 1011 vg, was intratracheally instilled. The detailed method for the AAV6.2‐TGF‐β1 model is described elsewhere (Strobel et al., 2015). Before starting treatment, the mice were randomly assigned to the different treatment groups. In a whole body exposure chamber, the animals were exposed to an aqueous solution of olodaterol aerosolized with a jet nebulizer (Pari Master; Pari GmbH, Starnberg, Germany) once daily for 5 min. To evaluate the efficacy of olodaterol, the compound was administered on either days 1–20 (preventive dosing) or days 7–20 (therapeutic dosing) after bleomycin or AAV6.2‐TGF‐β1 application.
Microcomputer tomography (μCT) of the lung
On day 21, lung density was assessed by μCT analysis. Animals were anesthetized with isoflurane (1.5%) and fixed in the prone position. μCT images were acquired with a Quantum FX μCT system (Perkin Elmer, Waltham, MA, USA) with cardiac gating (without respiratory gating), using the following parameters: 90 kV; 160 μA; FOV, 60 × 60 × 60 mm; spatial resolution, 0.11 mm. This resulted in a total acquisition time of 4.5 min. Images were analysed using MicroView 2.0 software (GE Healthcare, Amersham, UK). The Houndsfield unit (HU) corresponding to the peak of the HU‐histogram for the segmented pixels was used as a measure of fibrosis. After μCT analysis, animals were allowed to awaken from anaesthesia.
Assessment of lung function
Mice were anaesthetized by i.p. application of 60 mg·kg−1 pentobarbital (Narcoren; Merial GmbH, Hallbergmoos, Germany) and 2.32 mg·kg−1 xylazine hydrochloride (Rompun; Bayer Vital GmbH, Leverkusen, Germany) in a volume of 10 mL·kg−1. After cannulation of the trachea, mice received 0.64 mg·kg−1 pancuronium bromide (Pancuronium Inresa, Inresa Arzneimittel GmbH, Freiburg, Germany) by i.v. administration at a volume of 4 mL·kg−1. Lung function was measured with a FlexiVent system (SCIREQ, Montreal, PQ, Canada). Ventilation was conducted with 150 breaths·min−1, a tidal volume of 10 mL·kg−1 and an end‐expiratory pressure of 3 cmH2O. Data were analysed using the flexiWare 7 software (SCIREQ).
Bronchoalveolar lavage fluid (BALF) and lung preparation
Animals were killed with an overdose of pentobarbital (800 mg·kg−1, i.p.), and lungs were lavaged twice with 0.8 mL lavage buffer (1× PBS + protease inhibitor cocktail; Roche, Mannheim, Germany). Total BALF cell counts and differential cell counts were quantified by means of a Sysmex XT1800 iVet cell analyser (Sysmex Europe GmbH, Norderstedt, Germany). After lavage, left lung lobes were fixed with 4% paraformaldehyde (PFA) (Boster, China) and inflated under 20 cm water pressure for 20 min. Right lung lobes were weighed and shock frozen in liquid nitrogen for further analyses.
Ashcroft scoring
PFA fixed lungs were embedded in paraffin following standard procedures. Lung sections (3 μm) were prepared and used for Masson's trichrome staining. Stained para‐sagittal paraffin sections of the left lung lobe were systematically analysed in a microscope using a ×10 objective according to the scale defined by Ashcroft et al. (1988). All samples were blinded prior to scoring.
Protein determination in BALF supernatant and lung homogenate
For the quantification of TGF‐β, MMP‐9 and tissue inhibitor of metalloproteinase‐1 (TIMP‐1) Quantikine elisas from R&D Systems, Inc. (Minneapolis, MN, USA) were used. For the chemokine KC, TNF‐α (both determined in BALF supernatant) and IL‐1β (determined in lung homogenate), a mouse pro‐inflammatory panel (MSD) was used. The bicinchoninic acid (BCA) assay was used to quantify total protein in cell‐free BALF supernatant and lung homogenate. The BCA assay kit was purchased from Thermo Fisher Scientific (Waltham, MA, USA). A murine β2‐adrenoceptor elisa from Cusabio (Wuhan, China) was used to quantify levels of β2‐adrenoceptors in lung homogenates.
Isolation of total RNA from mouse lungs
Frozen right lungs from the AAV‐TGF‐β model were homogenized in RLT buffer (Qiagen) plus 1% β‐mercaptoethanol (Sigma‐Aldrich) using the Omni Prep Multi‐Sample Homogenizer (Omni International, Kennesaw, GA, USA). To isolate total RNA from formalin‐fixed paraffin‐embedded tissue slices in the bleomycin model, dewaxed 10 μm slices were incubated in PKD buffer plus Proteinase K (Qiagen) overnight at 56°C. Lung homogenate or tissue slice lysates were then mixed thoroughly with an equal volume of a phenol‐chloroform‐isoamyl‐alcohol mixture (Sigma‐Aldrich) and centrifuged in Phase Lock Gel Heavy 2 mL tubes (5Prime, Hamburg, Germany). The watery phase was then applied to the AllPrep DNA/RNA Mini Kit or the RNeasy FFPE kit (Qiagen), and RNA was purified according to the manufacturer's instructions.
TaqMan real‐time PCR‐analysis
Equal amounts of RNA of each sample were reversely transcribed to cDNA using the High‐Capacity cDNA Reverse Transcription Kit (Life Technologies, Carlsbad, CA, USA). qRT‐PCR reactions were set up using the TaqMan® Fast Advanced Master Mix (Applied Biosystems, Carlsbad, CA, USA) and species‐specific gene expression assays (Applied Biosystems: Hs00240532_s1, Hs00240532_s1, Mm00801666_g1, Mm01254476_m1, Mm00447142_m1). Gene expression analysis was performed with the ViiA7™ Real‐Time PCR System and software (Applied Biosystems). Gene expression was normalized to the expression of RNA polymerase II (PolA2), which was used as housekeeping gene.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as means ± SEM of n donors/animals. For the in vitro assays, technical replicates were used to ensure the reliability of single values. Unless otherwise noted, statistically significant differences between negative and positive controls were determined by an unpaired t‐test for parametric data. The comparison of the test groups was determined by one‐way ANOVA followed by Fisher's Least Significant Difference (LSD) test for parametric data. Statistical significance was accepted at P < 0.05. The tests were performed using GraphPad Prism version 7.00 for Windows, GraphPad Software, La Jolla, CA, USA, www.graphpad.com. To control for unwanted donor variances, the in vitro data were normalized as followed: the percentage inhibition was calculated according to the following: % inhibition = 100 − (Y/K1) * 100. K1 = mean rate of non‐stimulated, non‐compound‐treated control wells subtracted from the mean rate of stimulated, non‐compound‐treated control wells. Y = mean rate of non‐stimulated, non‐compound‐treated control wells subtracted from the mean rate of stimulated, compound‐treated wells. IC50 values were calculated by nonlinear regression of log (inhibitor concentration) versus % inhibition using a three‐ or four‐parameter fitting procedure of the Graph Pad Prism software package.
Materials
Olodaterol (BI 1744 CL) was synthesized at Boehringer Ingelheim (Biberach, Germany). ICI‐118,551 hydrochloride and CGP‐20712A methane sulfonate salt were obtained from Sigma‐Aldrich (St. Louis, MO, USA). Human recombinant PDGF‐BB, FGF‐basic, EGF and TGF‐β were purchased from R&D Systems, Inc. (Minneapolis, MN, USA). Human ET‐1 was purchased from Biotrend (BIOTREND Chemikalien GmbH, Cologne, Germany). FBS was purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). All primary antibodies (anti‐human phospho‐PLCγ, phospho‐SAPK/JNK, phospho‐c‐Raf, phospho‐ERK1/2, phospho‐Akt, phospho‐p38, and GAPDH) used for the Western blot analysis were produced in rabbit and purchased from Cell Signaling Technology (Danvers, MA, USA). Peroxidase‐conjugated mouse Anti‐Rabbit IgG antibody was purchased from Jackson Immuno Research (West Grove, PA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b,c).
Results
Olodaterol stimulates intracellular cAMP increase in primary human lung fibroblasts
To determine the expression of functional β2‐adrenoceptors on control and diseased primary HLF, we stimulated cells with various concentrations of olodaterol in the presence or absence of 30 nM ICI‐118,551 or 100 nM CGP‐20712A, which, at the concentrations used, are selective β2‐ and β1‐adrenoceptor antagonists, respectively (Chong et al., 2002). Olodaterol increased levels of intracellular cAMP, in a concentration‐dependent manner, in HLF and IPF‐LF with EC50 values of 740 and 690 pM, respectively. Pretreatment with ICI‐118,551 caused a 201‐fold EC50 shift in HLF and a 106‐fold shift in IPF‐LF. Pre‐incubation with CGP‐20712A did not affect the olodaterol‐induced cAMP increase in both cell types (Figure 1A, B). To further evaluate whether β2‐adrenoceptor expression levels on HLF and IPF‐LF correlate with the functional data, we measured β2‐adrenoceptor expression in cell lysates using elisa technique. Levels of the β2‐adrenoceptor protein were comparable in HLF and IPF‐LF (Figure 1C).
Figure 1.
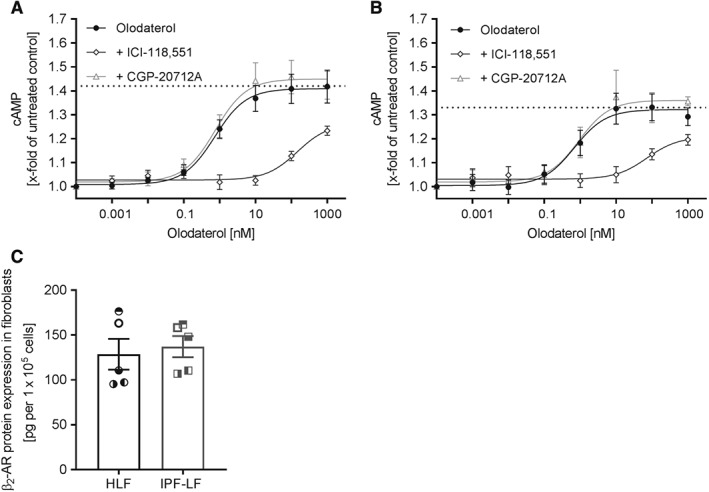
β2‐adrenoceptor function and protein expression in primary HLF. Intracellular cAMP increase was measured in fibroblasts from control donors (HLF) (A) and patients with IPF (IPF‐LF) (B) upon stimulation with different concentrations of olodaterol. Fibroblasts were pre‐incubated for 30 min with 30 nM ICI‐118,551 or 100 nM CGP‐20712A, respectively, before stimulation with olodaterol for another 30 min. Intracellular cAMP increase was measured by a bioluminescent cAMP assay. Dotted line represents the highest cAMP concentration reached. Control cells without stimulation were defined as 1. The EC50 value was calculated with a nonlinear regression fit by GraphPad Prism software. Levels of β2‐adrenoceptor (β2‐AR) protein in healthy and IPF fibroblasts (C). Expression of β2‐adrenoceptor protein was normalized to cell count. Each dot or square represents a different donor. Data shown are means ± SEM of n = 5 different donors each for HLF and IPF‐LF.
Olodaterol attenuates TGF‐β‐induced fibroblast‐to‐myofibroblast transition
We next investigated the effects of olodaterol on TGF‐β‐stimulated myofibroblast differentiation. The TGF‐β‐induced increase in α‐SMA protein expression in HLF and IPF‐LF was significantly inhibited in a concentration‐dependent manner by olodaterol with a maximal efficacy of 60% (HLF) and 42% (IPF‐LF) at 10 nM (Figure 2A). Furthermore, the expression of myofibroblast‐specific mediators and ECM proteins like pro‐collagen C‐peptide (PICP) or fibronectin increased by approximately twofold in TGF‐β‐treated cells. Olodaterol concentration‐dependently inhibited the PICP increase (from 20 to 40 ng·mL−1) with maximal efficacy of 70% at 10 nM (Figure 2B) in both HLF and IPF‐LF. To visualize this effect, Figure 2E shows representative images of the TGF‐β‐induced collagen I assembly in HLF and the inhibitory effect of olodaterol. The increase of fibronectin release (from approximately 1.5 to 2.5 μg·mL−1) was inhibited by olodaterol by 65% at 1 nM (Figure 2C) for both cell types. Levels of the pro‐fibrotic mediator ET‐1 were hardly detectable in untreated cells and increased 40‐ and 60‐fold in HLF and IPF‐LF (5 and 6 pg·mL−1, respectively), after TGF‐β‐stimulation. Olodaterol blocked this mediator release with an IC50 lower than 10 pM and a maximal inhibition of 100% and 50% at 1 nM, in HLFs and IPF‐LF respectively (Figure 2D).
Figure 2.
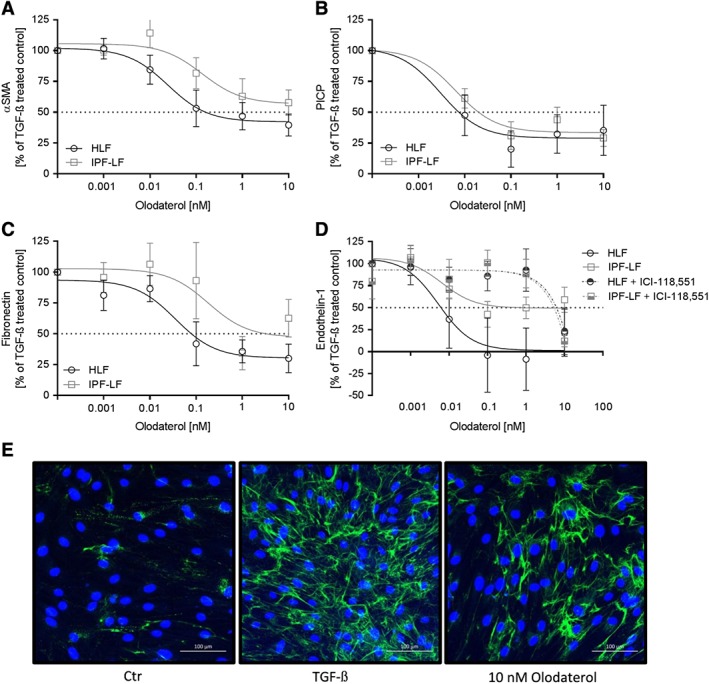
Olodaterol attenuates TGF‐β‐stimulated protein expression of primary HLF. Fibroblasts from control donors (HLF) and patients with IPF (IPF‐LF) were pre‐incubated with different concentrations of olodaterol and subsequently stimulated with TGF‐β (4 ng·mL−1) for 48 h in the presence of the compound. α‐SMA protein expression was measured in cell lysates by an MSD Western replacement assay (A). Pro‐collagen I C‐peptide (B), fibronectin (C) and ET‐1 (D) expression was measured in supernatants by elisa. Basal levels of 16 ng·mL−1, 1.5 μg·mL−1 and 0.2 pg·mL−1 increased to 40 ng·mL−1, 2.5 μg·mL−1 and 5 pg·mL−1 respectively. Effect of olodaterol on ET‐1 protein expression in HLF and IPF‐LF in the presence of ICI‐118,551 (30 nM) (D). Data are expressed as normalized protein expression (100% is expression with TGF‐β stimulation). Data shown are means ± SEM of n = 5 different donors for HLF and n = 5 different donors for IPF cells. Horizontal dotted line is 50% inhibition of the TGF‐β‐induced effect. Representative image of TGF‐β‐induced collagen I assembly and inhibition by 10 nM olodaterol in HLF in a ‘scar‐in‐a‐jar’ assay (E). Unstimulated cells (Ctr) were compared to TGF‐β‐stimulated cells (TGFβ) and TGFβ‐stimulated and olodaterol‐treated cells (10 nM Olodaterol).
To confirm that the inhibitory effects were driven by β2‐adrenoceptors, the β2‐adrenoceptor specific antagonist ICI‐118,551 was tested in the ET‐1 release assay. In the presence of ICI‐118,551, olodaterol showed no inhibitory effects on TGF‐β induced ET‐1 release in HLF and IPF‐LF (Figure 2D).
Olodaterol reduces FGF‐ and PDGF‐induced cell motility of primary human lung fibroblasts
To assess the ability of olodaterol to attenuate FGF‐ and PDGF‐induced migratory capacity of primary HLF, we performed migration assays using time‐lapse microscopy. FGF and PDGF each induced approximately a twofold increase in cell motility. Olodaterol reduced the FGF‐stimulated motility of HLF with an IC50 value of 4 pM and a maximal efficacy of 75% at 100 pM. For IPF‐LF, the inhibitory effect was not as marked and the maximal inhibition was reached at 10 nM (Figure 3A). With an IC50 of 1 pM and a maximal efficacy of 80%, olodaterol inhibited PDGF‐induced motility in HLF. In IPF‐LF, the IC50 shifted significantly to 34 pM and olodaterol reached a maximal inhibition of 50% at 10 nM (Figure 3B).
Figure 3.
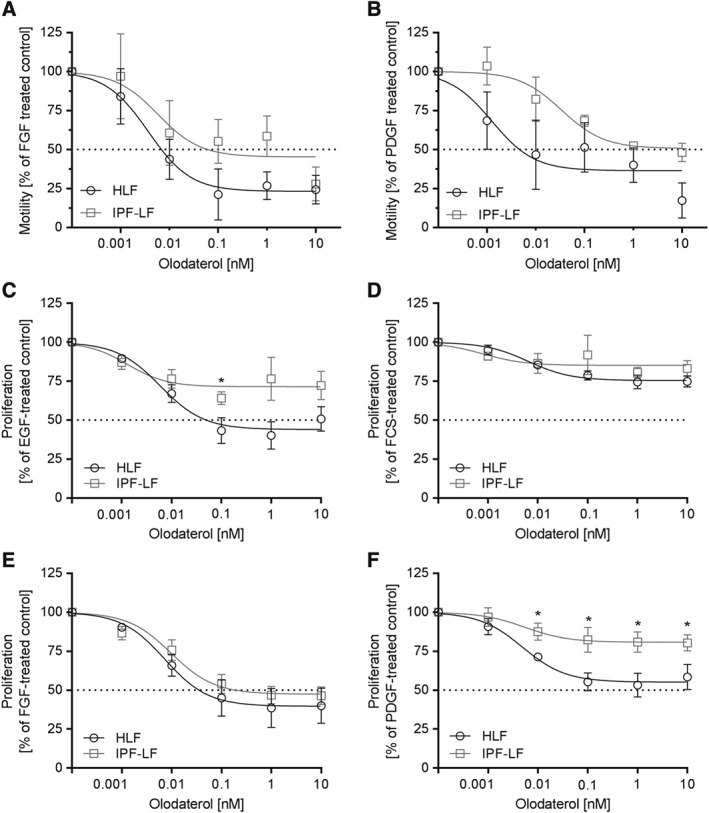
Olodaterol attenuates growth factor‐induced motility and proliferation of primary HLF. The motility of fibroblasts from control donors (HLF) and patients with IPF (IPF‐LF) after FGF or PDGF stimulation was measured by time‐lapse microscopy and manual single cell tracking. Cells were stimulated with FGF (20 ng·mL−1) or PDGF (50 ng·mL−1) for 72 h. Effect of olodaterol on FGF (A) and PDGF (B) induced motility. Data expressed as normalized migration (100% is defined as the migration measured after treatment with the respective stimulus). Data are shown as means ± SEM of n = 3 different donors for HLFs and IPFs. Cell proliferation was measured by BrdU incorporation. For proliferation, cells were stimulated with EGF (3 ng·mL−1), FCS (1%), FGF (20 ng·mL−1) or PDGF (50 ng·mL−1) for 92 h in the presence of the compound. Effect of olodaterol on EGF (C), FCS (D), FGF (E) or PDGF (F) induced proliferation. Data are shown as means ± SEM of n = 5 different donors for HLFs and n = 5 different donors for IPF‐LFs. Horizontal dotted line is 50% inhibition of the induced effect. *P < 0.05, significantly different from HLF data; unpaired Student's t‐test.
Olodaterol attenuates growth factor‐ and serum‐induced proliferation of fibroblasts
Stimulation of cells with either EGF, FCS, FGF or PDGF induced fibroblast proliferation as assessed by BrdU‐incorporation. Pretreatment with olodaterol attenuated the growth factor‐induced proliferation of fibroblasts. In HLF, the EGF‐stimulated proliferation was maximally inhibited (by approximately 50%) at 100 pM and an IC50 of 6 pM, while in IPF‐LF with an IC50 of 1.3 pM, the maximal inhibition was only 30% (Figure 3C). For FCS‐induced proliferation, olodaterol only showed trends towards an inhibition (Figure 3D), whereas FGF‐induced proliferation was potently attenuated in IPF‐LF with an IC50 of 10 pM and a maximal efficacy of 54% at 1 nM. Comparable values were measured for the inhibition in HLF (Figure 3E). At 100 pM, PDGF‐induced proliferation was inhibited by 40% in HLF and was significantly different, only by 20%, in IPF‐LF (Figure 3F).
Olodaterol prevents contraction of fibroblasts in collagen gels, induced by TGF‐β plus ET‐1
To study the effect of olodaterol in a functional bioassay, we used an in vitro floating collagen‐gel contraction model. For this purpose, fibroblasts were embedded in type I collagen. The gel matrix was detached from the wells, and the contraction of the cell‐gel matrix was stimulated with the pro‐fibrotic stimuli TGF‐β and ET‐1. While each stimulus alone induced a contraction of the fibroblasts, which reduced the gel size by approximately twofold, the combination of both stimuli resulted in a threefold reduction in gel size. As shown in Figure 4A at 10 nM, olodaterol inhibited the contractile properties of HLFs by approximately 85%. On the contrary, for IPF‐LF, the maximal inhibitory effect of olodaterol was 47% (Figure 4A). However, this numerical difference was not statistically significant.
Figure 4.
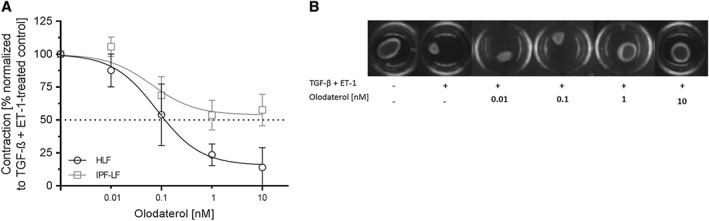
Olodaterol attenuates contraction of primary HLF induced by TGF‐β plus ET‐1. Free‐floating collagen lattices seeded with HLFs or IPF‐LFs were treated with different concentrations of olodaterol in the presence of TGF‐β (4 ng·mL−1), ET‐1 (100 ng·mL−1) or a combination of both. Inhibition of HLF or IPF‐LF contraction by olodaterol (A). Photographs of a representative experiment (B). Surface area of collagen gels was calculated in % of starting area and expressed as normalized contraction (100% is defined as the contraction measured after treatment with TGF‐β and ET‐1). Data shown are means ± SEM of n = 5 donors of HLFs or IPF‐LFs. Horizontal dotted line is 50% inhibition of the induced effect.
Olodaterol inhibits FGF‐induced phosphorylation of different fibrosis‐relevant kinases
To identify molecular pathways where olodaterol might interfere with pro‐fibrotic signalling, we stimulated fibroblasts with FGF for 5 min and checked the phosphorylation status of different fibrosis‐relevant kinases (PLCγ, JNK, Raf‐1, ERK1/2, Akt and p38 mitogen‐activated protein kinase). All kinases showed an increased phosphorylation signal after 5 min of FGF stimulation compared with unstimulated cells (Figure 5). Table 1 shows the inhibitory effect of olodaterol on the FGF‐induced phosphorylation of these kinases.
Figure 5.
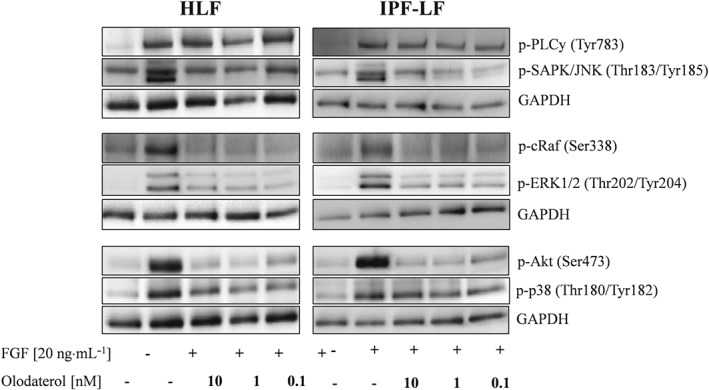
Olodaterol interferes with FGF‐induced phosphorylation of signalling cascades in primary HLF. Western blots show the FGF‐induced phosphorylation of different kinases. Western blot assays were performed for n = 3 different donors (HLF) and n = 5 different donors (IPF‐LF). Shown are blots of one representative experiment with HLF and IPF‐LF. Cells were stimulated with FGF (20 ng·mL−1) for 5 min in presence of different concentrations of olodaterol.
Table 1.
Densitometric analysis of Western blot assays of FGF‐stimulated HLF and IPF‐LF for different phospho‐proteins
| HLF | IPF‐LF | |||||
|---|---|---|---|---|---|---|
| 10 nM | 1 nM | 0.1 nM | 10 nM | 1 nM | 0.1 nM | |
| p‐PLCγ1 (Tyr783) | −7 ± 9.0ns | 7 ± 28.5ns | 19 ± 7.1ns | 10 ± 28.4ns | −2 ± 27.5ns | −27 ± 26.6ns |
| p‐SAPK/JNK (Thr183/Tyr185) | 76 ± 6.1 * | 60 ± 13.0 * | 50 ± 24.9 * | 92 ± 18.3 * | 94 ± 18.5 * | 80 ± 39.0ns |
| p‐c‐Raf (Ser338) | 120 ± 6.6 * | 127 ± 6.5 * | 129 ± 6.1 * | 115 ± 11.1 * | 92 ± 11.5 * | 92 ± 6.3 * |
| p‐ERK1/2 (Thr202/Tyr204) | 66 ± 8.9 * | 72 ± 5.9 * | 68 ± 5.0 * | 36 ± 24.1 * | 37 ± 18.4 * | 27 ± 27.5 * |
| p‐Akt (Ser473) | 98 ± 7.3 * | 96 ± 8.5 * | 88 ± 7.1 * | 100 ± 6.2 * | 93 ± 3.2 * | 72 ± 7.5 * |
| p‐p38 (Thr180/Tyr182) | 59 ± 15.6 * | 55 ± 4.4 * | 65 ± 12.9 * | 30 ± 9.0ns | 31 ± 10.1ns | 15 ± 18.0ns |
Data are mean values ± SEM from n = 3 HLF donors and n = 5 IPF‐LF donors. Densitometric evaluation of phospho‐proteins was normalized to loading control (GAPDH). Values are expressed as % inhibition of FGF‐stimulated effect.
P < 0.05, significantly different from FGF‐treated cells; ANOVA followed by Fisher's LSD test for parametric data: ns, not significant.
Olodaterol shows efficacy in a murine model of bleomycin‐induced lung fibrosis, after preventive or therapeutic treatment
In order to address the inhibitory activity of olodaterol to attenuate the fibrotic pathology in vivo, we used a mouse model of lung fibrosis induced by bleomycin. Treatment with inhaled olodaterol (1 mg·mL−1, q.d.) was either started at day 1 after bleomycin instillation for the preventive treatment or at day 7 for the therapeutic treatment. Bleomycin caused a maximal reduction of body weight at day 7 with a subsequent recovery phase. With preventive as well as therapeutic olodaterol treatment, the recovery back to control levels of body weight (at day 21) was accelerated (Figure 6A). On day 21, total cell counts were significantly increased in the BALF of mice stimulated with bleomycin (Figure 6B). The most prominent increase in cell number was found for lymphocytes and monocytic cells. The influx of white blood cells into the lung was significantly attenuated by olodaterol, by approximately 30%, in both treatment regimens. For lymphocytes, only the preventive treatment showed significant inhibitory effects, whereas in the therapeutic treatment regimen, we only observed a trend towards inhibition. For monocytes, only trends in the attenuation of the bleomycin‐induced effects were seen with both treatment strategies (Figure 6B). Regarding alterations in tissue density, bleomycin administration caused a twofold increase in lung density in treated animals compared to control animals, as measured by μCT analysis. Olodaterol treatment starting at day 1 attenuated the increase in lung density non‐significantly by approximately 60%. The therapeutic treatment regimen had no effect on this parameter (Figure 6C). Regarding the bleomycin‐induced increase of the Ashcroft score, olodaterol only showed a trend towards inhibition (by 17%) with preventive, but not with therapeutic, treatment (Figure 6D). Figure 6J, K show representative images of the μCT analysis and Masson's trichrome stained lung slices. With respect to lung function parameters, instillation of bleomycin increased lung resistance (Ra) and decreased dynamic lung compliance (Cdyn) and forced vital capacity (FVC), compared with control animals. Preventive olodaterol treatment inhibited the FVC decline significantly, by 45% (Figure 6E). These trends were also seen for the attenuation of the increase in resistance and the reduction in lung compliance by 57 and 71% respectively (data not shown). Other parameters such as the increase in relative lung weight (1.5‐fold) and the total protein content in BALF (9.3‐fold) were also significantly reduced in the preventive, as well as the therapeutic, treatment with olodaterol (33 and 43%, respectively) (Figure 6F, G). Furthermore, the fibrosis‐relevant mediators, MMP‐9, TGF‐β and TIMP‐1, were increased in BALF of bleomycin‐treated animals (4.3‐fold, 5.6‐fold and 35.3‐fold, respectively) and were significantly inhibited by olodaterol inhalation by about 50 to 70%, regardless of treatment start (Table 2). Other mediators, such as KC, TNF‐α, IL‐6, IFN‐γ and IL‐1β, were also significantly induced in bleomycin‐treated animals. With exception of IL‐6 and IFNγ, olodaterol reduced these mediators by about 60 to 80% (Table 2). Furthermore, bleomycin induced a 4.3‐fold increase of collagen I (Col1A1) mRNA in the lungs. This increase was inhibited, albeit not significantly, by olodaterol by 21 and 34% with therapeutic and preventive treatment respectively (Figure 6H). Expression of β2‐adrenoceptor protein in lung homogenates was reduced after bleomycin challenge (Figure 6I). No significant changes in levels of β2‐adrenoceptors were observed with olodaterol treatment.
Figure 6.
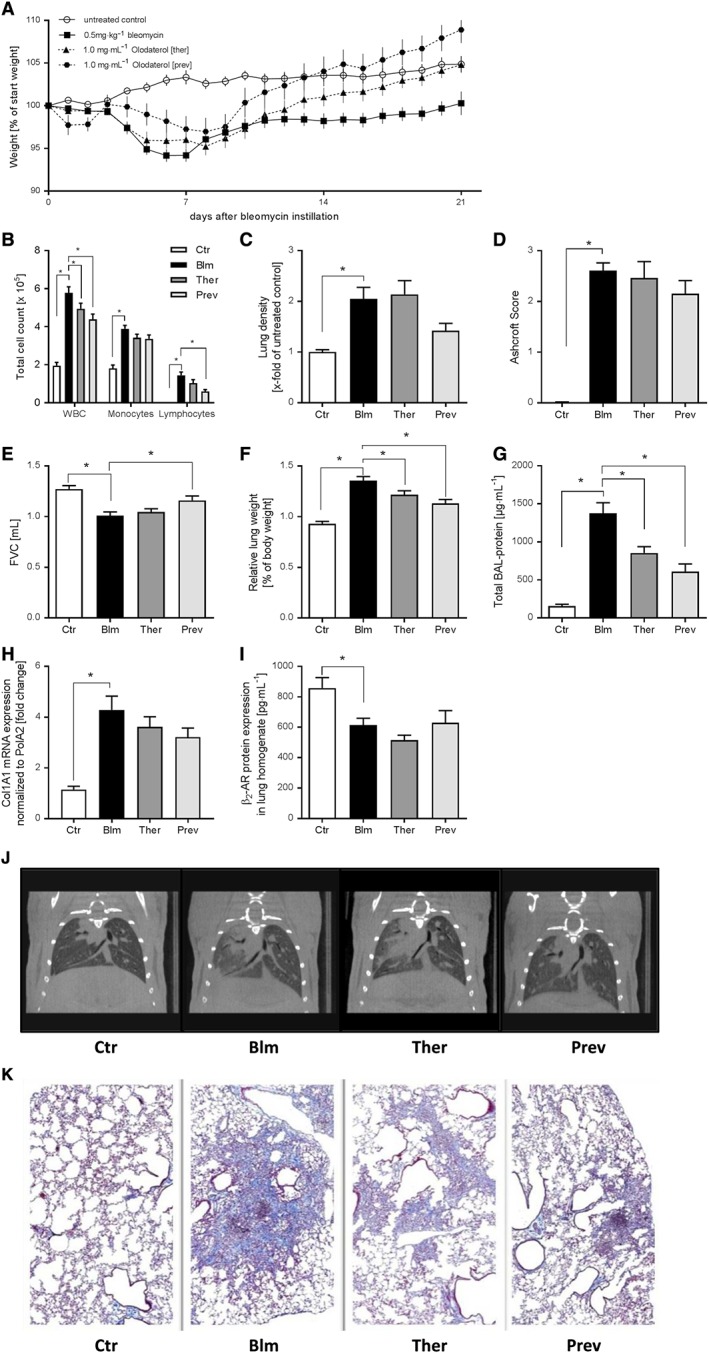
Olodaterol attenuates bleomycin‐induced lung fibrosis in mice. C57BL/6 mice received an intratracheal instillation of NaCl (Ctr, n = 18) or 0.5 mg·kg−1 bleomycin (Blm, n = 24). Olodaterol was administered each day by inhalation (1 mg·mL−1) either in a preventive regimen from day 1 to 20 (Prev, n = 12) or in a therapeutic mode from day 7 to 20 (Ther, n = 24). Analyses were performed at day 21. Body weight is expressed as % of starting weight (A). Cell count is expressed as total cell count per lung; WBC, white blood cells (B). Lung density was measured by μCT analysis (C). Ashcroft scoring was performed according to Ashcroft et al. (1988) (D). FVC was measured with flexiVent (E). Lungs were weighed before lavage. Relative lung weights are expressed as % of body weight (F). Total protein concentration in BALF was determined with a BCA assay (G). Col1A1 gene expression in lung was determined via qRT‐PCR (H). Expression of β2‐adrenoceptor (β2‐AR) protein expression was determined by elisa (I). Representative μCT pictures of mouse lungs (J). Representative images from mouse lungs (formalin‐fixed, paraffin embedded); Masson trichrome staining; 2.5× (K). Data shown are means ± SEM. *P < 0.05, significantly different as indicated. Bleomycin‐treated groups were compared with untreated groups, with unpaired Student's t‐test. Olodaterol‐treated groups were compared with Bleomycin‐treated control animals, by one‐way ANOVA followed by Fisher's LSD test for parametric data.
Table 2.
Analysis of BALF and tissue homogenate for pro‐fibrotic and pro‐inflammatory mediators in the bleomycin model
| Untreated | Bleomycin (0.5 mg·kg−1) | Olodaterol therapeutic | Olodaterol preventive | |
|---|---|---|---|---|
| Mediators in BALF (pg·mL−1) | ||||
| TGF‐β | 25 ± 3.2* | 140 ± 12.5 | 74 ± 7.4* | 77 ± 10.0* |
| MMP‐9 | 71 ± 9.5* | 283 ± 22.8 | 183 ± 19.6* | 208 ± 31.0* |
| TIMP‐1 | 20 ± 1.5* | 719 ± 125.5 | 439 ± 115.9ns | 236 ± 61.9* |
| mKC/GRO | 4 ± 0.5* | 15 ± 2.1 | 8 ± 0.9* | 10 ± 1.2ns |
| TNF‐α | 4 ± 0.5* | 10 ± 1.3 | 6 ± 0.9* | 6 ± 0.9* |
| IL‐6 | 17 ± 2.0* | 43 ± 7.0 | 35 ± 2.6ns | 29 ± 3.0ns |
| IFN‐γ | 78 ± 14.4* | 188 ± 22.0 | 188 ± 24.8ns | 211 ± 30.4ns |
| Mediators in tissue homogenate (pg·mL−1) | ||||
| IL‐1β | 53 ± 6.3* | 213 ± 33.9 | 124 ± 13.7* | 78 ± 18.0* |
Bleomycin‐treated groups were compared with untreated groups by Student's unpaired t‐test. Olodaterol‐treated groups (n = 10–24 animals per group) were compared with bleomycin‐treated animals by one‐way ANOVA followed by Fisher's LSD test for parametric data. Values are presented as mean ± SEM
P < 0.05, significantly different from bleomycin‐treated animals: ns, not significant.
Olodaterol attenuates pro‐fibrotic mechanism in a murine model of TGF‐β1‐induced lung fibrosis
To confirm the efficacy of olodaterol on TGF‐β‐induced mechanisms, we used a mechanistic mouse model of AAV6.2m‐TGF‐β1‐induced lung fibrosis. Olodaterol treatment was performed according to the treatment regimen in the bleomycin study. Overexpression of TGF‐β1 in mouse lungs caused a reduction in body weight by 5 ‐ 10%, from day 10 to 21 after virus instillation. This weight loss was inhibited by olodaterol with both preventive and therapeutic treatment (Figure 7A). Furthermore, TGF‐β1‐overexpression induced a significant extravasation of immune cells into the lungs as measured in BALF (Figure 7B). Challenge with AAV6.2m‐TGF‐β1 caused an increase in lymphocyte, neutrophil and monocytic cell counts. Preventive olodaterol treatment significantly attenuated the influx of monocytes and lymphocytes by about 40%, whereas therapeutic treatment only resulted in 20% inhibition. Neutrophil numbers in BALF were reduced by 43% with preventive treatment, but non‐significantly increased with the therapeutic regimen (Figure 7B). With regard to fibrotic remodelling, AAV6.2m‐TGF‐β1 instillation resulted in a fourfold increase in lung density as measured by μCT. Therapeutic olodaterol treatment had no effect on the increase in tissue density, although preventive treatment resulted in 36% inhibition (Figure 7C). Figure 7H shows representative images of the μCT analysis. These TGF‐β1‐induced changes in lung parameters altered functional parameters, as the FVC decreased in AAV6.2m‐TGF‐β1‐treated mice. However, olodaterol treatment did not show significant effects on this decline of FVC (Figure 7D). Furthermore, AAV6.2m‐TGF‐β1 treatment induced an increase in relative lung weight and protein content in BALF. With preventive treatment, olodaterol attenuated the increase in lung weight and BALF protein by over 50%. Further, therapeutic treatment caused a decrease of about one third in both parameters (Figure 7E, F). TGF‐β overexpression also resulted in a 4.5‐fold increase of collagen III mRNA (Col3A1). This increase was significantly inhibited by 37 and 50% with therapeutic and preventive treatment respectively (Figure 7G). Furthermore, AAV6.2m‐TGF‐β1 instillation caused an increase in MMP9, TGF‐β and TIMP‐1 in BALF. Preventive treatment again attenuated this mediator release significantly up to 70%, while therapeutic treatment had no inhibitory effect (Table 3).
Figure 7.
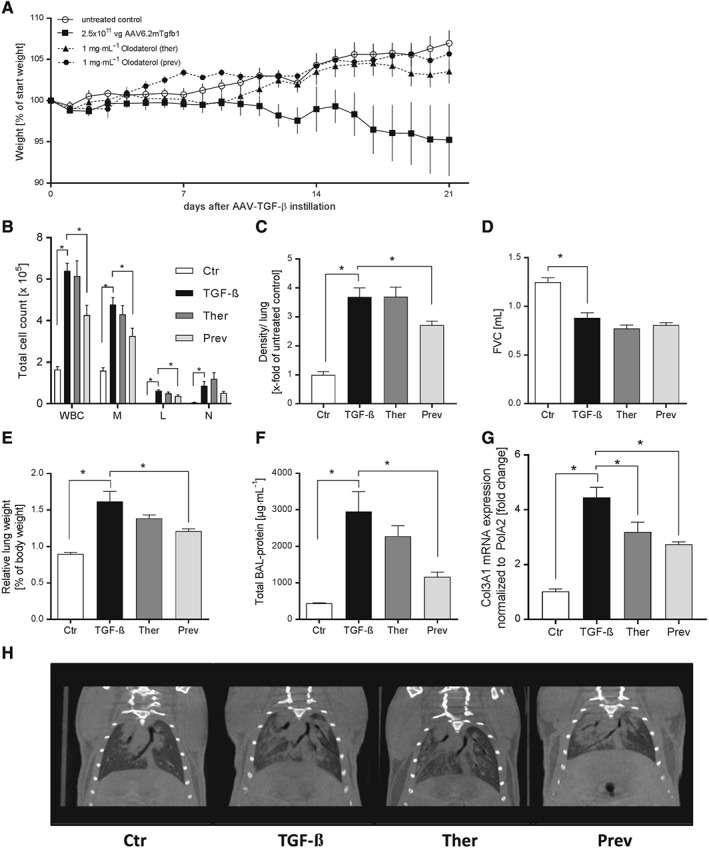
Olodaterol attenuates TGF‐β‐ induced lung fibrosis in mice. C57BL/6 mice received an intratracheal instillation of 2.5 × 1011 vg virus (TGF‐β, n = 8) or PBS (Ctr, n = 8). Olodaterol was administered each day by inhalation (1 mg·mL−1) either in a preventive regimen from day 1 to 20 (Prev, n = 8) or in a therapeutic mode from day 7 to 20 (Ther, n = 8). Analyses were performed at day 21. Body weight is expressed as % of starting weight (A). Cell count is expressed as total cell count per lung; WBC, white blood cells; M, monocytic cells; L, lymphocytes; N, neutrophils (B). Lung density was measured by μCT analysis (C). FVC was measured with flexiVent (D). Lungs were weighed before lavage. Relative lung weights are expressed as % of body weight (E). Total protein concentration in BALF was determined with a BCA assay (F). Col3A1 gene expression in lung was determined via qRT‐PCR (G). Representative μCT pictures of mouse lungs (H). Data shown are means ± SEM. *P < 0.05, significantly different as indicated. AAV6.2m‐TGF‐β1‐treated groups were compared with untreated groups, with unpaired Student's t‐test. Olodaterol‐treated groups were compared with AAV6.2m‐TGF‐β1‐treated control animals, by one‐way ANOVA followed by Fisher's LSD test for parametric data.
Table 3.
Analysis of BALF for pro‐fibrotic mediators in the AAV‐TGF‐β1 overexpression model
| Untreated | AAV6.2m‐TGF‐β1 (2.5 × 1011 vg) | Olodaterol therapeutic | Olodaterol preventive | |
|---|---|---|---|---|
| Mediators in BALF (pg·mL−1) | ||||
| TGF‐β | 24 ± 4.4* | 4831 ± 1110 | 4404 ± 735ns | 1659 ± 118* |
| MMP‐9 | 31 ± 4.6* | 930 ± 227 | 1348 ± 330ns | 365 ± 79ns |
| TIMP‐1 | 23 ± 3.2* | 6417 ± 1363 | 6662 ± 1304ns | 1934 ± 370* |
AAV6.2m‐TGF‐β1‐treated groups were compared with untreated groups by an unpaired Student's t‐test. Olodaterol‐treated groups (n = 8 animals per group) were compared with AAV6.2m‐TGF‐β1‐treated animals by one‐way ANOVA followed by Fisher's LSD test for parametric data. Data shown are means ± SEM.
P < 0.05, significantly different from animals treated with AAV6.2m‐TGF‐β1; ns, not significant.
Discussion and conclusions
Olodaterol (Striverdi®) is a long‐acting β2‐adrenoceptor agonist marketed as a bronchodilator for the maintenance treatment of COPD (Bouyssou et al., 2010b). Besides its well‐proven bronchodilatory activity, olodaterol has recently been demonstrated to elicit anti‐inflammatory activity in mouse models of cigarette smoke and lipopolysaccharide‐induced lung inflammation (Wex et al., 2015) and to have anti‐tussive properties in guinea pigs (Wex and Bouyssou, 2015). However, its anti‐fibrotic activity has not been systematically evaluated. Thus, the objective of our study was to test the efficacy of the long‐acting β2‐adrenoceptor agonist olodaterol to inhibit pro‐fibrotic mechanisms in primary HLF from control donors and IPF patients and to evaluate whether our findings translate into the in vivo situation.
In a first set of experiments, we demonstrated that olodaterol stimulates cAMP release in HLF from control and IPF donors in a β2‐adrenoceptor‐dependent manner. Some differences were seen regarding the maximal cAMP release in HLF and IPF‐LF, but these results were not significantly different. Furthermore, the EC50 values of intracellular cAMP increase after olodaterol stimulation was in good agreement with the EC50 values determined in CHO cells (Bouyssou et al., 2010a).
With TGF‐β being one of the major activators of the transition of fibroblasts to myofibroblasts, many treatments aim to inhibit TGF‐β‐induced signalling pathways. Our data demonstrate that olodaterol blocks the TGF‐β‐induced fibroblast‐to‐myofibroblast differentiation in both HLF and IPF‐LF, as indicated by the inhibition of α‐SMA protein expression, as well as the attenuated release of fibronectin, PICP (a surrogate marker reflecting type I collagen synthesis) and the pro‐fibrotic mediator ET‐1. These findings are in good agreement with an abstract by Wang et al. (2012), showing the inhibitory effects of olodaterol on collagen synthesis by human fibroblasts. Additionally, indacaterol and isoprenaline were shown to inhibit TGF‐β‐induced pro‐fibrotic protein expression in normal human lung and cardiac fibroblasts (Liu et al., 2006; Tannheimer et al., 2012). In another functional bioassay, the gel contraction assay, olodaterol, inhibited the TGF‐β plus ET‐1‐stimulated contraction of HLF and, to a lesser extent, of IPF‐LF. In accordance with our findings, Schiller et al. (2010) showed inhibitory effects of db‐cAMP (a stable cAMP analogue) on TGF‐β‐induced contraction of NIH3T3 cells. However, the exact mechanism of how cAMP interferes with the TGF‐β signalling cascade is unclear. One possible explanation is the interference of cAMP with TGF‐β‐specific Smad3/4‐dependent gene expression. The sequestration of the transcriptional co‐activators CBP and p300 by phospho‐CREB would make them unavailable for recruitment to Smad transcriptional complexes (Schiller et al., 2010). Additionally, cAMP may further oppose fibrotic responses via the PKA or Epac‐mediated inhibition of TGF‐β stimulated ERK1/2 and JNK activation (Liu et al., 2006). Nevertheless, the final clarification of the pivotal mechanism in the inhibition of TGF‐β‐mediated signalling will need further exploration.
Besides TGF‐β, additional mediators are found to be elevated in fibrotic tissues, such as PDGF, FGF or EGF (Bonner, 2004; Hetzel et al., 2005; Wollin et al., 2014). They are believed to play important roles in the pathogenesis of IPF by stimulating fibroblast survival, proliferation and migration. Thus, reducing fibroblast proliferation and migration in IPF will reduce spreading and accumulation of myofibroblasts. In this study, we demonstrated that olodaterol inhibits the growth factor‐induced motility and proliferation of human primary lung fibroblasts with significantly different potency in HLF compared to IPF‐LF. Accordingly, other β2‐adrenoceptor agonists like isoprenaline or formoterol were shown to inhibit proliferation and migration upon FGF‐ and PDGF‐stimulation in different cell lines (Chen et al., 2008; Lamyel et al., 2011). There are several studies suggesting a link between cAMP‐dependent PKA activation and cellular migration (Howe, 2004; Chen et al., 2008). Furthermore, various articles show an inhibition of cell proliferation by cAMP elevating agents, in a PKA or Epac‐1 dependent manner (Hafner et al., 1994; Stork and Schmitt, 2002; Huang et al., 2008).
To evaluate the inhibitory activity of olodaterol on relevant kinases, activated by pro‐fibrotic growth‐factors, we performed Western blot analysis after FGF stimulation. Our results show distinct inhibitory effects of olodaterol on FGF‐induced Akt, c‐Raf and JNK phosphorylation, while the phosphorylation of other kinases like p38 or ERK1/2 was only attenuated or unaffected (PLCγ).
Summarizing the in vitro part of our data, we found no significant differences regarding the efficacy of olodaterol in inhibiting pro‐fibrotic mechanisms in HLF compared to IPF‐LF. This indicates that β2‐adrenoceptors are still functional in cells from fibrotic patients and builds a rationale for the efficacy of olodaterol in IPF. The effects of olodaterol in healthy cells is not a matter of concern to us, with regard to altered wound healing, as olodaterol has been used in clinical practice for several years without showing detrimental effects on physiological wound healing processes. Additionally, further research will be needed to elucidate how olodaterol interferes with signal cascades, activated by other growth factors, and whether this interference is stimulus‐, cell‐ and/or disease status‐dependent. Moreover, it will be necessary to identify the anti‐fibrotic effects of olodaterol in other fibrosis relevant cell types, such as epithelial cells (mediator release and EMT) or macrophages (mediator release, migration and differentiation).
Having shown the efficacy of olodaterol to inhibit pro‐fibrotic mechanisms in several disease‐relevant functional fibroblast assays, we explored whether these effects translated into in vivo efficacy. With regard to the dosing, we based our dose estimation for the fibrosis models on functional data generated in a model of acetylcholine‐induced bronchoconstriction (Wex et al., 2015). In this model, a concentration of 1 mg·mL−1 olodaterol given by inhalation was determined as the first dose showing maximal bronchodilation. Therefore, we used the same experimental set‐up with regard to the olodaterol inhalation in our fibrosis models.
In the bleomycin‐induced lung fibrosis model, intratracheal instillation of bleomycin results in an initial lung injury, followed by an inflammatory response and the subsequent development of fibrosis. Even though this model shows some limitations regarding the transferability to human IPF, it is still a good tool to assess efficacy of potential compounds in general as proof of principle (Moeller et al., 2008). We show that weight loss induced by bleomycin challenge was diminished with olodaterol treatment in both treatment regimens. Furthermore, in line with its anti‐inflammatory efficacy, olodaterol reduced the pulmonary influx of white blood cells into the lung and attenuated the levels of pro‐inflammatory and pro‐fibrotic mediators in BALF. These data demonstrate an additional dimension of the inhibitory effects of olodaterol. In contrast to our in vitro assays, not only the inhibition of mediator‐induced effects, but also the release of those pro‐fibrotic mediators and the influx of disease‐relevant cell types (for example macrophages) can be investigated in vivo. Furthermore, preventive treatment also attenuated the decline in FVC and showed trends towards an inhibition of the increase in lung density (the two main parameters used in the diagnosis of IPF in the clinical situation) and collagen I deposition. However, with therapeutic treatment, no significant effects on lung density or Ashcroft score were seen.
To confirm our results from the bleomycin model and to further dissect effects of olodaterol on inflammation and direct anti‐fibrotic effects, we chose to test olodaterol in another, more mechanistic, lung fibrosis model. For this purpose, we chose an AVV6.2‐TGF‐β1 overexpression model. Compared to the common adenovirus‐5 technique, no measurable inflammation after transduction with the AAV6.2 is present, and thus, the model has a lower risk to misinterpret anti‐fibrotic findings due to an anti‐inflammatory mechanism of a drug. Lung epithelial cells secrete large amounts of TGF‐β after being transduced by AAV6.2‐TGF‐β1. This overexpression of TGF‐β results in fibrotic conditions as determined by a decrease in FVC and an increase in lung density without an initial inflammatory phase (Strobel et al., 2015). In this model, preventive olodaterol treatment significantly reduced lung density (as determined by μCT analysis), Col3A1 mRNA expression and the release of pro‐fibrotic mediators. With the early inflammation phase being absent in this model, the inhibitory effects of olodaterol given in a preventive manner are interfering with TGF‐β‐driven mechanisms and cannot be attributed to the anti‐inflammatory properties of the compound.
In summary, our in vivo experiments support anti‐fibrotic actions of olodaterol both, in the bleomycin model, as well as in a model of TGFß‐induced fibrosis. In both models, the most prominent effects were seen with a preventive dosing regimen compared to the therapeutic approach. As fibrosis in IPF is often described as being ‘patchy’ in its distribution with intervening areas of unaffected lung, a preventive approach confining disease progression by inhibiting fibroblast activation at early stages might attenuate spreading of fibrotic regions (King et al., 2011).
Having seen trends towards a reduced efficacy of olodaterol to inhibit pro‐fibrotic mechanisms in IPF compared to healthy fibroblasts, we investigated the β2‐adrenoceptor expression in cell lysates and in lung homogenate after bleomycin challenge. Protein expression analysis of cell lysates revealed comparable expression levels of the β2‐adrenoceptor in HLF and IPF fibroblasts. However, we found reduced β2‐adrenoceptor protein expression in bleomycin‐challenged mice. With this observation in mind, we checked the expression of β2‐adrenoceptors in other data sets. Our in‐house next‐generation sequencing (NGS) data of murine bleomycin models, the mouse AAV6.2m‐TGF‐β1 overexpression model and TGF‐β‐treated primary HLF and the analysis of external NGS studies (GSE1066; Konishi et al., 2009; GSE52463; Nance et al., 2014) together with current and earlier literature (Giri et al., 1987; Bauer et al., 2015) revealed a general down‐regulation of β2‐adrenoceptors in IPF lung samples and IPF‐related in vitro and in vivo models. This is in contrast to our observation that, in vitro, cultures of IPF‐LF and HLF showed no significant differences in expression of β2‐adrenoceptors. It is possible that the absence of differences in β2‐adrenoceptor protein between healthy and IPF fibroblasts might be caused by an assimilation of the receptor expression due to the in vitro culture of the primary cells on hard plastic dishes and in standardized culture media. However, this will need further investigation. Additionally, besides the levels of β2‐adrenoceptors, other proteins involved in the downstream signalling of the β2‐adrenoceptor, such as PDE‐4, might also be differentially expressed and thereby might influence the responsiveness of IPF‐LF to cAMP‐increasing drugs. However, to evaluate the validity of this hypothesis, additional experiments need to be done. Whether other cAMP‐elevating agents (not relying on β2‐adrenoceptors) show more pronounced inhibitory effects in fibrotic in vitro and in vivo models will also need further investigation.
Taken together, our in vitro data demonstrated that olodaterol attenuated pro‐fibrotic mechanisms involved in the onset and progression of IPF, including the proliferation, trans‐differentiation, migration, contraction, ECM production and pro‐fibrotic mediator release of primary HLF. In a mechanistic TGF‐β‐driven fibrosis model in mice, we demonstrated that olodaterol significantly attenuated the increase in lung density, Col3A1 mRNA expression and the release of pro‐fibrotic mediators when given preventively. Likewise, olodaterol attenuated the FVC decline and reduced the release of pro‐fibrotic mediators in a model of bleomycin‐induced lung fibrosis when given in a prophylactic manner. Our data provided clear evidence that stimulation of the β2‐adrenoceptor attenuates pro‐fibrotic events both in healthy and diseased settings and, thus, supports the hypothesis that cAMP‐increasing drugs might be beneficial in the treatment of IPF. As IPF is known to be a heterogeneous disease, with alternating preserved and fibrotic areas, a preventive treatment and effects on non‐diseased cells might inhibit the transition of normal to fibrotic lung fibroblasts and thus halt disease progression in yet unaffected regions. However, due to the possible down‐regulation of the β2‐adrenoceptor under fibrotic conditions, it might be assumed that cAMP‐elevating mechanisms independent of the β2‐adrenoceptor could be an even more effective choice in clinical conditions. Whether the robust inhibition of pro‐fibrotic mechanisms in HLF or the weaker anti‐fibrotic activity demonstrated only in the preventive treatment regimen in animal models of lung fibrosis is the better predictor of effective translation to a clinical treatment option is still an open question.
Author contributions
F.E.H. conducted the experiments. F.E.H., L.W., F.G. and E.W. designed the experiments, analysed the data and wrote the manuscript. J.W. performed the immunocytochemistry. B.L. performed the NGS analyses. All authors contributed to scientific discussions and approved the manuscript.
Conflict of interest
All authors are employees of Boehringer Ingelheim Pharma GmbH & Co. KG.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The authors would like to thank Helene Lichius, Janine Beier and Andrea Vögtle (Boehringer Ingelheim Pharma GmbH & Co. KG, Biberach, Germany) for their excellent technical assistance. Furthermore, we thank Benjamin Strobel for his support with the AAV6.2m‐TGF‐β model and Birgit Stierstorfer for the Ashcroft score analysis. This paper contains parts of the PhD thesis of F.E.H.
Herrmann, F. E. , Wollin, L. , Wirth, J. , Gantner, F. , Lämmle, B. , and Wex, E. (2017) Olodaterol shows anti‐fibrotic efficacy in in vitro and in vivo models of pulmonary fibrosis. British Journal of Pharmacology, 174: 3848–3864. doi: 10.1111/bph.13982.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Catalytic receptors. Br J Pharmacol 172: 5979–6023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen JT, Spiteri MA (2002). Growth factors in idiopathic pulmonary fibrosis: relative roles. Respir Res 3: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft T, Simpson JM, Timbrell V (1988). Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol 41: 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer Y, Tedrow J, de BS , Birker‐Robaczewska M, Gibson KF, Guardela BJ et al (2015). A novel genomic signature with translational significance for human idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol 52: 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner JC (2004). Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15: 255–273. [DOI] [PubMed] [Google Scholar]
- Borie R, Justet A, Beltramo G, Manali ED, Pradere P, Spagnolo P et al (2016). Pharmacological management of IPF. Respirology 21: 615–625. [DOI] [PubMed] [Google Scholar]
- Bouyssou T, Casarosa P, Naline E, Pestel S, Konetzki I, Devillier P et al (2010a). Pharmacological characterization of olodaterol, a novel inhaled beta2‐adrenoceptor agonist exerting a 24‐hour‐long duration of action in preclinical models. J Pharmacol Exp Ther 334: 53–62. [DOI] [PubMed] [Google Scholar]
- Bouyssou T, Hoenke C, Rudolf K, Lustenberger P, Pestel S, Sieger P et al (2010b). Discovery of olodaterol, a novel inhaled beta2‐adrenoceptor agonist with a 24 h bronchodilatory efficacy. Bioorg Med Chem Lett 20: 1410–1414. [DOI] [PubMed] [Google Scholar]
- Chen CZ, Peng YX, Wang ZB, Fish PV, Kaar JL, Koepsel RR et al (2009). The Scar‐in‐a‐Jar: studying potential antifibrotic compounds from the epigenetic to extracellular level in a single well. Br J Pharmacol 158: 1196–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhang JJ, Huang XY (2008). cAMP inhibits cell migration by interfering with Rac‐induced lamellipodium formation. J Biol Chem 283: 13799–13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong LK, Chess‐Williams R, Peachell PT (2002). Pharmacological characterisation of the beta‐adrenoceptor expressed by human lung mast cells. Eur J Pharmacol 437: 1–7. [DOI] [PubMed] [Google Scholar]
- Cortijo J, Iranzo A, Milara X, Mata M, Cerda‐Nicolas M, Ruiz‐Sauri A et al (2009). Roflumilast, a phosphodiesterase 4 inhibitor, alleviates bleomycin‐induced lung injury. Br J Pharmacol 156: 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G (1993). Transforming growth factor‐beta 1 induces alpha‐smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122: 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkern TR, Feurstein D, Rossi GA, Sabatini F, Hatzelmann A (2007). Inhibition of TGF‐beta induced lung fibroblast to myofibroblast conversion by phosphodiesterase inhibiting drugs and activators of soluble guanylyl cyclase. Eur J Pharmacol 572: 12–22. [DOI] [PubMed] [Google Scholar]
- Giri SN, Sanford DA Jr, Robison TW, Tyler NK (1987). Impairment in coupled beta‐adrenergic receptor and adenylate cyclase system during bleomycin‐induced lung fibrosis in hamsters. Exp Lung Res 13: 401–416. [DOI] [PubMed] [Google Scholar]
- Hafner S, Adler HS, Mischak H, Janosch P, Heidecker G, Wolfman A et al (1994). Mechanism of inhibition of Raf‐1 by protein kinase A. Mol Cell Biol 14: 6696–6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzel M, Bachem M, Anders D, Trischler G, Faehling M (2005). Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 183: 225–237. [DOI] [PubMed] [Google Scholar]
- Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch‐Grunt U et al (2008). BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res 68: 4774–4782. [DOI] [PubMed] [Google Scholar]
- Howe AK (2004). Regulation of actin‐based cell migration by cAMP/PKA. Biochim Biophys Acta 1692: 159–174. [DOI] [PubMed] [Google Scholar]
- Huang SK, Wettlaufer SH, Chung J, Peters‐Golden M (2008). Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac‐1. Am J Respir Cell Mol Biol 39: 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King TE Jr, Pardo A, Selman M (2011). Idiopathic pulmonary fibrosis. Lancet 378: 1949–1961. [DOI] [PubMed] [Google Scholar]
- Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R et al (2009). Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 180: 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana M, Okazaki Y, Kodama H, Izumi K, Yasuoka H, Ogawa Y et al (2003). Human circulating CD14+ monocytes as a source of progenitors that exhibit mesenchymal cell differentiation. J Leukoc Biol 74: 833–845. [DOI] [PubMed] [Google Scholar]
- Lamyel F, Warnken‐Uhlich M, Seemann WK, Mohr K, Kostenis E, Ahmedat AS et al (2011). The beta2‐subtype of adrenoceptors mediates inhibition of pro‐fibrotic events in human lung fibroblasts. Naunyn Schmiedebergs Arch Pharmacol 384: 133–145. [DOI] [PubMed] [Google Scholar]
- Liu X, Ostrom RS, Insel PA (2004). cAMP‐elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J Physiol Cell Physiol 286: C1089–C1099. [DOI] [PubMed] [Google Scholar]
- Liu X, Sun SQ, Hassid A, Ostrom RS (2006). cAMP inhibits transforming growth factor‐beta‐stimulated collagen synthesis via inhibition of extracellular signal‐regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol 70: 1992–2003. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller A, Ask K, Warburton D, Gauldie J, Kolb M (2008). The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol 40: 362–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore B, Lawson WE, Oury TD, Sisson TH, Raghavendran K, Hogaboam CM (2013). Animal models of fibrotic lung disease. Am J Respir Cell Mol Biol 49: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M et al (2014). Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One 9: e97550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D et al (2011). Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 377: 1760–1769. [DOI] [PubMed] [Google Scholar]
- van Noord JA, Smeets JJ, Drenth BM, Rascher J, Pivovarova A, Hamilton AL et al (2011). 24‐hour bronchodilation following a single dose of the novel beta(2)‐agonist olodaterol in COPD. Pulm Pharmacol Ther 24: 666–672. [DOI] [PubMed] [Google Scholar]
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK et al (2011). An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence‐based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U et al (2014). Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 370: 2071–2082. [DOI] [PubMed] [Google Scholar]
- Schiller M, Dennler S, Anderegg U, Kokot A, Simon JC, Luger TA et al (2010). Increased cAMP levels modulate transforming growth factor‐beta/Smad‐induced expression of extracellular matrix components and other key fibroblast effector functions. J Biol Chem 285: 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi‐Wen X, Rodriguez‐Pascual F, Lamas S, Holmes A, Howat S, Pearson JD et al (2006). Constitutive ALK5‐independent c‐Jun N‐terminal kinase activation contributes to endothelin‐1 overexpression in pulmonary fibrosis: evidence of an autocrine endothelin loop operating through the endothelin A and B receptors. Mol Cell Biol 26: 5518–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork PJ, Schmitt JM (2002). Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol 12: 258–266. [DOI] [PubMed] [Google Scholar]
- Strobel B, Duechs MJ, Schmid R, Stierstorfer BE, Bucher H, Quast K et al (2015). Modeling pulmonary disease pathways using recombinant adeno‐associated virus 6.2. Am J Respir Cell Mol Biol 53: 291–302. [DOI] [PubMed] [Google Scholar]
- Swigris JJ, Brown KK (2010). The role of endothelin‐1 in the pathogenesis of idiopathic pulmonary fibrosis. BioDrugs 24: 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannheimer SL, Wright CD, Salmon M (2012). Combination of roflumilast with a beta‐2 adrenergic receptor agonist inhibits proinflammatory and profibrotic mediator release from human lung fibroblasts. Respir Res 13: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd NW, Luzina IG, Atamas SP (2012). Molecular and cellular mechanisms of pulmonary fibrosis. Fibrogenesis Tissue Repair 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Nelson A, Gunji Y, Farid M, Basma H, Ikari J et al (2012). Olodaterol inhibits type i collagen synthesis by human lung fibroblasts. Am J Respir Crit Care Med 185: A1923. [Google Scholar]
- Wex E, Bouyssou T (2015). Olodaterol attenuates citric acid‐induced cough in naive and ovalbumin‐sensitized and challenged guinea pigs. PLoS One 10: e0119953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wex E, Kollak I, Duechs MJ, Naline E, Wollin L, Devillier P (2015). The long‐acting beta2 ‐adrenoceptor agonist olodaterol attenuates pulmonary inflammation. Br J Pharmacol 172: 3537–3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollin L, Maillet I, Quesniaux V, Holweg A, Ryffel B (2014). Antifibrotic and anti‐inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J Pharmacol Exp Ther 349: 209–220. [DOI] [PubMed] [Google Scholar]