

Abstract
Background and Purpose
Tissue plasminogen activator (tPA) is the only approved pharmacological therapy for acute brain ischaemia; however, a major limitation of tPA is the haemorrhagic transformation that follows tPA treatment. Here, we determined whether nicotinamide mononucleotide (NMN), a key intermediate of nicotinamide adenine dinucleotide biosynthesis, affects tPA‐induced haemorrhagic transformation.
Experimental Approach
Middle cerebral artery occlusion (MCAO) was achieved in CD1 mice by introducing a filament to the left MCA for 5 h. When the filament was removed for reperfusion, tPA was infused via the tail vein. A single dose of NMN was injected i.p. (300 mg·kg−1). Mice were killed at 24 h post ischaemia, and their brains were evaluated for brain infarction, oedema, haemoglobin content, apoptosis, neuroinflammation, blood–brain barrier (BBB) permeability, the expression of tight junction proteins (TJPs) and the activity/expression of MMPs.
Key Results
In the mice infused with tPA at 5 h post ischaemia, there were significant increases in mortality, brain infarction, brain oedema, brain haemoglobin level, neural apoptosis, Iba‐1 staining (microglia activation) and myeloperoxidase staining (neutrophil infiltration). All these tPA‐induced alterations were significantly prevented by NMN administration. Mechanistically, the delayed tPA treatment increased BBB permeability by down‐regulating TJPs, including claudin‐1, occludin and zonula occludens‐1, and enhancing the activities and protein expression of MMP9 and MMP2. Similarly, NMN administration partly blocked these tPA‐induced molecular changes.
Conclusions and Implications
Our results demonstrate that NMN ameliorates tPA‐induced haemorrhagic transformation in brain ischaemia by maintaining the integrity of the BBB.
Abbreviations
- BBB
blood–brain barrier
- ECA
external carotid artery
- MCAO
middle cerebral artery occlusion
- MPO‐1
myeloperoxidase‐1
- NAD
nicotinamide adenine dinucleotide
- NMN
nicotinamide mononucleotide
- NSC
neural stem cell
- TJP
tight junction proteins
- ZO‐1
zonula occludens‐1
- tPA
tissue plasminogen activator
Introduction
Currently, intravenous treatment with tissue plasminogen activator (tPA) is the only Food and Drug Administration‐approved pharmacological treatment for acute ischaemic stroke (Docagne et al., 2015). Haemorrhagic transformation is the most feared complication of intravenous tPA treatment. Generally, the time window of tPA treatment is within 0–4.5 h and several studies have shown that a delayed tPA treatment (>4.5 h) does not decrease brain infarction but instead worsens haemorrhagic conversion (Murata et al., 2008; Zhang et al., 2009). Many parameters, including the severity of the stroke, blood pressure, cardiac function, the extent of parenchymal hypoattenuation and increasing age of the patient, contribute to the occurrence of haemorrhagic transformation with tPA (Larrue et al., 2001).
Disruption of the blood–brain barrier (BBB) is thought to be one of the main causes of tPA‐associated haemorrhagic transformation (Kastrup et al., 2008). The BBB is formed by the interaction of cerebral capillary endothelial cells with pericytes and astrocytes. The tight junctions of the BBB have a very low permeability that restricts the entry of substances in the peripheral circulation from entering the brain (Yang and Rosenberg, 2011). The tPA activates matrix metalloproteinases (MMPs) that then cause the destruction of tight junction proteins (TJPs), including claudins, occludin and zonula occludens‐1 (ZO‐1). This results in the disruption in the integrity of the BBB (Yang et al., 2007). Thus, the prevention or amelioration of tPA‐associated haemorrhagic transformation by increasing the resistance of the cerebral vascular system to ischaemic damage is a great challenge and yet unsolved problem.
Nicotinamide adenine dinucleotide (NAD) is a coenzyme, which is essential for mitochondrial electron transfer reactions (Rich, 2003). Recently, several lines of evidence have implicated the critical roles of NAD in a broad range of biological functions. NAD participates in the transduction of numerous important intracellular signalling pathways and controls cell proliferation, death, metabolism, circadian rhythms and ageing (Garten et al., 2015; Wang and Miao, 2015). The effects of NAD on a group of NAD‐dependent proteins, such as the sirtuin family of proteins and PARP1, are the main mechanisms behind NAD's biological functions (Garten et al., 2015; Wang and Miao, 2015; Zhang et al., 2016; Jokinen et al., 2017). Depletion of intracellular NAD is a common pathway in many pathophysiological events, including ageing, metabolic disorders and cerebral ischaemia (Yang et al., 2002; Massudi et al., 2012; Zhang et al., 2016; Zhou et al., 2016; Wang et al., 2016a). The administration of nicotinamide mononucleotide (NMN) or nicotinamide riboside, two NAD precursors, as a supplement is able to enhance NAD levels and is used to treat high‐fat–induced obesity/fatty liver (Yoshino et al., 2011; Uddin et al., 2016; Zhou et al., 2016). We and other groups have demonstrated that NMN protects against post‐ischaemic NAD degradation and decreases brain damage after cerebral ischaemia (Wang et al., 2011; 2012; Zhao et al., 2014; 2015b; Park et al., 2016).
Since tPA is an important pharmacological therapy for acute cerebral ischaemia, the potential effect of NMN on tPA treatment needed be investigated. In the present study, we showed that adjuvant treatment with NMN dramatically ameliorated tPA‐induced brain injury via maintaining BBB integrity in the mouse middle cerebral artery occlusion (MCAO) model.
Methods
Drug and animals
NMN was purchased from Bontac‐Biotech Synthesis Corp. (Shenzhen, China). The purity of NMN was >98%, which was confirmed by HPLC analysis. The chemical structure of NMN is shown in Figure 1A. Male 8‐week‐old CD1 mice (22–30 g) were purchased from Sino‐British SIPPR/BK Lab Animal Ltd (Shanghai, China). They were kept at a constant temperature of 23 ± 3°C and relative humidity of 30–70% under a 12 h light/dark cycle with free access to chow and water. The experimental procedures were approved by the Laboratory Animal Care and Use Committee of the Second Military Medical University, China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Every effort was taken to minimize the number of animals used and their suffering.
Figure 1.
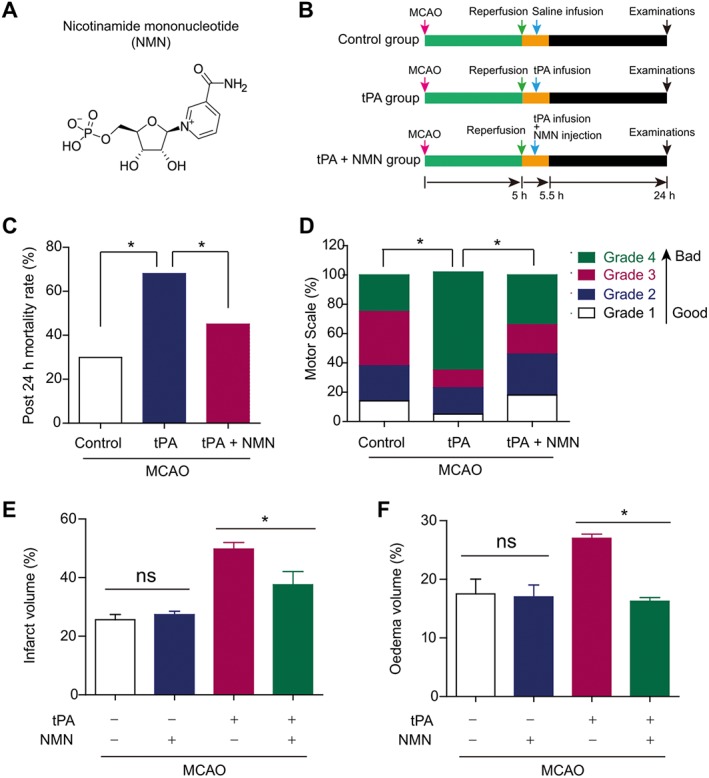
Experimental design and effects of NMN on animal survival in delayed tPA‐treated mice. (A) Chemical structure of NMN. (B) Experimental design. (C) Effects of NMN on mice mortality at 24 h post MCAO. (D) Effects of NMN of motor scale distribution at 24 h post MCAO. (E–F) Effects of NMN on brain infarct volume (E) and oedema volume (F). *P < 0.05. ns, no significance; n = 10 per group.
Randomization and blinding
Mice in the present study were divided into different groups randomly. All the experiments were performed blind; the evaluator did not know from which group the mice or samples came from.
Mouse MCAO model
The experimental model of cerebral ischaemia used in this study was the mouse MCAO. The MCAO model was prepared exactly as described by us previously (Wang et al., 2011; 2012; Li et al., 2017). Briefly, the mice were anaesthetized with 4% choral hydrate (350 mg·kg−1, i.p.), and the external carotid artery (ECA) and internal carotid artery were exposed respectively. A nylon suture (Sunbio Bioteck, Beijing, China) with a diameter of 0.38 mm (Sunbio Bioteck) was inserted via the ECA, advanced to a distance of 20 mm and kept there for 5 h, then withdrawn for reperfusion and delayed tPA treatment. Cortical blood flow was measured with a laser‐doppler flowmeter (Moor Instruments LTD, Millwey, Axminster, UK) to ensure that cerebral blood flow was reduced by more than 80% and the rectal temperature was maintained at 36.5–37.5°C by a homeothermic heating pad. Mice that showed convulsions, sustained impaired consciousness or had no apparent contralateral limb dysfunction were excluded from subsequent experiments.
Delayed tPA treatment and NMN administration
The mice were subjected to the MCAO operation and then divided into three groups: Control group, tPA group and tPA + NMN group. The experimental design is illustrated in Figure 1B. At 5 h post ischaemia, the nylon suture was withdrawn for reperfusion. Immediately, the mice in the tPA group were infused with recombinant human tPA (9 mg·kg−1 in 1 mL saline, Actilyse®, Boehringer Ingelheim, Ingelheim am Rhein, Germany) via the tail vein for 30 min with a minipump. In the tPA + NMN group, the mice were infused with tPA as described above and injected i.p. with a single dose of NMN (300 or 100 mg·kg−1) dissolved in saline. In the control group, mice were infused and injected with the same volumes of saline. At 24 h after ischaemia, the mortality was calculated. The mice that died before the 24 h time point were included only in the experiment of post 24 h mortality rate; these mice were not included in the other analyses. In previous studies, NMN was administered to mice at doses of ~100 mg·kg−1 (Park et al., 2016) to 500 mg·kg−1 (Long et al., 2015; Uddin et al., 2016). However, most studies applied the dose of 300 mg·kg−1 (Wang et al., 2011; Yoshino et al., 2011; Zhang et al., 2011; Stein and Imai, 2014; Zhao et al., 2015b; de Picciotto et al., 2016; Wei et al., 2017); therefore, we used 300 mg·kg−1 in this study.
Motor function evaluation
Outcomes at 24 h after ischaemia were scored using the 5‐point motor function scale as described previously (Wang et al., 2011), in a non‐blinded manner: 0 = no motor deficit; 1 = flexion of the forelimb contralateral to the ischaemic hemisphere; 2 = reduced resistance against push toward the paretic side; 3 = spontaneous circling toward the paretic side; and 4 = death.
Infarct and oedema volume
Mice were transcardially perfused with PBS and killed at 24 h after ischaemia, and brains were removed and cut into six 2‐mm‐thick consecutive sections followed by staining with 1.5% 5‐tri‐phenyl‐2H‐tetrazolium chloride (Sigma, T8877) as described previously (Wang et al., 2011; Wang et al., 2012). The white area was thought to represent infarcted brain tissue and the red area non‐infarcted tissue. The infarct and oedema volume were acquired by digital cameras and analysed using Image J software (NIH).
Haemoglobin assay
A colourimetric haemoglobin assay (#700540, Cayman Chemical, Ann Arbor, MI) was used to assess the haemoglobin content of brains tissues. Mice were killed under deep anaesthesia and infused with cold PBS (45 mL). The brains were removed immediately and divided into right and left hemispheres. Haemorrhagic hemispheres were isolated and washed in ice‐cold PBS solution for three times. Then, 1 g tissue was homogenized with 1 mL PBS solution with 0.16 g·L−1 heparin. After being centrifuged at 10 000 g for 10 min, the supernatant was collected for determination of haemoglobin using the commercial kit according to the manufacturer's instructions. The optical absorbance of each the haemoglobin standards was measured using a microplate reader to obtain a standard curve. Then, the concentrations of haemoglobin in the tissue extracts were calculated using their absorbance and the standard curve. Finally, the average brain haemoglobin levels were measured as haemoglobin content relative to the tissue protein concentration.
Assessment of BBB integrity
Evans Blue extravasation was used to determine BBB integrity as described previously (Liu et al., 2016). At 24 h after MCAO, 2% Evans Blue dye (4 mL·kg−1, Sigma) was injected via the tail vein. The mice were anaesthetized using 4% choral hydrate (350 mg·kg−1, i.p.) at 2 h after injection of Evans Blue and then perfused with 0.01 M PBS solution. The brains were rapidly harvested and imaged. Then the brain tissue was weighed, homogenized in 50% ice‐cold trichloracetic acid and then centrifuged at 12 000 g at 4°C for 15 min to remove the debris. The supernatants were transferred to a new tube. To determine the quantity of Evans Blue in the supernatants, the absorbance at 620 nm was measured spectrophotometrically using an Infinite M200 plate reader (Tecan, Research Triangle Park, NC). Evans Blue leakage into the brain tissue was assessed by reference to a standard curve and expressed as μg.g‐1 wet brain tissue.
Histology examination
The histology was examined as described previously (Wang et al., 2014a,b, 2016b). Briefly, brain tissues were embedded in paraffin, cut into 8 μm sections. The sections were deparaffinized and stained with haematoxylin and eosin and examined under a light microscope (Leica Microsystems, Berlin, Germany). The haemorrhage was visible under the microscope and calculated.
elisa
The levels of TNF‐α and IL‐1β in brain tissues were determined with commercial elisa kits (RD system, Minneapolis, MN, USA). The lowest sensitivity of the TNF‐α elisa kit is 7.21 pg·mL−1, while the sensitivity for IL‐1β elisa kit is 4.8 pg·mL−1. Briefly, brain tissue was homogenized in ice‐cold PBS (pH 7.4) containing a mixture of protease inhibitors first and then centrifuged at 12 000 g for 10 min at 4°C. The supernatant was collected and used to assess the levels of TNF‐α and IL‐1β. The elisa kits were used to assess the levels in accordance with the manufacturer's instructions. The protein concentration in each sample was determined using the Bradford method (Beyotime, Haimen, China). Then, a total of 50 μL tissue samples were loaded into the 96‐well plate. Experiments were performed with duplicate wells for each sample. The standard curve was produced by serial dilutions of standard recombinant TNF‐α and IL‐1β samples in the kits and calculated using a linear regression programme. The concentrations of TNF‐α and IL‐1β in each sample were calculated according to the standard curve and normalized to the protein concentration.
Caspase‐3 activity
Caspase‐3 activity was determined using a commercial kit purchased from BioVision Inc (Mountain View, CA, USA) according the manufacturer's instruction. This kit was generated based on a specific tetrapeptide substrate DEVD‐para‐nitroaniline (pNA). The activity of caspase‐3 was quantified by spectrophotometric detection of free pNA after cleavage from the peptide substrate DEVD‐pNA, using an M200 microplate reader (Tecan, Group Ltd., Switzerland).
Immunohistochemistry staining
Immunohistochemistry staining was performed as described in our previous studies (Wang et al., 2012). Each primary antibody was tested before the formal experiments for immunohistochemistry were performed. Moreover, normal IgG was used as a negative control to validate the specific staining in these experiments. At 24 h after MCAO, anaesthetized mice were perfused under deep with 20 mL cold PBS (pH = 7.4), followed by an infusion of 4% paraformaldehyde for 10 min. The brains were then removed and fixed in 4% paraformaldehyde at 4°C overnight. The brains were dehydrated with 30% sucrose in formalin (pH = 7.4), and the frozen coronal slices (8 μm thick) were then sectioned using a cryostat (CM3050S; Leica Microsystems, Bannockburn, IL, USA). The sections were blocked in 8% normal goat serum for 4 h and incubated in specific primary antibodies overnight at 4°C. After being washed three times with PBS, the sections were incubated with horseradish peroxidase‐conjugated secondary antibodies. Staining was visualized using the chromogenic substrate DAB. Images were obtained with a digital microscope (Leica Microsystems, Berlin, Germany) and analysed with a computerized image system (Quantimet 500 Image Processing and Analysis System, Qwin V0200B software; Leica, Berlin, Germany). The following antibodies were used for immunohistochemistry staining: Iba‐1 (#ab5076, Abcam, 1: 1000 dilution), myeloperoxidase‐1 (MPO‐1, #BA0544, Boster, Wuhan, China, 1: 200 dilution), TNF‐α (#ab6671, Abcam, 1: 200 dilution) and IL‐1β (#ab8320, Abcam, 1: 500 dilution). For quantification of immunohistochemistry staining, eight sections per animal and five random microscope fields per section were chosen. The average was then calculated.
TUNEL staining
We used TUNEL staining kit (DeadEnd Colorimetric TUNEL system; Promega) to detect apoptosis in brain ischaemic areas according to the manufacturer's instructions. The main procedure of TUNEL assay has been described in detail in our previous studies (Wang et al., 2009; 2011). For quantification of immunohistochemistry staining, five sections per animal and five random microscope fields per section were chosen. The average was then calculated.
Protein extraction and immunoblotting
Protein extraction and immunoblotting were performed as described previously (Wang et al., 2011; 2014c). Briefly, brain tissues were washed in PBS (0.1 mmol·L−1) and homogenized with the RIPA buffer (Beyotime Biotechnology, Haimen, China) with a protease inhibitor cocktail (Pierce, Rockford, IL). The peri‐haemorrhagic brain tissues were isolated for determination of protein expression of MMP2 and MMP9. Immunoblotting was performed in Odyssey Infrared Fluorescence Imaging System (Li‐Cor, Lincoln, NE). The protein concentration was determined by the Bradford assay. About 30 μg samples were run on 10% SDS‐PAGE. The proteins were electrotransferred to nitrocellulose membranes, probed with primary antibodies against MMP‐2 (1: 800, Abcam, #ab37150), MMP‐9 (1: 800, Abcam, #ab38898), claudin‐1 (1: 600, Santa Cruz Biotechnology, sc‐17658), occludin (1: 600, Santa Cruz Biotechnology, sc‐5562) and ZO‐1 (1: 600, Santa Cruz Biotechnology, sc‐8146) overnight and then incubated with corresponding secondary antibodies conjugated with Infrared‐Dye (Li‐Cor, Lincoln, NE, USA). The images were obtained with an Odyssey Infrared Fluorescence Imaging System (Li‐Cor, Lincoln, NE). All immunoblotting experiments were repeated at least three times.
MMP activity
MMP activity was assayed using a Fluorometric‐Green Probe® Kit from Abcam (#112146). This kit uses a FRET peptide as an indicator of generic MMP activity. In the intact FRET peptide, the fluorescence of one part is quenched by another. After cleavage into two separate fragments by MMPs, the fluorescence signal is detected. In our experiments, the brain samples were homogenized with 300 μL distilled water with protease inhibitor. After being centrifuged at 3000 g for 10 min, the supernatant was collected and adjusted to similar protein concentrations and then examined using this kit with a fluorescence microplate reader at Ex/Em = 490/525 nm for 60 min. The fluorescence signal was recorded every 5 min.
Assessment of tPA activity
The tPA activity was measured using a commercial kit (Abcam). In this kit, the amount of plasmin produced is quantified using a highly specific plasmin substrate that releases a yellow para‐nitroaniline (pNA) chromophore. The change in absorbance of the pNA in the reaction solution at 405 nm is directly proportional to the tPA enzymatic activity. Briefly, the same amounts of assay mix (80 μL) and tPA (20 μL) were prepared. Then, 100 μL distilled water or NMN solution was added. The final concentration of NMN in the mixture was 300 μM. Then, the entire mixture was incubated at 37°C in a humid incubator to form the colourful pNA for detection. The absorbance of 405 nm was recorded and analysed.
Statistical analysis
All values are presented as the mean ± SEM. The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data were tested for normality by applying the Kolmogorov–Smirnov test. If normality was agreed and there were no significant differences in variance between groups (F test), Student's two‐tailed t‐test or one‐way ANOVA followed by Tukey's post hoc test was used. Otherwise, the nonparametric Kruskal–Wallis test followed by Mann–Whitney U‐test was used. Data were analysed with SPSS11.0 software (SPSS Inc.,Chicago, IL) and GraphPad Prism‐5 statistic software (La Jolla, CA). P < 0.05 was considered statistically significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Administration of NMN prevents delayed tPA treatment‐induced brain damage
The chemical structure of NMN and the experimental process are shown in Figure 1A–B respectively. The mortality in MCAO mice (control group) was 29% (Figure 1C). Delayed tPA treatment caused a very high mortality (67.1%) in MCAO mice (Figure 1C). We tested the effects of NMN in this mouse model. Our preliminary results showed that NMN was effective in preventing tPA‐induced haemorrhagic transformation at 300 mg·kg−1 (i.p.) but not at 100 mg·kg−1 (i.p., data no shown). Thus, we used NMN at 300 mg·kg−1 (i.p.) in this study. In the tPA‐treated mice, NMN administration significantly reduced the mortality from 67.1 to 42.4% (Figure 1C). Motor scale data also showed delayed tPA treatment markedly aggravated the neurological deficits in MCAO mice, which was largely prevented by NMN administration (Figure 1D). Moreover, both the brain infarction volume (Figure 1E) and brain oedema (Figure 1F) in tPA‐treated mice were higher than those in the control group, suggesting that delayed tPA treatment exacerbated ischaemia‐induced brain injury. NMN administration, not only inhibited the delayed tPA treatment‐induced enhancement of brain infarction volume (Figure 1E) but also attenuated the worsened brain oedema (Figure 1F). These results indicate that administration of NMN prevents delayed tPA treatment‐induced brain damage.
Administration of NMN restrains delayed tPA treatment‐induced cerebral haemorrhage
The brains from the mice of three groups were isolated and sectioned into several segments. As shown in Figure 2A, there was slight cerebral haemorrhage in the ipsilateral hemisphere in MCAO mice without tPA treatment (control group). In the delayed tPA‐treated mice, the cerebral haemorrhage was serious, while adjuvant administration of NMN significantly decreased the cerebral haemorrhage induced by tPA (Figure 2A). The quantitative results confirmed that the haemoglobin level in the ipsilateral hemisphere of tPA‐treated mice was about ~2‐fold greater than that in mice of the control group, and NMN administration significantly attenuated this change (Figure 2B). We also determined the cerebral haemorrhage using histology. As shown in Figure 2C–D, there were a large number of haemorrhagic areas in brain sections from tPA‐treated mice, and NMN administration significantly attenuated the brain haemorrhage (Figure 2C–D). To rule out the possibility that NMN reduces tPA thrombolytic activity directly, we measured the effect of NMN (300 μM) on tPA thrombolytic activity in vitro. As shown in Supporting Information Figure S1, NMN did not alter the tPA thrombolytic activity. These results suggest that the cerebral haemorrhage induced by delayed tPA treatment can be partly prevented by NMN.
Figure 2.
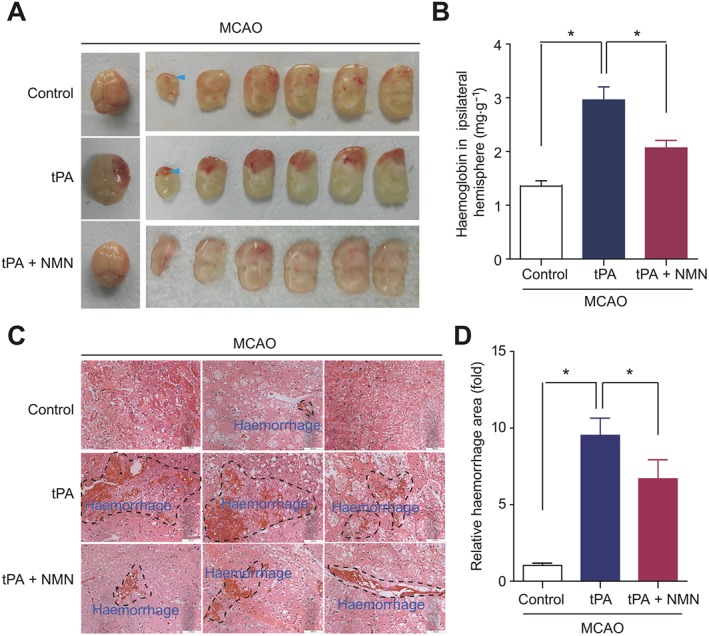
Effects of NMN on brain haemorrhage in delayed tPA‐treated mice. (A) Representative images of intact brain and corresponding brain sections showing the haemorrhagic transformation induced by delayed tPA treatment and the effects of NMN on brain haemorrhagic transformation. (B) Haemoglobin levels in ipsilateral brain hemisphere determined with a commercial kit. (C) Representative images of haematoxylin and eosin staining showing the haemorrhage area (black dotted line) in mouse brain sections. (D) Quantitative analysis of haemorrhage area in mice brain sections. For quantification of haemorrhage, 10 sections per animal and at least five random microscope fields per section were chosen. The average number was then calculated. *P < 0.05; n = 10 per group.
Administration of NMN inhibits delayed tPA treatment‐induced neural apoptosis
TUNEL staining demonstrated that there were few apoptotic cells (TUNEL positive, brown) in the brain sections from mice that had not been subjected to a MCAO operation (Figure 3A). In the MCAO mice (control group), neural apoptosis was significantly increased (Figure 3A). Delayed tPA treatment further increased the proportion of apoptotic neural cells. This action of tPA was largely inhibited by NMN administration (Figure 3A). Quantitative analysis confirmed this result (Figure 3B). Cleaved caspase‐3 is a well‐established indicator of apoptotic pathway activation (Porter and Janicke, 1999). In line with the data from the TUNEL staining, NMN administration substantially inhibited the increased caspase‐3 activity induced by delayed tPA treatment in ischaemic brain tissues (Figure 3C). Thus, these data indicate that NMN is able to inhibit delayed tPA treatment‐induced neural apoptosis.
Figure 3.
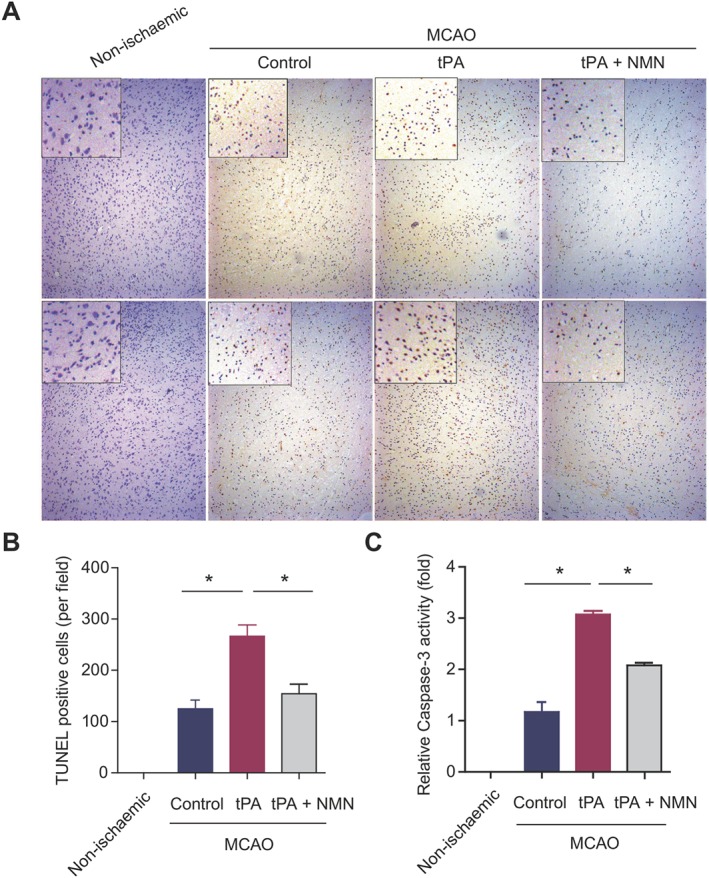
Effects of NMN on neural apoptosis in delayed tPA‐treated mice. (A) The global and magnified images of TUNEL staining in brain sections. Brown staining (positive) was scarce in the brain sections from mice without MCAO operation (non‐ischaemic). In the MCAO mice, the number of brown‐stained cells was largely increased, which was reduced by NMN administration. (B) Quantitative analysis of TUNEL‐positive cells in mouse brain sections. For quantification of TUNEL staining, eight sections per animal and five random microscope fields per section were chosen. The average was then calculated. (C) Comparison of caspase‐3 activities in brain tissues from different groups. *P < 0.05; n = 8 per group.
Administration of NMN suppresses delayed tPA treatment‐induced neuroinflammation
Treatment with tPA has been reported to trigger the recruitment of microglia and macrophage and induce pro‐inflammatory M1 polarization leading to neuroinflammation (Won et al., 2015). So we used immunohistochemistry to examine the expression of Iba‐1, a microglia‐specific protein marker. We found all the microglia cells in brain sections from the three groups of mice displayed activated phenotypes (Figure 4A). However, the number of Iba‐1‐positive microglia in tPA‐treated mice was significantly higher than that in the control group (Figure 4A–B). This tPA‐induced microglia activation was partly prevented by NMN administration (Figure 4A–B). The expression of MPO‐1, a marker of infiltrated neutrophils, was also determined using immunohistochemistry. Similarly, delayed tPA treatment caused a significant increase in the number of MPO‐1 positive cells, suggesting tPA enhances the infiltration of neutrophils (Figure 4C–D). NMN administration, slightly but significantly, reduced the tPA‐induced up‐regulation of MPO‐1 (Figure 4C–D).
Figure 4.
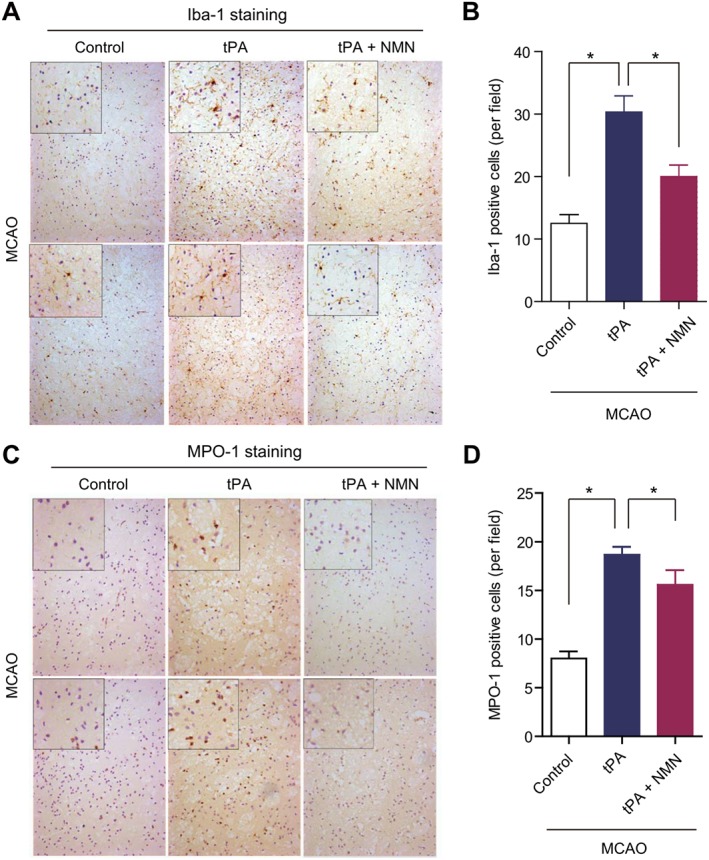
Effects of NMN on microglia activation and neutrophil infiltration in delayed tPA‐treated mice. (A) The global and magnified images of microglial marker Iba‐1 immunohistochemical staining. (B) Quantitative analysis of Iba‐1 positive microglia in mouse brain sections. (C) The global and magnified images of neutrophil marker MPO‐1 immunohistochemical staining. (D) Quantitative analysis of MPO‐1‐positive neutrophils in mouse brain sections. For quantification of immunohistochemical staining, eight sections per animal and five random microscope fields per section were chosen. The average was then calculated. *P < 0.05; n = 8 per group.
In agreement with these results, the level of TNF‐α in brain tissue from tPA‐treated mice was higher than that in control mice, and this increase was partly blocked by NMN administration (Figure 5A). In contrast, there was no difference in the IL‐1β level in the brain tissues among the three groups of mice (Figure 5B). Immunohistochemistry of TNF‐α and IL‐1β in the mouse brain sections from three groups also confirmed these results (Figure 5C–D). These results demonstrate that NMN ameliorates delayed tPA treatment‐induced microglia activation and neuroinflammation.
Figure 5.
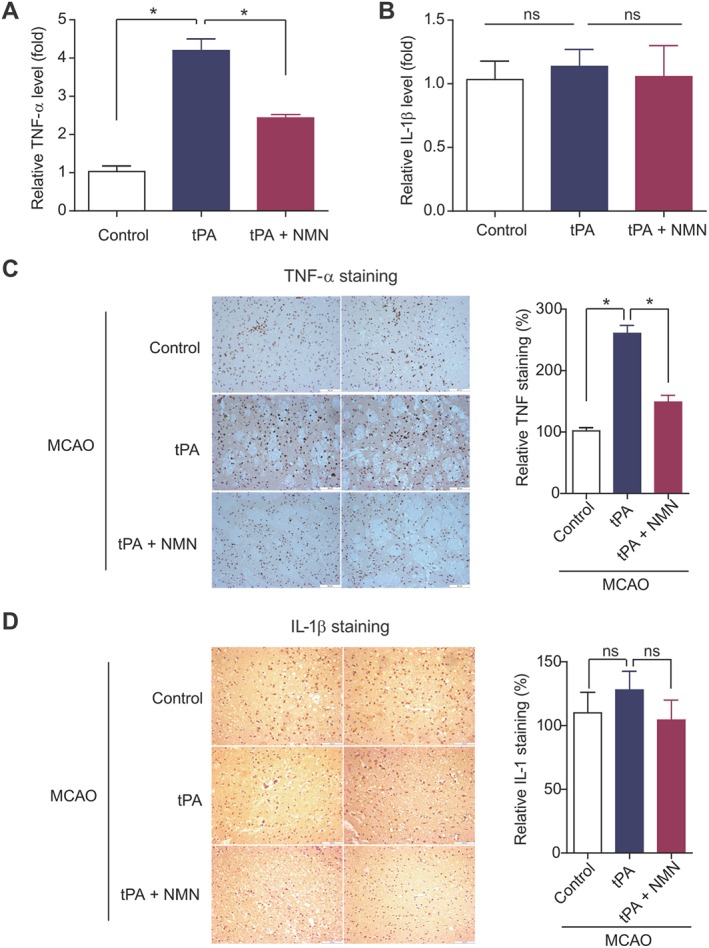
Effects of NMN on pro‐inflammatory factors in delayed tPA‐treated mice. (A–B) Comparison of tissue TNF‐α (A) and IL‐1β (B) concentrations by elisa kits. (C) Representative images of immunohistochemical staining of TNF‐α and IL‐1β in mouse brain sections. (D) Quantitative analysis of immunohistochemical staining of TNF‐α and IL‐1β in mouse brain sections. Eight sections per animal and five random microscope fields per section were chosen. The average was then calculated. *P < 0.05. ns, no significance; n = 8 per group.
Protection of BBB integrity contributes to the beneficial effects of NMN against tPA‐induced haemorrhagic transformation
The ischaemic brain hemisphere of tPA‐treat mice displayed a much severer leakage of Evans Blue dye compared with the control group of mice (Figure 6A), suggesting tPA disrupts the BBB. NMN administration obviously prevented the tPA‐induced increase in Evans Blue extravasation (Figure 6A). Quantitative analysis of the Evans Blue leakage confirmed this result. Delayed tPA treatment increased the amount of Evans Blue extravasation by ~5‐fold, which was markedly depressed by NMN administration (Figure 6B). Immunoblotting analysis demonstrated that the protein levels of three types of TJPs (claudin‐1, occludin and ZO‐1) were down‐regulated by delayed tPA treatment (Figure 6C–F). NMN administration, at least partly, restored the protein expression of claudin‐1, occludin and ZO‐1 (Figure 6C–F). These data indicate that NMN protects BBB integrity.
Figure 6.
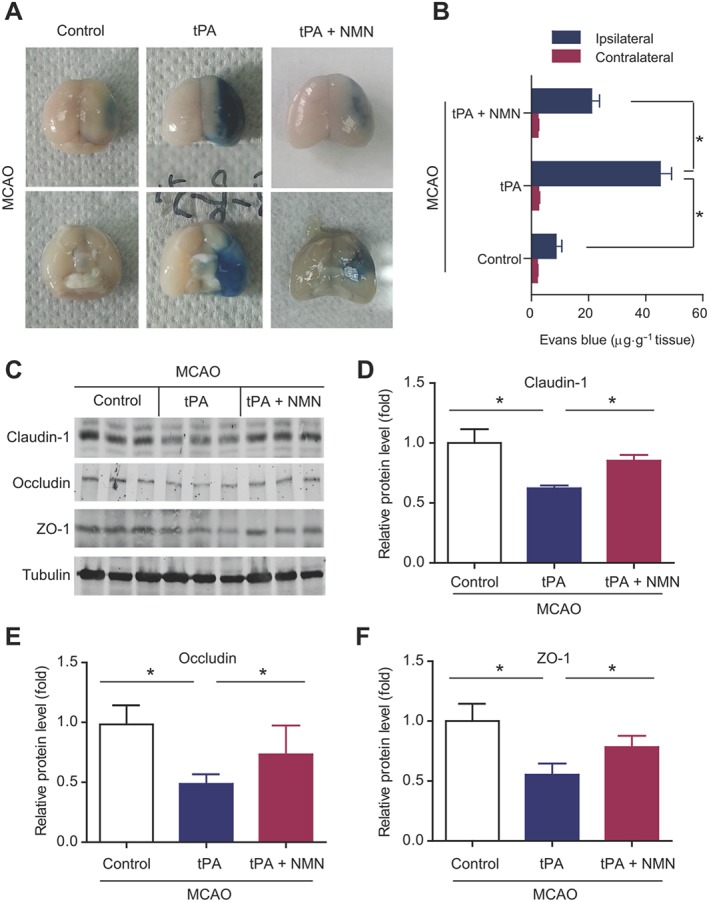
Effects of NMN on BBB integrity in delayed tPA‐treated mice. (A) Representative images of extravasation of Evans Blue in mouse brains. (B) Quantitative analysis of the extravasation of Evans Blue dye into brain hemisphere. *P < 0.05; n = 8 per group. (C) Representative images of immunoblotting of claudin‐1, occludin and ZO‐1. (D–F) Quantitative analysis of protein expressions of claudin‐1, occludin and ZO‐1 in mouse brain tissues. *P < 0.05; n = 6 per group.
NMN protects BBB integrity by suppressing tPA‐induced activation of MMPs
In addition, we explored how NMN protects BBB integrity. MMP9 and MMP2 are two structurally‐similar MMPs regulating TJPs destruction and BBB integrity in tPA‐associated haemorrhage (Rempe et al., 2016). The kinetic analysis based on a FRET‐peptide fluorometric kit showed that delayed tPA treatment significantly enhanced the activities of MMP9 (Figure 7A) and MMP2 (Figure 7B). NMN administration almost totally abolished the increases in MMP9 and MMP2 activities induced by tPA (Figure 7A–B). Delayed tPA treatment also enhanced the protein expressions of pro‐MMP9 and active‐MMP9 (Figure 7C), both of which were suppressed by NMN administration (Figure 7C). Interestingly, delayed tPA treatment up‐regulated active‐MMP2 but did not alter the expression of pro‐MMP2 (Figure 7D). NMN administration significantly inhibited the tPA‐induced up‐regulation of active‐MMP2 (Figure 7D). All these data suggest that the suppression of tPA‐induced activation of MMPs by NMN may be an important mechanism underlying the protective action of NMN on BBB integrity.
Figure 7.
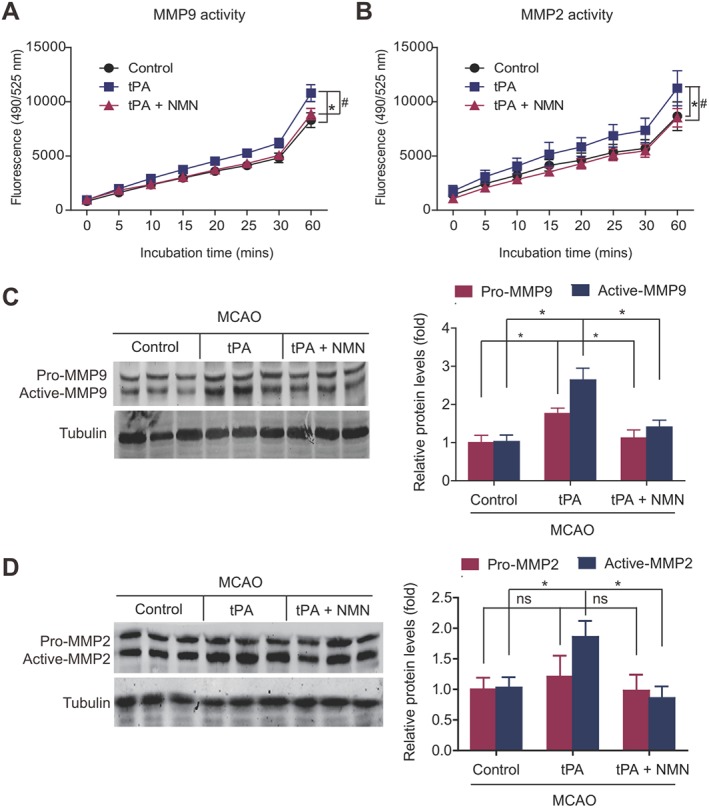
Effects of NMN on activation of MMPs in delayed tPA‐treated mice. (A–B) Kinetic readouts of MMP9 and MMP2 activities in brain tissue samples. The fluorescence (excitation 490 nm and emission 525 nm) of brain tissue samples was monitored via a kinetic reading for 60 min. *P < 0.05 tPA versus control. # P < 0.05 tPA + NMN versus tPA; n = 6 per group. (C) Representative images and quantitative analysis of immunoblotting of MMP9. The primary antibody recognizes both pro‐MMP9 and active‐MMP9. *P < 0.05; n = 6. (D) Representative images and quantitative analysis of immunoblotting of MMP2. The primary antibody recognizes both pro‐MMP2 and active‐MMP2. *P < 0.05. ns, no significance; n = 6.
Discussion
Previously, we showed that NMN had a potent neuroprotective effect against acute ischaemia‐induced neuron death (Wang et al., 2011) and facilitated post‐ischaemia neurogenesis (Wang et al., 2015). As NMN affects NAD levels and thus participates in multiple intracellular biological functions, we proposed that it may also alter the effects of tPA therapy in cerebral ischaemia. If NMN worsens the tPA‐induced haemorrhagic transformation, there is no question that the therapeutic value of NMN in cerebral ischaemia would be greatly compromised because it is the only approved thrombolysis therapy for most patients with acute cerebral ischaemia. If NMN does not affect or even reduces the tPA‐induced haemorrhagic transformation, NMN would be a promising chemical compound with multiple functions in cerebral ischaemia treatment. As expected, our results demonstrated that NMN administration decreased the delayed tPA treatment‐induced increase in brain infarction volume and oedema and, most importantly, reduced the high mortality in mice subjected to the delayed treatment with tPA.
As NMN is able to limit the tPA‐induced brain haemorrhage, we therefore investigated whether NMN affects the haemoglobin‐induced neuronal toxicity that followed the delayed tPA infusion. It is well accepted that brain haemorrhage results in cell death around the margins of the haemorrhage, induced by the toxic effects of haemoglobin (Yang and Rosenberg, 2011). In addition, tPA further amplifies the haemoglobin‐induced neurotoxicity in PC12 neuronal cells and primary cultured neurons (Wang et al., 1999), suggesting that the deterioration of patients with haemorrhagic transformation may be due to both the effects of haemorrhage per se as well as the additional toxicity of tPA‐amplified haemoglobin neurotoxicity. Extensive neuroinflammation, caspase activation and an increase in oxidative stress are the predominating factors of haemoglobin‐induced toxicity (Wang et al., 2002). In our study, NMN treatment was found to attenuate the tPA‐induced neural apoptosis, microglia activation, neutrophil infiltration and release of pro‐inflammatory factors. In addition, delayed NMN treatment, significantly improved the post‐ischaemic neural regenerative process through promoting the proliferation and differentiation of neural progenitor cells in the brain subventricular zone (Zhao et al., 2015b). Thus, these data support the notion that NMN may be a promising multi‐functional pharmacological agent for treatment of cerebral ischaemia in both early and late phases, rather than just a neuroprotector. In our study, delayed tPA administration (5 h post ischaemia) caused a significant increase in brain infarct volume. Many previous studies have shown that brain infarct volume was not affected by delayed tPA treatment in both a thromboembolic and filament model (Kanazawa et al., 2011; Garcia‐Culebras et al., 2017). However, delayed tPA treatment has also been found to tend to aggravate brain infarct volume (Zuo et al., 2014). This discrepancy may be due to differences in the dose of tPA, manufacturer of tPA or animal model used, or even the method used to measure of infarct volume. Further research is needed to resolve these disparities.
After discovering that NMN has a potent inhibitory effect on the tPA‐induced haemorrhagic transformation and haemoglobin neurotoxicity, we sought to find the molecular mechanisms underlying these effects of NMN. The results from the Evans Blue assay showed that delayed tPA treatment significantly disrupted the integrity of the BBB. We also examined the expression of TJPs, a group of integral membrane proteins, including claudin‐1, occludin and ZO‐1, which are necessary for cell‐to‐cell contacts and responsible for the barrier function of brain vessels. Immunoblots of claudin‐1, occludin and ZO‐1 clearly showed that delayed tPA treatment down‐regulated the expression of claudin‐1, occludin and ZO‐1, which was prevented by NMN administration. As MMPs are major enzymes involved in mediating the destruction of TJPs during tPA treatment (Yang et al., 2007; Kastrup et al., 2008), we further studied the activities and protein expression of MMP9 and MMP2. As expected, NMN significantly inhibited the tPA‐induced increase in MMP2/MMP9 activity and protein expression. These results were in line with those from the Evans Blue assay and TJP immunoblotting. Thus, we suggest that NMN affects the activity/expression of MMP9 and MMP2, and the resulting attenuation of the destruction of TJPs may contribute to the protective effect of NMN on BBB integrity.
Sirtuins, PARP1 and CD38 use NAD to critically regulate nutrient sensing and signalling pathway transduction (Imai and Guarente, 2014; Garten et al., 2015; Wang and Miao, 2015). Although all of them are major users of NAD, increasing NAD levels result in the activation of sirtuins but inhibition of PARP1/CD38 (Imai and Guarente, 2014; Garten et al., 2015; Wang and Miao, 2015). SIRT1 is a key regulator of cellular metabolism and mediates beneficial metabolic effects (Dagon et al., 2016). Chemical inhibition of SIRT1 led to BBB breakdown and aggravated brain oedema after experimental subarachnoid haemorrhage (Zhou et al., 2014). Conversely, inhibition of PARP1 in brain endothelium protects the BBB under physiological and neuroinflammatory conditions (Rom et al., 2015). Moreover, SIRT1 and SIRT3 seemed to be involved in the protective effects of melatonin (Zhao et al., 2015a) and minocycline (Yang et al., 2015) on BBB integrity respectively. As NMN is converted to NAD in vivo and then positively regulates sirtuin activity but negatively affects PARP1 activity, we propose that the regulatory action of NMN‐mediated NAD pool on sirtuins/PARP1 may replicate the protective effect of NMN on BBB integrity in this tPA‐induced haemorrhagic transformation model. Notably, the effects of NAD on the biological functions of cells are likely to be a result of the collaborative effects of several sirtuin proteins (Imai and Guarente, 2014). For example, we previously observed that SIRT1 and SIRT2 cooperatively regulate the proliferation of neural stem cells (NSCs), while SIRT1, SIRT2 and SIRT6 cooperatively regulate the differentiation of NSCs after ischaemic stroke (Zhao et al., 2015b). Thus, further studies may be warranted to clarify which sirtuin protein is involved in the protective effect of NMN on BBB integrity.
A major limitation in the present study lies in the fact that the animal model used does not mimic human embolic stroke very well. The filament model, photo‐thrombosis model and thromboembolic model are nearly always used for studying the therapeutic and side effects of tPA in cerebral ischaemia. The autologous thromboembolic model best mimics the pathophysiological state of ischaemic stroke; however, the instability of this model creates unavoidable problems (Zhang et al., 2015). Although the filament model is not directly relevant to the tPA‐induced thrombolysis during ischaemic stroke in patients, this stable model would allow us to compare the different effects of NMN on the complications associated with tPA under identical and controlled ischaemia and reperfusion conditions. Moreover, the main purpose of the present study was to investigate the influence of NMN on tPA‐induced BBB permeability and haemorrhagic transformation, rather than the potential influence of NMN on the thrombolytic activity of tPA. The filament model was used in many recent studies focusing on the disruption of the BBB induced by tPA (Wang et al., 2013; Liang et al., 2015; Hafez et al., 2016). After considering all these factors, we chose the filament model to induce MCAO in this study. Nevertheless, the limitations of this model should be kept in mind, in that our data only reflect the protective action of NMN on BBB integrity under this specific setting.
In conclusion, our results provide the first evidence that NAD replenishment with NMN ameliorates tPA‐induced haemorrhagic transformation by maintaining the integrity of the BBB. These results, together with previous findings on the effects of NMN in brain ischaemic injury (Wang et al., 2011; 2012; Zhao et al., 2014; 2015b; Park et al., 2016), suggest that NMN may be a promising agent for the treatment of ischaemic stroke.
Author contributions
C.C.W., Y.Y.K., X.H., Q.G.L., S.L.Z. and M.H.C. carried out the experiments. P.W. and C.Y.M. were grant awardees. P.W. and C.Y.M. designed the study, drafted the manuscript and coordinated the whole project. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 No influence of NMN (300 μM) on tPA thrombolytic activity in vitro.
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (nos 81422049, 81473208, 81673485, 81373414 and 81130061), National 863 Plan Young Scientist Program of China (no. 2015AA020943), Shanghai Innovation Action Plan (nos 16431901400 and 16JC1405100) and Yantai University Key Laboratory of Molecular Pharmacology and Drug Evaluation of Ministry of Education (P201603).
Wei, C.‐C. , Kong, Y.‐Y. , Hua, X. , Li, G.‐Q. , Zheng, S.‐L. , Cheng, M.‐H. , Wang, P. , and Miao, C.‐Y. (2017) NAD replenishment with nicotinamide mononucleotide protects blood–brain barrier integrity and attenuates delayed tissue plasminogen activator‐induced haemorrhagic transformation after cerebral ischaemia. British Journal of Pharmacology, 174: 3823–3836. doi: 10.1111/bph.13979.
Contributor Information
Pei Wang, Email: pwang@smmu.edu.cn.
Chao‐Yu Miao, Email: cymiao@smmu.edu.cn.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagon Y, Mantzoros CS, Kim YB (2016). AMPK<–>Sirt1: from a signaling network to a combination drug. Metabolism: clinical and experimental 65: 1692–1694. [DOI] [PubMed] [Google Scholar]
- de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF et al (2016). Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15: 522–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docagne F, Parcq J, Lijnen R, Ali C, Vivien D (2015). Understanding the functions of endogenous and exogenous tissue‐type plasminogen activator during stroke. Stroke; a journal of cerebral circulation 46: 314–320. [DOI] [PubMed] [Google Scholar]
- Garcia‐Culebras A, Palma‐Tortosa S, Moraga A, Garcia‐Yebenes I, Duran‐Laforet V, Cuartero MI et al (2017). Toll‐like receptor 4 mediates hemorrhagic transformation after delayed tissue plasminogen activator administration in in situ thromboembolic stroke. Stroke; a journal of cerebral circulation 48: 1695–1699. [DOI] [PubMed] [Google Scholar]
- Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W (2015). Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol 11: 535–546. [DOI] [PubMed] [Google Scholar]
- Hafez S, Abdelsaid M, El‐Shafey S, Johnson MH, Fagan SC, Ergul A (2016). Matrix metalloprotease 3 exacerbates hemorrhagic transformation and worsens functional outcomes in hyperglycemic stroke. Stroke 47: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L (2014). NAD+ and sirtuins in aging and disease. Trends Cell Biol 24: 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen R, Pirnes‐Karhu S, Pietilainen KH, Pirinen E (2017). Adipose tissue NAD+−homeostasis, sirtuins and poly(ADP‐ribose) polymerases – important players in mitochondrial metabolism and metabolic health. Redox Biol 12: 246–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa M, Igarashi H, Kawamura K, Takahashi T, Kakita A, Takahashi H et al (2011). Inhibition of VEGF signaling pathway attenuates hemorrhage after tPA treatment. J Cereb Blood Flow Metab 31: 1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastrup A, Groschel K, Ringer TM, Redecker C, Cordesmeyer R, Witte OW et al (2008). Early disruption of the blood‐brain barrier after thrombolytic therapy predicts hemorrhage in patients with acute stroke. Stroke; a journal of cerebral circulation 39: 2385–2387. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrue V, von Kummer RR, Muller A, Bluhmki E (2001). Risk factors for severe hemorrhagic transformation in ischemic stroke patients treated with recombinant tissue plasminogen activator: a secondary analysis of the European‐Australasian Acute Stroke Study (ECASS II). Stroke; a journal of cerebral circulation 32: 438–441. [DOI] [PubMed] [Google Scholar]
- Li DJ, Li YH, Yuan HB, Qu LF, Wang P (2017). The novel exercise‐induced hormone irisin protects against neuronal injury via activation of the Akt and ERK1/2 signaling pathways and contributes to the neuroprotection of physical exercise in cerebral ischemia. Metabolism: clinical and experimental 68: 31–42. [DOI] [PubMed] [Google Scholar]
- Liang J, Qi Z, Liu W, Wang P, Shi W, Dong W et al (2015). Normobaric hyperoxia slows blood‐brain barrier damage and expands the therapeutic time window for tissue‐type plasminogen activator treatment in cerebral ischemia. Stroke 46: 1344–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang Y, Xiao Y, Hua Z, Cheng J, Jia J (2016). Hydrogen sulfide attenuates tissue plasminogen activator‐induced cerebral hemorrhage following experimental stroke. Translational stroke research 7: 209–219. [DOI] [PubMed] [Google Scholar]
- Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA (2015). Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer's disease‐relevant murine model. BMC Neurol 15: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massudi H, Grant R, Braidy N, Guest J, Farnsworth B, Guillemin GJ (2012). Age‐associated changes in oxidative stress and NAD+ metabolism in human tissue. PLoS One 7 e42357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata Y, Rosell A, Scannevin RH, Rhodes KJ, Wang X, Lo EH (2008). Extension of the thrombolytic time window with minocycline in experimental stroke. Stroke; A Journal of Cerebral Circulation 39: 3372–3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Long A, Owens K, Kristian T (2016). Nicotinamide mononucleotide inhibits post‐ischemic NAD(+) degradation and dramatically ameliorates brain damage following global cerebral ischemia. Neurobiol Dis 95: 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter AG, Janicke RU (1999). Emerging roles of caspase‐3 in apoptosis. Cell Death Differ 6: 99–104. [DOI] [PubMed] [Google Scholar]
- Rempe RG, Hartz AM, Bauer B (2016). Matrix metalloproteinases in the brain and blood‐brain barrier: versatile breakers and makers. J Cereb Blood Flow Metab 36: 1481–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich PR (2003). The molecular machinery of Keilin's respiratory chain. Biochem Soc Trans 31: 1095–1105. [DOI] [PubMed] [Google Scholar]
- Rom S, Zuluaga‐Ramirez V, Dykstra H, Reichenbach NL, Ramirez SH, Persidsky Y (2015). Poly(ADP‐ribose) polymerase‐1 inhibition in brain endothelium protects the blood‐brain barrier under physiologic and neuroinflammatory conditions. J Cereb Blood Flow Metab 35: 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LR, Imai S (2014). Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J 33: 1321–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin GM, Youngson NA, Sinclair DA, Morris MJ (2016). Head to head comparison of short‐term treatment with the NAD(+) precursor nicotinamide mononucleotide (NMN) and 6 weeks of exercise in obese female mice. Front Pharmacol 7: 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Fan W, Cai P, Fan M, Zhu X, Dai Y et al (2013). Recombinant ADAMTS13 reduces tissue plasminogen activator‐induced hemorrhage after stroke in mice. Ann Neurol 73: 189–198. [DOI] [PubMed] [Google Scholar]
- Wang P, Du H, Zhou CC, Song J, Liu X, Cao X et al (2014a). Intracellular NAMPT‐NAD+−SIRT1 cascade improves post‐ischaemic vascular repair by modulating Notch signalling in endothelial progenitors. Cardiovasc Res 104: 477–488. [DOI] [PubMed] [Google Scholar]
- Wang P, Guan YF, Du H, Zhai QW, Su DF, Miao CY (2012). Induction of autophagy contributes to the neuroprotection of nicotinamide phosphoribosyltransferase in cerebral ischemia. Autophagy 8: 77–87. [DOI] [PubMed] [Google Scholar]
- Wang P, Guan YF, Li WL, Lu GC, Liu JM, Miao CY (2015). Nicotinamide phosphoribosyltransferase facilitates post‐stroke angiogenesis. CNS Neurosci Ther 21: 475–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Miao CY (2015). NAMPT as a therapeutic target against stroke. Trends Pharmacol Sci 36: 891–905. [DOI] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Su DF, Fan GR, Miao CY (2009). Perivascular adipose tissue‐derived visfatin is a vascular smooth muscle cell growth factor: role of nicotinamide mononucleotide. Cardiovasc Res 81: 370–380. [DOI] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Tian WW, Viollet B, Rui YC et al (2011). Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1‐dependent adenosine monophosphate‐activated kinase pathway. Ann Neurol 69: 360–374. [DOI] [PubMed] [Google Scholar]
- Wang P, Xu TY, Guan YF, Zhao Y, Li ZY, Lan XH et al (2014b). Vascular smooth muscle cell apoptosis is an early trigger for hypothyroid atherosclerosis. Cardiovasc Res 102: 448–459. [DOI] [PubMed] [Google Scholar]
- Wang P, Xu TY, Wei K, Guan YF, Wang X, Xu H et al (2014c). ARRB1/beta‐arrestin‐1 mediates neuroprotection through coordination of BECN1‐dependent autophagy in cerebral ischemia. Autophagy 10: 1535–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Yang X, Zhang Z, Song J, Guan YF, Zou DJ et al (2016a). Depletion of NAD pool contributes to impairment of endothelial progenitor cell mobilization in diabetes. Metabolism: clinical and experimental 65: 852–862. [DOI] [PubMed] [Google Scholar]
- Wang SN, Xu TY, Wang X, Guan YF, Zhang SL, Wang P et al (2016b). Neuroprotective efficacy of an aminopropyl carbazole derivative P7C3‐A20 in ischemic stroke. CNS Neurosci Ther 22: 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Asahi M, Lo EH (1999). Tissue type plasminogen activator amplifies hemoglobin‐induced neurotoxicity in rat neuronal cultures. Neurosci Lett 274: 79–82. [DOI] [PubMed] [Google Scholar]
- Wang X, Mori T, Sumii T, Lo EH (2002). Hemoglobin‐induced cytotoxicity in rat cerebral cortical neurons: caspase activation and oxidative stress. Stroke; a journal of cerebral circulation 33: 1882–1888. [DOI] [PubMed] [Google Scholar]
- Wei CC, Kong YY, Li GQ, Guan YF, Wang P, Miao CY (2017). Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO‐1 signaling pathway. Sci Rep 7: 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Lee JK, Stein DG (2015). Recombinant tissue plasminogen activator promotes, and progesterone attenuates, microglia/macrophage M1 polarization and recruitment of microglia after MCAO stroke in rats. Brain Behav Immun 49: 267–279. [DOI] [PubMed] [Google Scholar]
- Yang F, Zhou L, Wang D, Wang Z, Huang QY (2015). Minocycline ameliorates hypoxia‐induced blood‐brain barrier damage by inhibition of HIF‐1alpha through SIRT‐3/PHD‐2 degradation pathway. Neuroscience 304: 250–259. [DOI] [PubMed] [Google Scholar]
- Yang J, Klaidman LK, Nalbandian A, Oliver J, Chang ML, Chan PH et al (2002). The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci Lett 333: 91–94. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA (2007). Matrix metalloproteinase‐mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 27: 697–709. [DOI] [PubMed] [Google Scholar]
- Yang Y, Rosenberg GA (2011). Blood‐brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke; a journal of cerebral circulation 42: 3323–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S (2011). Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet‐ and age‐induced diabetes in mice. Cell Metab 14: 528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Zhang JP, Hu JY, Huang YS (2016). The potential regulatory roles of NAD(+) and its metabolism in autophagy. Metabolism: clinical and experimental 65: 454–462. [DOI] [PubMed] [Google Scholar]
- Zhang L, Chopp M, Jia L, Cui Y, Lu M, Zhang ZG (2009). Atorvastatin extends the therapeutic window for tPA to 6 h after the onset of embolic stroke in rats. J Cereb Blood Flow Metab 29: 1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang RL, Jiang Q, Ding G, Chopp M, Zhang ZG (2015). Focal embolic cerebral ischemia in the rat. Nat Protoc 10: 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang RY, Qin Y, Lv XQ, Wang P, Xu TY, Zhang L et al (2011). A fluorometric assay for high‐throughput screening targeting nicotinamide phosphoribosyltransferase. Anal Biochem 412: 18–25. [DOI] [PubMed] [Google Scholar]
- Zhao L, An R, Yang Y, Yang X, Liu H, Yue L et al (2015a). Melatonin alleviates brain injury in mice subjected to cecal ligation and puncture via attenuating inflammation, apoptosis, and oxidative stress: the role of SIRT1 signaling. J Pineal Res 59: 230–239. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Guan YF, Zhou XM, Li GQ, Li ZY, Zhou CC et al (2015b). Regenerative neurogenesis after ischemic stroke promoted by nicotinamide phosphoribosyltransferase‐nicotinamide adenine dinucleotide cascade. Stroke 46: 1966–1974. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Liu XZ, Tian WW, Guan YF, Wang P, Miao CY (2014). Extracellular visfatin has nicotinamide phosphoribosyltransferase enzymatic activity and is neuroprotective against ischemic injury. CNS Neurosci Ther 20: 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CC, Yang X, Hua X, Liu J, Fan MB, Li GQ et al (2016). Hepatic NAD(+) deficiency as a therapeutic target for non‐alcoholic fatty liver disease in ageing. Br J Pharmacol 173: 2352–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XM, Zhang X, Zhang XS, Zhuang Z, Li W, Sun Q et al (2014). SIRT1 inhibition by sirtinol aggravates brain edema after experimental subarachnoid hemorrhage. J Neurosci Res 92: 714–722. [DOI] [PubMed] [Google Scholar]
- Zuo W, Chen J, Zhang S, Tang J, Liu H, Zhang D et al (2014). IMM‐H004 prevents toxicity induced by delayed treatment of tPA in a rat model of focal cerebral ischemia involving PKA‐and PI3K‐dependent Akt activation. Eur J Neurosci 39: 2107–2118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 No influence of NMN (300 μM) on tPA thrombolytic activity in vitro.