

Abstract
Background and Purpose
5‐fluorouracil (5FU) and its prodrug, capecitabine, can damage endothelial cells, whilst endothelial integrity is preserved by glucagon‐like peptide 1 (GLP‐1). Here, we studied the effect of 5FU on endothelial senescence and whether GLP‐1 antagonizes it.
Experimental Approach
EA.hy926 cells were exposed to 5FU or sera from patients taking capecitabine, with or without pre‐incubation with GLP‐1. Senescence was identified by expression of senescence‐associated β‐galactosidase and p16INK4a and reduced cell proliferation. Soluble vascular cell adhesion molecule‐1 (sVCAM‐1), soluble intercellular adhesion molecule‐1 (sICAM‐1) and CD146 (marker of endothelial injury) were measured by ELISA before and at completion of capecitabine chemotherapy. RT‐PCR, western blotting, functional experiments with signalling inhibitors and ERK1/2 silencing were performed to characterize 5FU‐induced phenotype and elucidate the pathways underlying 5FU and GLP‐1 activity.
Key Results
Both 5FU and sera from capecitabine‐treated patients stimulated endothelial cell senescence. 5FU‐elicited senescence occurred via activation of p38 and JNK, and was associated with decreased eNOS and SIRT‐1 levels. Furthermore, 5FU up‐regulated VCAM1 and TYMP (encodes enzyme activating capecitabine and 5FU), and sVCAM‐1 and CD146 concentrations were higher after than before capecitabine chemotherapy. A non‐significant trend for higher ICAM1 levels was also observed. GLP‐1 counteracted 5FU‐initiated senescence and reduced eNOS and SIRT‐1 expression, this protection being mediated by GLP‐1 receptor, ERK1/2 and, possibly, PKA and PI3K.
Conclusions and Implications
5FU causes endothelial cell senescence and dysfunction, which may contribute to its cardiovascular side effects. 5FU‐triggered senescence was prevented by GLP‐1, raising the possibility of using GLP‐1 analogues and degradation inhibitors to treat 5FU and capecitabine vascular toxicity.
Linked Articles
This article is part of a themed section on New Insights into Cardiotoxicity Caused by Chemotherapeutic Agents. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.21/issuetoc
Abbreviations
- 5FU
5‐fluorouracil
- E9
exendin (9–39)
- ICAM‐1
intercellular adhesion molecule
- SA β‐gal
senescence‐associated β‐galactosidase
- SB
SB203580
- TP
thymidine phosphorylase
- VCAM‐1
vascular cell adhesion molecule
Tables of Links
| LIGANDS | |
|---|---|
| 5‐fluorouracil | L‐NAME |
| Capecitabine | LY‐294002 |
| Exendin (9‐39) (E9) | PD98059 |
| GLP‐1 | SB203580 |
| H‐89 | SP600125 |
| ICAM‐1 | VCAM‐1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
5‐fluorouracil (5FU) and capecitabine are fluoropyrimidine chemotherapeutic agents widely employed to treat solid cancers, especially of the colon‐rectum (Cunningham et al., 2010). Whilst 5FU is immediately active but must be administered i.v., capecitabine is an oral prodrug that is converted to 5FU preferentially within tumours (Walko and Lindley, 2005). The antineoplastic activity of 5FU relies on inhibition of DNA synthesis and interference with RNA processing, eventually leading to cell death (Thomas and Zalcberg, 1998). Furthermore, induction of senescence of cancer cells has been reported (Tato‐Costa et al., 2016).
Both 5FU and capecitabine have been associated with cardiac side effects, with the most common one being myocardial ischaemia (Polk et al., 2014). The incidence of 5FU and capecitabine symptomatic cardiotoxicity varies depending on chemotherapy schedule and the presence or not of cardiovascular comorbidities: this was reported in as many as 35% of treated patients, and in larger studies, it was 1.2–4.3% (Polk et al., 2013). Severe events, such as acute myocardial infarction, cardiogenic shock and cardiac arrest, may occur in up to 2% of subjects. Therefore, cardiac complications of treatment with 5FU and capecitabine should not be overlooked, even more so considering that these drugs are frequently used in oncology.
The pathophysiology of myocardial ischaemia secondary to 5FU and capecitabine therapy has repeatedly been related to epicardial coronary artery spasm and/or coronary microvascular dysfunction (Polk et al., 2014), which both may result from disrupted endothelial cell homeostasis (Lanza et al., 2011; Recio‐Mayoral et al., 2013). Here, we focused on senescence of endothelial cells exposed to 5FU, since stress‐induced senescence is a hallmark of the toxicity of anticancer drugs on cardiovascular cells (Spallarossa et al., 2010; Hodjat et al., 2013; Lazzarini et al., 2016). To increase the translational value of our work, we also assessed senescence of endothelial cells incubated with sera collected from patients receiving capecitabine.
After demonstrating that 5FU elicits senescence of endothelial cells, we sought for a way to antagonize this effect and tested glucagon‐like peptide 1 (GLP‐1), a hormone with beneficial cardiovascular actions for which both analogues and degradation inhibitors are clinically available (Tate et al., 2015) and which has already been shown to prevent the endothelial senescence response to stressors other than oncological treatments (Oeseburg et al., 2010; Matsubara et al., 2012; Chien et al., 2014).
Methods
Cells and treatments
The EA.hy926 human endothelial cell line (American Type Culture Collection, Manassas, FL, USA). was cultured in DMEM with 10% fetal calf serum. Experiments were always started when cells covered no more than 70% of the culture substrate, in order to avoid senescence due to confluence.
Cells were treated for 4 h with serum‐free DMEM alone, serum‐free DMEM supplemented with 100 μg·mL−1 5FU (Teva, Milan, Italy) or serum‐free DMEM added with 10% human serum from a patient receiving capecitabine (see below), left another 44 h in complete medium and then analysed for senescence. The 5FU concentration of 100 μg·mL−1 was chosen because it was the most pro‐senescent, yet sub‐apoptotic in preliminary assessments (Figure S1); of note, it is comparable with the concentration that has been shown to stimulate senescence of cancer cells (Adamsen et al., 2011). Protection by GLP‐1 was evaluated by pre‐incubating for 15 min with this hormone at 100 nmol·L−1 before exposure to 5FU.
The intracellular signalling pathways implicated in 5FU and GLP‐1 action were explored by pretreating the cells before the addition of 5FU or GLP‐1 with: SB203580 (inhibitor of p38; 45 min, 10 μmol·L−1); SP600125 (inhibitor of JNK; 45 min, 20 μmol·L−1); LY‐294002 (inhibitor of PI3K, Calbiochem, San Diego, CA, USA; 45 min, 20 μmol·L−1); PD98059 (inhibitor of ERK1/2, Calbiochem; 45 min, 40 μmol·L−1); L‐NAME (inhibitor of NOS; 30 min, 100 μmol·L−1); exendin (9–39) (E9; antagonist of GLP‐1 receptor; 30 min, 100 nmol·L−1); H‐89 (inhibitor of PKA; 30 min, 1 μmol·L−1); or vehicle.
ERK 1/2 expression was silenced by means of ERK1/2 siRNA (Cell Signaling Technology, Danvers, MA, USA) transfected with Lipofectamine RNAiMAX Reagent 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol (final siRNA concentration 100 nmol·L−1); a sham siRNA (Cell Signaling Technology) was used as control. The transfection reagent was removed after 12 h, and the cells were harvested after 48 h.
Each experiment included control and test cells (5FU, GLP‐1, the inhibitors listed above and/or siRNA), which were cultured, exposed to the different treatments and eventually lysed or fixed in parallel; thus, there was no need of randomization. Senescence, cell morphology, gene and protein expression and oxidative stress were evaluated as described in the following paragraphs by investigators other than those who performed the cell experiments, in a blinded manner.
Assessment of senescence
Senescence was demonstrated by increased expression of the markers senescence‐associated β‐galactosidase (SA β‐gal) and p16INK4a and reduced cell proliferation.
SA β‐gal characterizes senescence because in this condition, the lysosomal content increases and has residual activity at suboptimal pH 6 (van der Loo et al., 1998). To detect SA β‐gal, EA.hy926 cells were fixed in 0.5% glutaraldehyde and incubated overnight at 37°C in a solution containing 10 mM citric acid, 5 mM potassium ferricyanide, 150 mM NaCl, 2 mM MgCl2 and 1 mg·mL−1 5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactopyranoside (Lazzarini et al., 2016). SA β‐gal positive cells were counted in 100 randomly chosen low‐power fields (×100) and expressed as % of total cells.
Immunocytochemistry for p16INK4a was performed by using a primary rabbit polyclonal antibody against p16INK4a (Proteintech, Rosemont, IL, USA) and Vectastain secondary antibody, HRP‐streptavidin and 3,3′‐diaminobenzidine (all from Vector Laboratories, Burlingame, CA, USA) to reveal the bound primary antibody.
To quantify proliferation, cells were seeded on a 96‐well plate, treated with 100 μg·mL−1 5FU in serum‐free DMEM (control was serum‐free DMEM alone) and then incubated for another 20 h in complete medium. A BrdU labelling solution (Roche Diagnostics, Indianapolis, IN, USA) was added during the last 4 h in complete DMEM. Cells were then fixed, and BrdU incorporation was measured with the Cell Proliferation ELISA BrdU kit (Roche) following the manufacturer's instructions. Signal was read as intensity of absorbance at 450 nm with a Labsystems iEMS Reader MF9 plate reader (Labsystems, Helsinki, Finland).
Cell morphology
To examine morphology, EA.hy926 cells were grown on a 96‐well plate, treated or not with 100 μg·mL−1 5FU in serum‐free medium for 4 h, and then cultured for another 20 h in complete DMEM. Cells were then fixed in 4% paraformaldeyde for 10 min, permeabilized with 0.1% triton, and stained with 5 U·mL−1 AlexaFluo488‐conjugated phalloidin (Thermo Fisher Scientific, Waltham, MA, USA) for 20 min. Nuclei were counterstained with DAPI. Images were acquired with a Leica DM2000 fluorescence microscope (Leica Microsystems GmbH, Wetzlar, Germany) coupled to a CCD high resolution cooled camera, using the Leica Application Suite software (Leica Microsystems).
RT‐PCR
Total RNA was extracted using Quick‐RNA TM MiniPrep (Zymo Research, Irvine, CA, USA) and reverse‐transcribed into cDNA with iSCRIPT RT Supermix (Bio‐Rad Laboratories, Hercules, CA, USA). VCAM1, ICAM1 and TYMP were amplified by using the primers listed in Table S1 from Primerdesign (Southampton, UK) and Tibmolbiol (Genova, Italy), and their relative mRNA levels were calculated from cycle threshold (Ct) values using GAPDH as the internal control.
Western blot
Cells were lysed in lysis buffer [20 mM Tris HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP40, 2.5 mM Na2P2O7 and 1 mM β‐glycerophosphate] supplemented with 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF and a protease inhibitor cocktail (Thermo Fisher Scientific).
Cell lysates were separated on a tris‐glycine gel (8–16%, Thermo Fisher Scientific), transferred onto a PVDF membrane (Bio‐Rad Laboratories) and incubated overnight with the following primary antibodies: anti‐endothelial NOS (eNOS; mouse monoclonal antibody, clone 6H2, Cell Signaling Technology); anti‐SIRT‐1 (rabbit monoclonal antibody, clone D739, Cell Signaling Technology); anti‐GLP‐1 receptor (rabbit polyclonal antibody, Abcam, UK); and anti‐actin (mouse monoclonal antibody, clone C4, Santa Cruz Biotechnology, Dallas, TX, USA). After incubation with a HRP‐conjugated secondary antibody (Santa Cruz Biotechnology), blots were visualized with the Clarity Western ECL Substrate (Bio‐Rad). Band densitometry was carried out by using an image analyser system (UVItec Limited, Cambridge, UK); the signal intensity of eNOS, SIRT‐1 and GLP‐1 receptors was normalized to the one of actin to control for unwanted variations.
Evaluation of oxidative stress
After incubation with 5FU and/or GLP‐1, EA.hy926 cells were stained with 10 μM 2′,7′‐dichlorodihydrofluorescein diacetate (DCFH‐DA) in PBS for 20 min at 37°C. Fluorescent 2′,7′‐dichlorofluorescein, which is formed by oxidation of DCFH‐DA, was measured by using an Attune Acoustic Focusing cytometer and the Attune cytometry software (Thermo Fisher Scientific).
Patients
Serum samples were collected from patients who had undergone complete resection of stages II–III colorectal cancer and met the following inclusion criteria: indication for adjuvant chemotherapy with capecitabine; non‐metastatic disease; performance status 01; normal renal and hepatic function; no dyslipidaemia or diabetes mellitus; and no known heart disease or previous chemotherapy or radiation therapy. Six such patients were recruited; they were treated every 21 days for a total of six cycles, each including 130 mg·m−2 oxaliplatin on day 1 and 2000 mg·m−2 capecitabine b.i.d. from days 1 to 15.
Venous blood samples were taken at baseline, before starting chemotherapy, and in the morning of day 15 of the sixth cycle, after the second last dose of capecitabine. Serum was obtained by centrifuging at 1800 x g for 15 min at 4°C and stored −80°C until use.
The study was approved by the local Ethical Committee (protocol 464REG2016), and all patients signed an informed consent approving the utilization of their anonymized clinical information for medical research purposes.
Measurement of 5FU concentrations
5FU in the post‐chemotherapy serum was quantified as previously described (Chen and Zhou, 2010) with slight modifications. Briefly, samples (0.1 mL) were extracted with acetonitrile (1:3 in volume) spiked with 5‐bromo‐uracil (5BrU, 100 ng·mL−1) as internal standard, vortexed for 1 min, and centrifuged for 10 min at 3500 x g. Five microlitres of supernatant were then loaded on a Xevo TQ ultra‐performance liquid chromatography – tandem mass spectrometry (UPLC‐MS/MS) triple quad system (Waters, Milford, MA, USA), equipped with a HILIC 2.1 × 100 mm column. The flow rate was set at 0.4 mL·min−1, and the eluent composition was 75:25 acetonitrile: 10 mM ammonium bicarbonate pH 8. 5FU and 5BrU were detected by negative ion mode electrospray ionization and quantified using their MRM (multiple reaction monitoring) transitions, which were preliminarily selected and optimized. 5FU was quantified using an external calibration curve prepared in naïve human serum and prepared as described for the samples.
Assays
Serum concentrations of soluble vascular cell adhesion molecule‐1 (sVCAM‐1) and soluble intercellular adhesion molecule‐1 (sICAM‐1) were measured by colorimetric ELISA (R&D Systems) according to the manufacturer's instructions. The limits of detection were 15.625 and 31.25 pg·mL−1 respectively. Mean intra‐ and inter‐assay coefficients of variation were <8% for both assays.
Levels of CD146, a marker of endothelial damage (Bardin et al., 2003), were measured by means of a capture ELISA (CY‐QUANT ELISA sCD146, Biocytex, Marseille, France). The limit of detection and mean intra‐assay coefficient of variation were 10 ng·mL−1 and 7% respectively.
Statistical analysis
Statistical analysis was performed by means of GraphPad Prism Version 6.0a (GraphPad Software) and complied with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
With the exception of the RT‐PCR results, data are presented as mean ± SEM of at least five independent experiments and were compared by Student's unpaired or paired t‐test or one‐way ANOVA, as specified in Figure legends. The post hoc Tukey's multiple comparisons test was run after ANOVA when F achieved P < 0.05, and there was no significant variance inhomogeneity. RT‐PCR data are shown as fold change versus control and were compared by the non‐parametric Mann–Whitney test. Statistical significance was set at P < 0.05.
Materials
When not otherwise indicated, materials were supplied by Sigma‐Aldrich (Sigma‐Aldrich, St. Louis, MO, USA).
Results
5FU elicits senescence of endothelial cells
As presented in Figure 1, short‐term exposure to 5FU caused profound alterations in EA.hy926 cells, overall indicating senescence. Positivity for SA β‐gal and p16INK4a significantly increased (Figure 1A, B), whilst proliferation was inhibited (Figure 1C). Morphologically, 5FU‐treated cells displayed an enlarged, flattened cytoplasm with a ‘fried egg’ appearance (Figure 1D). These phenotypic changes were associated with the up‐regulation of the genes encoding VCAM‐1 (Figure 1E) and – although not to a significant extent – ICAM‐1 (P = 0.30, Figure 1F), consistent with earlier evidence of a dysfunctional activation of endothelial cells undergoing senescence (Yokoi et al., 2006). Interestingly, 5FU also significantly induced the expression of TYMP (Figure 1G), encoding the enzyme responsible for the conversion of capecitabine into 5FU as well as of 5FU into its active metabolite.
Figure 1.
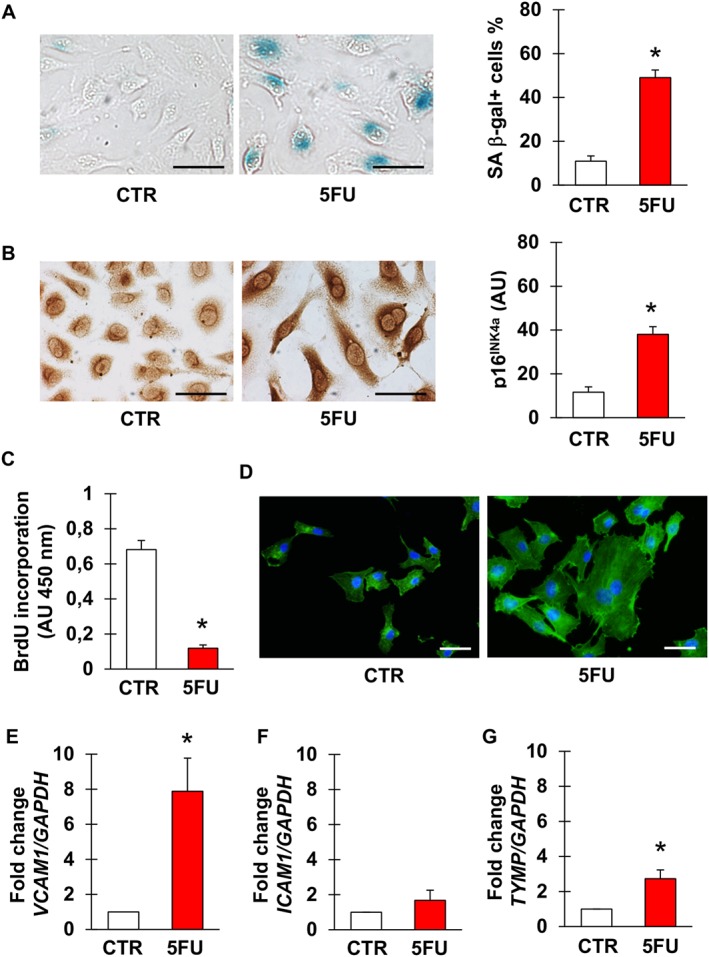
5FU induces senescence of endothelial cells. (A, B) Representative pictures and percentage of EA.hy926 cells stained for the senescence markers SA β‐gal (A) and p16INK4a (B) after no treatment (CTR) or exposure to 5FU. (C) Percentage of BrdU positive EA.hy926 cells in CTR condition and after incubation with 5FU. (D) Representative images of CTR and 5FU‐treated EA.hy926 cells stained for actin to visualize morphology and cytoplasm size. Nuclei are counterstained with DAPI. (E–G) Expression of VCAM1, ICAM1 and TYMP in CTR or 5FU‐treated cells. n = 7 for the experiments depicted in (A, B) and 5 for the other ones. * Statistically significant versus CTR (unpaired t‐test for A–C, Mann–Whitney test for E–G). Magnification of pictures is 400×, bars correspond to 50 μm.
5FU‐initiated senescence is mediated by p38 and JNK and involves a decrease in the expression of eNOS and SIRT‐1
To gain insight into the pathways underlying 5FU‐triggered endothelial cell senescence, EA.hy926 cells were exposed to 5FU after pre‐incubation with inhibitors of intracellular signalling mediators that we previously found to be involved in chemotherapy‐induced cardiovascular cell senescence (Spallarossa et al., 2010; Altieri et al., 2016). Induction of senescence by 5FU, as assessed by staining for SA β‐gal, was significantly reduced by inhibiting p38 and JNK, suggesting the involvement of these kinases (Figure 2A, B). Conversely, blockade of PI3K and ERK1/2 did not modify the effects of 5FU.
Figure 2.
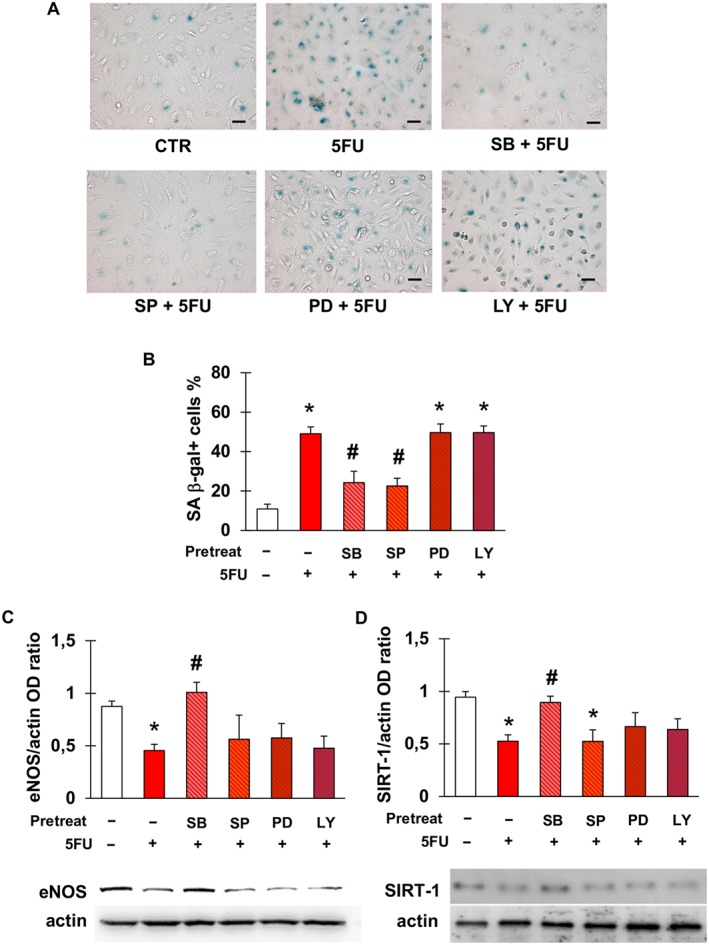
5FU‐elicited senescence is mediated by p38 and JNK and is associated with a down‐regulation of eNOS and SIRT‐1. (A) Representative pictures of the staining for SA β‐gal after no treatment (CTR) or exposure of EA.hy926 cells to 5FU with or without pre‐incubation with the following signalling inhibitors: SB203580 (inhibitor of p38); SP600125 (inhibitor of JNK); PD98059 (inhibitor of ERK1/2); or LY‐294002 (inhibitor of PI3K). Magnification is 200×, and bars are 50 μm. (B) Percentage of SA β‐gal‐positive EA.hy926 cells in the same conditions as in (A). (C–D) Densitometry analysis and representative western blots for eNOS (C) and SIRT‐1 (D) after the treatments described in (A). eNOS and SIRT‐1 protein levels were expressed as optical density (OD) ratio with actin as internal standard. Data are presented as mean ± SEM of five independent replicates. In all graphs, * indicates statistically significant versus CTR and # statistically significant versus 5FU (ANOVA with post hoc Tukey's multiple comparisons test).
Next, we assessed eNOS and SIRT‐1 protein levels following treatment with 5FU, since their down‐regulation has been related to endothelial senescence (Ota et al., 2010). Compared with control, 5FU decreased the expression of both eNOS (Figure 2D) and SIRT1 (Figure 2E). Among the same signalling inhibitors used to explore the mediators of 5FU‐elicited senescence, only SB significantly reversed the effects of 5FU on eNOS and SIRT‐1, indicating a primary role of p38, whilst the other molecules had a minimal effect, if any (Figure 2D, E). None of the signalling inhibitors, when used alone, modified the % of senescent cells compared with control (Figure S2).
Oxidative stress has previously been reported to occur in endothelial cells incubated with 5FU for 48 h or longer (Focaccetti et al., 2015); however, it was not detected after the 4 h‐treatment protocol used for the present work (Figure S3).
Evidence for endothelial cell senescence and dysfunction after chemotherapy with capecitabine
In an effort to achieve more translational results, EA.hy926 cells were exposed to sera withdrawn from patients before and at the end of adjuvant chemotherapy with capecitabine. The mean age of these patients was 71 ± 5.4 years, and five were male patients. Four had arterial hypertension and were being treated chronically with an ACE‐inhibitor (one subject), angiotensin‐receptor blocker (two subjects) or dihydropyridine calcium channel blocker (one subject). According to the enrolment criteria, none had diabetes mellitus, dyslipidaemia or known cardiovascular disease. Consistent with an earlier study in which 5FU was detectable in plasma and healthy tissues 2 to 12 h after the last dose of a 5–7 day course of capecitabine (Schüller et al., 2000), we could demonstrate the presence of 5FU in the sera obtained at completion of chemotherapy by UPLC‐MS/MS (median concentration 43.6 ng·mL−1, range 5.5–257.5 ng·mL−1).
Figure 3A shows that, compared with sera obtained prior to starting capecitabine, those samples taken at the end of treatment significantly increased the frequency of SA β‐gal positive cells. Similar to what was observed with 5FU, senescence promoted by post‐chemotherapy sera was attenuated by pre‐incubation with inhibitors of p38 and JNK, whilst it was not affected by blockade of ERK1/2 or PI3K (Figure 3A). Measurement of sVCAM‐1 and sICAM‐1 concentrations in the same sera revealed that levels of sVCAM‐1 were significantly higher after than before chemotherapy (Figure 3B). By contrast, there was no statistically significant elevation in sICAM‐1 values, although a tendency for that direction was noted (P = 0.26, Figure 3C). Furthermore, concentrations of CD146 significantly increased after treatment (Figure 3D).
Figure 3.
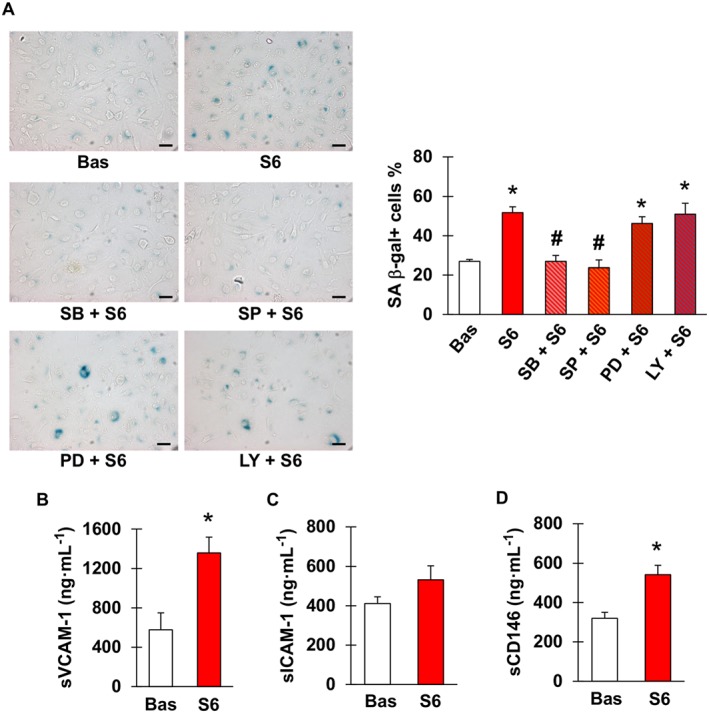
Sera from patients receiving capecitabine cause endothelial cell senescence and contain higher concentrations of sVCAM‐1 and CD146. (A) Representative images (left) and percentage (right) of SA β‐gal positive EA.hy926 cells after incubation with sera obtained from patients with radically resected colorectal cancer either before (Bas) or at the end of six cycles of chemotherapy with capecitabine (S6), in the latter case with or without pretreatment with SB203580 (inhibitor of p38), SP600125 (inhibitor of JNK), PD98059 (inhibitor of ERK1/2) or LY‐294002 (inhibitor of PI3K). Magnification 200×, bars correspond to 50 μm. (B–D) Concentrations of sVCAM‐1, sICAM‐1 and CD146 in sera of patients used in the experiment depicted in (A); n = 5. * and # indicate statistically significant versus Bas and S6, respectively (ANOVA with post hoc Tukey's multiple comparisons test for A, paired t‐test for B–D).
GLP‐1 antagonizes endothelial cell senescence and the down‐regulation of eNOS and SIRT‐1 triggered by 5FU
After confirming that EA.hy926 cells expressed GLP‐1 receptor and that they were not modulated by 5FU (Figure S4), we evaluated whether GLP‐1 protected against 5FU toxicity. Pre‐incubation with GLP‐1 significantly reduced 5FU‐elicited senescence of EA.hy926 cells (Figure 4). This effect occurred through GLP‐1 receptor, as it was attenuated by the GLP‐1 receptor antagonist, E9 (Figure 4A). This was also the case with H‐89, suggesting that GLP‐1 antagonism of senescence was also mediated by PKA (Figure 4A). Moreover, inhibition of ERK1/2, PI3K and NOS also eliminated this protective effect of GLP‐1 (Figure 4B), consistent with previous studies demonstrating that these signalling mediators lie downstream of GLP‐1 (Favaro et al., 2012; Chien et al., 2014).
Figure 4.
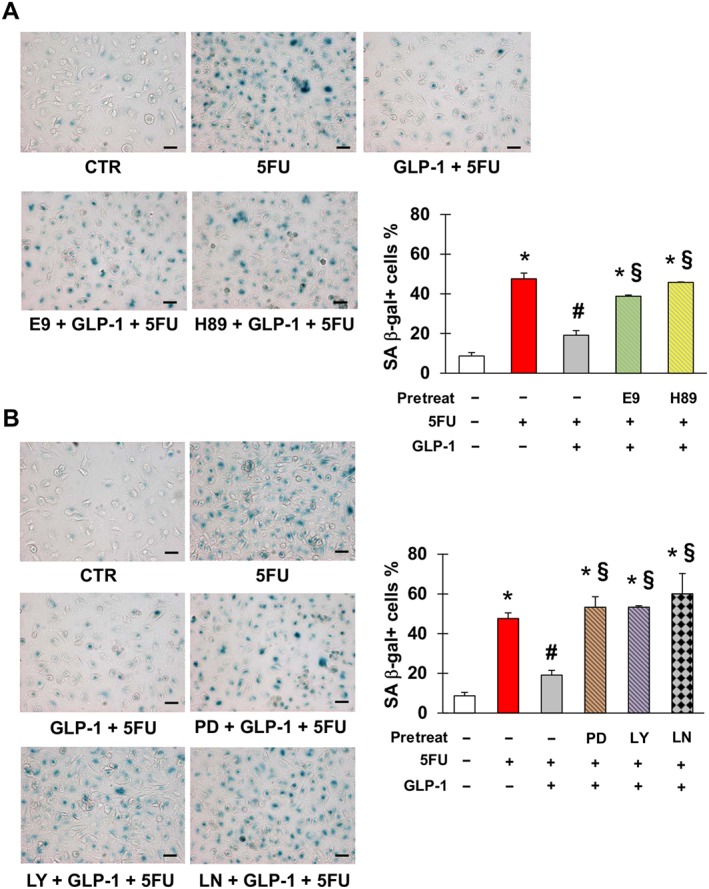
Glucagon‐like peptide 1 (GLP‐1) protects against 5FU‐initiated senescence. (A) Representative pictures (left) and percentage (right) of SA β‐gal positive EA.hy926 cells after no treatment (CTR) or exposure to 5FU, GLP‐1, E9 (GLP‐1 receptor antagonist) and/or H‐89 (inhibitor of PKA) as indicated. (B) Representative pictures (left) and percentage (right) of EA.hy926 positive for SA β‐gal after CTR or exposure to 5FU, GLP‐1, PD98059 (inhibitor of ERK1/2), LY‐294002 (inhibitor of PI3K) and/or L‐NAME (inhibitor of NOS) as indicated. Magnification of pictures is 200×, and bars are equal to 50 μm. Data are mean ± SEM of five independent experiments; * indicate statistically significant versus CTR, # statistically significant versus 5FU and § statistically significant versus GLP‐1 + FU.
Functional experiments also indicated that GLP‐1 could restore the expression of eNOS and SIRT‐1 through GLP‐1 receptors and PKA (Figure 5A, B), as well as ERK1/2 and PI3K (Figure 5C, D). Restoration of SIRT‐1 by GLP‐1 also appeared to be NOS‐dependent, since it was lost after pre‐treating cells with L‐NAME (Figure 5D). Nonetheless, our blot data did not allow us to perform a post hoc comparison of the levels of eNOS and SIRT‐1 in cells untreated or incubated with 5FU, GLP‐1 and/or the signalling inhibitors tested. Importantly, these latter molecules did not have any effect on eNOS and SIRT‐1 when employed alone (Figure S5).
Figure 5.
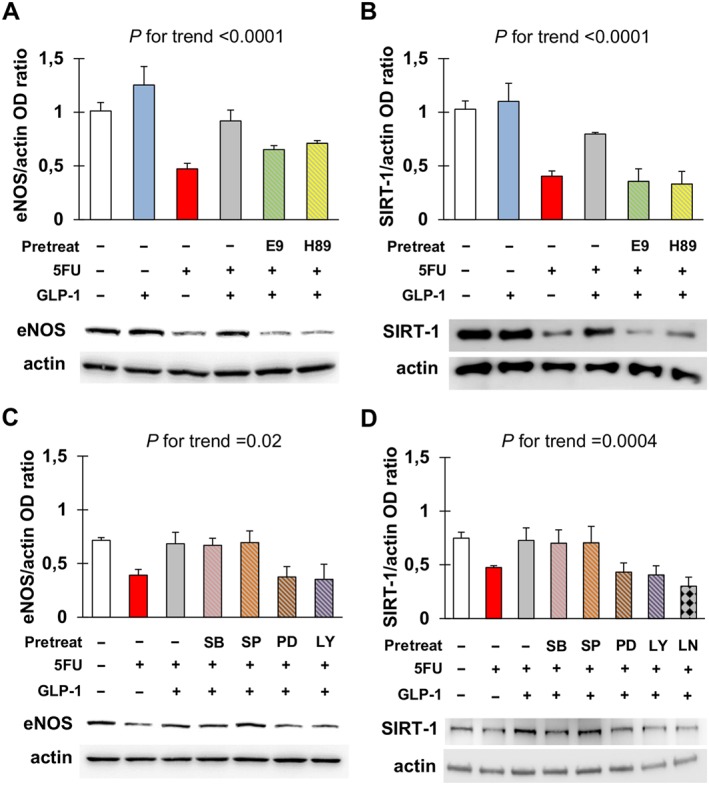
Glucagon‐like peptide 1 (GLP‐1) antagonizes eNOS and SIRT‐1 down‐regulation in response to 5FU. (A, B) Densitometry analysis and representative western blots for eNOS (A) and SIRT‐1 (B) in EA.hy926 cells after no treatment or exposure to 5FU, GLP‐1, E9 (GLP‐1 receptor antagonist) and/or H‐89 (inhibitor of PKA) as indicated. For each condition, optical density (OD) of eNOS and SIRT‐1 bands was normalized to that of actin as internal control. (C, D) Densitometry analysis and representative western blots for eNOS (C) and SIRT‐1 (D) in EA.hy926 cells after no treatment or exposure to 5FU, GLP‐1, SB203580 (inhibitor of p38), SP600125 (inhibitor of JNK), PD98059 (inhibitor of ERK1/2), LY‐294002 (inhibitor of PI3K) and/or L‐NAME (inhibitor of NOS) as indicated. Data were analysed by ANOVA, and the P for trend values are shown above the graphs. Post hoc comparisons were not drawn because of F > 0.05 and significant variance inhomogeneity.
Given the established role of ERK1/2 in GLP‐1 signalling (Favaro et al., 2012; Kang et al., 2015; Zhu et al., 2016), we explored the effect of silencing ERK1/2 on the protective effect of GLP‐1. ERK1/2 silencing prevented the GLP‐1‐induced inhibition of 5FU‐induced senescence (Figure 6A). Moreover, in cells in which ERK1/2 was silenced, GLP‐1 failed to prevent the decrease in eNOS caused by 5FU (Figure 6B, C). Interestingly, the silencing of ERK1/2 alone led to a reduction in the expression of eNOS, although not to a significant extent, indicating a role for this kinase in maintaining eNOS levels in EA.hy926 cells.
Figure 6.
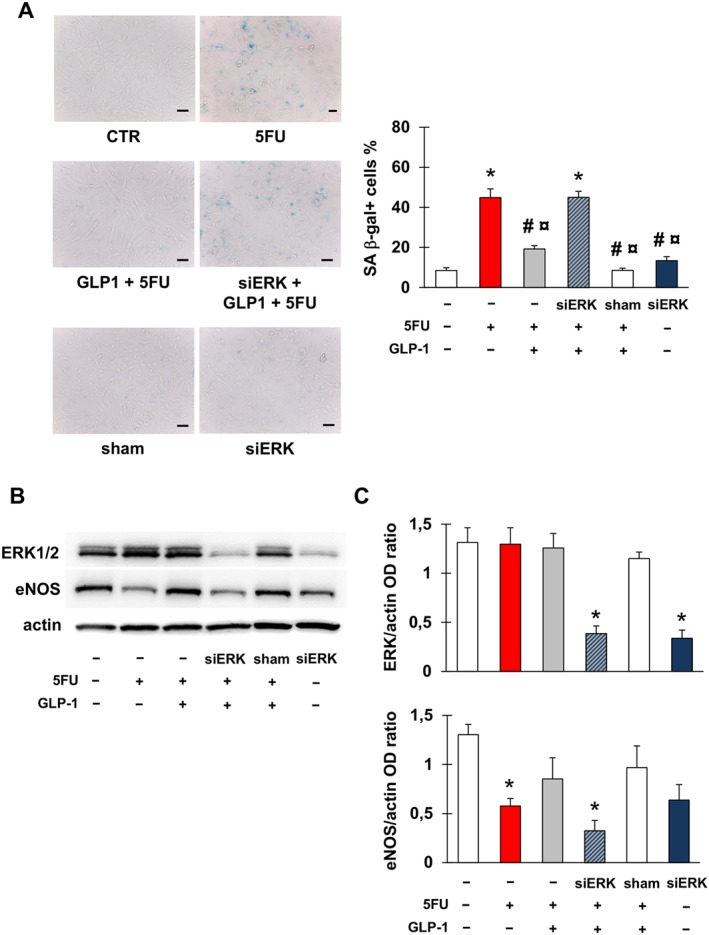
ERK 1/2 is implicated in 5FU toxicity on endothelial cells. (A) Representative images and percentage of EA.hy926 cells stained for SA β‐gal after the following treatments: no treatment (CTR); exposure to 5FU; pre‐incubation with GLP‐1 followed by exposure to 5FU; transfection with ERK1/2 siRNA (siERK), pre‐incubation with GLP‐1 and then exposure to 5FU; transfection with sham siRNA (sham); transfection with siERK. Magnification of pictures is 200×, and bars correspond to 50 μm. (B, C) Representative western blots for ERK1/2 and eNOS (B) and optical density (OD) of the protein bands normalized to that of actin (C) after the treatments described in (A). Graphs show mean ± SEM of five independent experiments. Comparisons were made by means of ANOVA followed by post hoc Tukey's multiple comparisons test. * Statistically significant versus CTR, # statistically significant versus 5FU and ¤ statistically significant versus siERK + GLP‐1 + FU.
Finally, GLP‐1 prevented endothelial cells senescence initiated by sera from patients taking capecitabine, with this effect being eliminated by antagonism of GLP‐1 receptors or inhibition of PKA, ERK1/2, PI3K and NOS (Figure 7).
Figure 7.
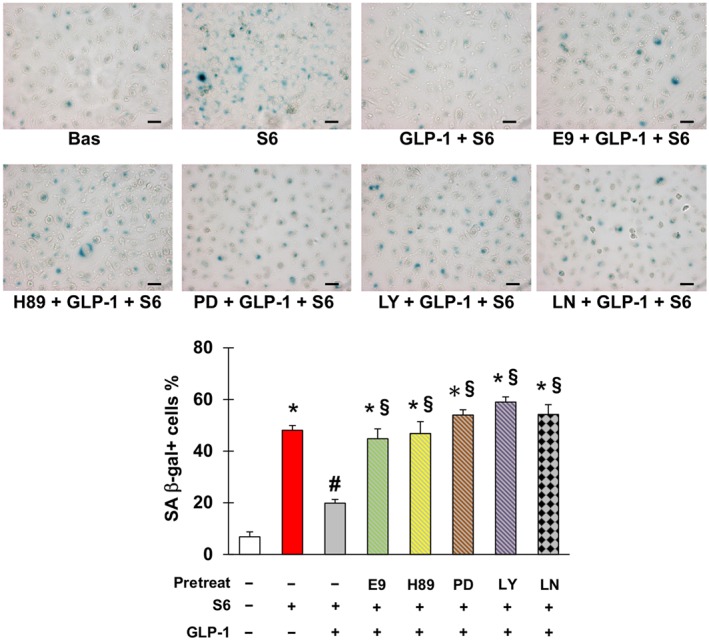
Glucagon‐like peptide 1 (GLP‐1) counteracts senescence triggered by sera from patients taking capecitabine. Representative pictures (upper part) and percentage (lower part) of SA β‐gal positive EA.hy926 cells after no treatment or incubation with sera obtained from patients with radically resected colorectal cancer either before (Bas) or at the end of six cycles of chemotherapy with capecitabine (S6), in the latter case with or without pretreatment with GLP‐1, E9 (GLP‐1 receptor antagonist), H‐89 (inhibitor of PKA), PD98059 (inhibitor of ERK1/2), LY‐294002 (inhibitor of PI3K) and/or L‐NAME (inhibitor of NOS) as indicated. Magnification 200×, bars 50 μm. * Statistically significant versus CTR, # statistically significant versus S6 and § statistically significant versus GLP‐1 + S6.
Discussion
Myocardial ischaemia is a potentially life‐threatening and not uncommon complication of chemotherapy with 5FU or capecitabine (Polk et al., 2014; Layoun et al., 2016). In most 5FU or capecitabine‐related acute coronary syndromes for which coronary angiography was timely performed, no significant obstructive epicardial coronary artery disease was found, suggesting that microvascular dysfunction or coronary artery spasm are the anatomical substrate of 5FU or capecitabine‐associated myocardial ischaemia (Layoun et al., 2016). Coronary artery spasm has in fact been demonstrated in anecdotal cases (Goldsmith et al., 2008). However, the underlying mechanisms of this kind of 5FU toxicity remain quite elusive.
In the present study, we have shown that 5FU potently causes senescence of endothelial cells. In addition, it enhances the expression of the adhesion molecule VCAM‐1, consistent with the experimental evidence that endothelial cell senescence triggered by stressors other than chemotherapy, such as hydrogen peroxide or high glucose, entails a dysfunctional pro‐inflammatory phenotype (Yokoi et al., 2006; Freund et al., 2010). Since induction of senescence was also seen when endothelial cells were incubated with sera from patients taking capecitabine, we propose that 5FU‐initiated endothelial senescence may occur in the clinical setting. This concept is also supported by the observation that concentrations of sVCAM‐1, as well as of CD146, increased in patients' sera following chemotherapy with capecitabine.
Only a recent study has specifically addressed the issue of senescence of endothelial cells exposed to 5FU (Focaccetti et al., 2015). The authors observed that the number of SA β‐gal cells was significantly higher in response to 5FU, in good agreement with our results. Furthermore, they reported that endothelial cells from the heart and kidneys of a xenograft model of colon cancer treated with 5FU displayed nuclear alterations, cytoplasmic vacuolization and membrane breakage, confirming that 5FU damages the endothelium in vivo. In an earlier investigation it was shown that 5FU at a concentration comparable with the one used by us led to reduced [3H]‐thymidine incorporation and release of prostacyclin by endothelial cells without overt cell death (Cwikiel et al., 1996), which is again consistent with our findings.
Thus, endothelial cell senescence is a feature of 5FU vascular toxicity. This information adds to the knowledge about the effects of 5FU on the cardiovascular system. More generally, it highlights that anticancer treatment‐induced senescence of endothelial cells, which so far has mainly been described and discussed for doxorubicin (Spallarossa et al., 2010; Hodjat et al., 2013; De Falco et al., 2016) and ionizing radiation (Azimzadeh et al., 2015; Park et al., 2016), may also be relevant for other oncological therapies. In the heart, 5FU‐initiated senescence might at least in part contribute to coronary spasm and/or microvascular disease and might promote the formation and progression of coronary atherosclerotic lesions, as endothelial senescence has been implicated in atherogenesis (Yin and Pickering, 2016). However, these hypotheses need to be verified.
At the intracellular level, 5FU affected eNOS and SIRT‐1. These enzymes are signalling nexuses pivotal to endothelial cell homeostasis and are linked to each other by a positive feedback loop, where eNOS stimulates the expression of SIRT‐1 (Nisoli et al., 2005) and SIRT‐1 activates eNOS (Mattagajasingh et al., 2007). We found that both eNOS and SIRT‐1 are down‐regulated during 5FU‐triggered senescence, as was previously demonstrated for hydrogen peroxide‐initiated endothelial cell senescence (Ota et al., 2010). Our data also suggest that 5FU acts through p38 and JNK. Interestingly, these kinases also mediate the induction of senescence by doxorubicin (Spallarossa et al., 2009; 2010).
By contrast, 5FU up‐regulated the expression of TYMP. Thymidine phosphorylase (TP), the product of this gene, is fundamental for the activity of capecitabine and 5FU, since it converts capecitabine into 5FU and 5FU into 2′‐deoxy‐5‐fluorouridine. This latter compound is further metabolized by thymidine kinase to 5‐fluorodeoxyuridine monophosphate, which in turn inhibits thymidylate synthase and, thereby, blocks de novo thymidine biosynthesis and DNA replication (Thomas and Zalcberg, 1998). Therefore, TP dictates the rate of production of the active metabolite actually responsible for 5FU cytotoxicity. Although preferentially expressed by cancer cells, TP is also present in normal tissues, including the endothelium (Fox et al., 1995; Schüller et al., 2000). Furthermore, TP (also known as platelet‐derived endothelial cell growth factor) has been demonstrated in coronary atherosclerotic plaques where it may play a pathogenetic role (Ignatescu et al., 1999). Based on our results, we propose that 5FU induces TP in endothelial cells and thus sustains its own activation. This phenomenon may amplify the vascular injury induced by 5FU, by rendering endothelial cells more susceptible to 5FU and promoting atherosclerosis.
Because of major advances in diagnosis and therapy, some types of cancer, such as colorectal, are now curable in a substantial proportion of subjects, and this is very likely to grow in the next few years. As a consequence, the side effects of oncological drugs may become a major health problem in cancer patients and have a dramatic impact on prognosis, even higher than that of the tumour for which treatment is initially given. Therefore, there is an urgent need for effective approaches to prevent the complications of antineoplastic therapy. In the present study, we evaluated whether GLP‐1 might counteract 5FU‐induced senescence of endothelial cells. The interest in a potential protective activity of GLP‐1 was prompted by the clinical availability of GLP‐1 analogues and degradation inhibitors, which have been reported to exert beneficial cardiovascular actions, including preventing endothelial cell senescence stimulated by reactive oxygen species (Matsubara et al., 2012; Chien et al., 2014). Indeed, GLP‐1 antagonized the senescence of endothelial cells evoked by 5FU. ERK1/2 was required for this protective effect of GLP‐1 against 5FU‐triggered senescence and eNOS down‐regulation, consistent with the previous data reporting the positioning this kinase downstream of GLP‐1 (Favaro et al., 2012; Kang et al., 2015; Zhu et al., 2016). Experiments with multiple signalling inhibitors also suggested that the effect of GLP‐1's action was secondary to PKA and PI3K, which have both been shown to mediate GLP‐1's effects previously (Erdogdu et al., 2010; Oeseburg et al., 2010; Favaro et al., 2012; Chien et al., 2014; Zhu et al., 2016). The inhibitory effect of L‐NAME indicated that GLP‐1's action was also dependent on NOS, which may be downstream of GLP‐1‐recruited PI3K/Akt (Chien et al., 2014) or may be activated in a GLP‐1 receptor‐independent manner by a truncated form of GLP‐1, produced by cleavage of the full hormone by dipeptidyl peptidase‐4 (Ban et al., 2008).
We acknowledge that the demonstration that GLP‐1 prevents senescence due to 5FU in vitro does not mean that GLP‐1 analogues or degradation inhibitors may prevent 5FU‐induced cardiovascular toxicity in vivo and, particularly, in the clinical scenario. Moreover, the effects of GLP‐1 on cancer should be thoroughly analysed. Nonetheless, we believe that the results presented here provide a good basis for future studies, especially considering the promising results obtained with GLP‐1‐based drugs in the setting of experimental ischaemia/reperfusion injury (Timmers et al., 2009) and in patients with acute myocardial infarction (Woo et al., 2013). Of note, GLP‐1 has also been reported to facilitate recovery from the toxic effects of oxaliplatin (Fujita et al., 2015), which is combined with capecitabine in some chemotherapy regimens, such as the one given to the patients from whom we collected serum samples.
Our work has limitations. Firstly, we assumed that endothelial cell senescence induced by post‐capecitabine sera was promoted by 5FU. Nonetheless, the possibility that other factors may increase in the circulation following chemotherapy with capecitabine and promote senescence of endothelial cells should not be discounted. This may be also the case with oxaliplatin, even though the presence of this drug in serum and thus its direct effect can be reasonably excluded, as blood samples were taken 2 weeks after oxaliplatin infusion. Secondly, other aspects of endothelial function besides expression of adhesion molecules may be altered by 5FU. Thirdly, chemical inhibitors of signalling mediators may not be specific, and additional experiments are required to distinguish the pathways implicated in 5FU toxicity and GLP‐1 protection.
In conclusion, 5FU induces senescence of endothelial cells, which may account for the vascular complications of this drug and of its precursor, capecitabine. The ability of GLP‐1 to antagonize 5FU‐elicited endothelial senescence raises the possibility of using GLP‐1 analogues and degradation inhibitors to treat 5FU‐ and capecitabine‐mediated vascular toxicity. Further mechanistic studies are needed to investigate this hypothesis.
Author contributions
P.Al. designed and performed the experiments in vitro; R.M. and S.B. recruited and followed the patients; E.L., P.F., C.B., A.A. and F.C. performed experiments; S.G. and C.R. analysed the data; M.C., A.B., C.B. and F.M. provided critical discussions; P.S. conceived the study and supervised the recruitment and follow‐up of patients; P.A. supervised the experiments in vitro, analysed the data and wrote the manuscript. All authors approved the version to be published.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Primers used in the study.
Figure S1 Senescence of EA.hy926 cells in response to increasing concentrations of 5FU. EA.hy926 cells were incubated for 4 h with the indicated concentrations of 5FU. After this treatment, senescence was assessed as percentage of SA β‐gal positive cells. No apoptosis, as evaluated by immunocytochemistry using a rabbit monoclonal anti‐cleaved caspase‐3 antibody (clone 5A1E, Cell Signalling Technologies), was observed. The experiment was repeated 5 times.
Figure S2 Effect of the signalling inhibitors used for functional experiments on senescence of EA.hy926 cells. EA.hy926 cells were treated with the following molecules (see Methods for times and concentrations): SB203580 (inhibitor of p38); SP600125 (inhibitor of JNK); PD98059 (inhibitor of ERK1/2); LY‐294002 (inhibitor of phosphatidylinositol‐3 kinase); L‐NAME (inhibitor of nitric oxide synthase); exendin (9–39) (antagonist of GLP‐1 receptor); or H‐89 (inhibitor of PKA). Senescence was evaluated by staining for SA β‐gal and compared to the one found in untreated cells (CTR). The upper part of the Figure shows representative pictures of SA β‐gal staining, whilst the percentage of positive cells (mean ± SEM of 5 independent replicates) is presented in the graph in the lower part.
Figure S3 Oxidative stress in EA.hy926 cells treated with 5FU and/or GLP‐1. Mean fluorescence intensity (MFI) of dichlorofluorescein in EA.hy926 cells after no treatment (CTR) or incubation with 5FU, 5FU preceded by GLP‐1, or GLP‐1 alone. N = 5.
Figure S4 GLP‐1R protein levels in EA.hy926 cells. Representative western blot and densitometry analysis of the expression of GLP‐1R protein in EA.hy926 cells after no treatment (CTR) or exposure to 5FU.
Figure S5 Effect of the signalling inhibitors used for functional experiments on eNOS and SIRT‐1 expression by EA.hy926 cells. Densitometry analysis and representative western blots for eNOS and SIRT‐1 in EA.hy926 after no treatment (CTR) or incubation with the following molecules (see Methods for times and concentrations): SB203580 (inhibitor of p38); SP600125 (inhibitor of JNK); PD98059 (inhibitor of ERK1/2); LY‐294002 (inhibitor of phosphatidylinositol‐3 kinase); L‐NAME (inhibitor of nitric oxide synthase); exendin (9–39) (antagonist of GLP‐1 receptor); or H‐89 (inhibitor of PKA). The optical density of the eNOS and SIRT‐1 bands was normalized for the one of actin. Data are mean ± SEM of 5 independent experiments.
Acknowledgements
The authors thank Daniela Verzola, Luca Liberale and Aldo Bonaventura for helping with some experiments.
Altieri, P. , Murialdo, R. , Barisione, C. , Lazzarini, E. , Garibaldi, S. , Fabbi, P. , Ruggeri, C. , Borile, S. , Carbone, F. , Armirotti, A. , Canepa, M. , Ballestrero, A. , Brunelli, C. , Montecucco, F. , Ameri, P. , and Spallarossa, P. (2017) 5‐fluorouracil causes endothelial cell senescence: potential protective role of glucagon‐like peptide 1. British Journal of Pharmacology, 174: 3713–3726. doi: 10.1111/bph.13725.
References
- Adamsen BL, Kravik KL, De Angelis PM (2011). DNA damage signaling in response to 5‐fluorouracil in three colorectal cancer cell lines with different mismatch repair and TP53 status. Int J Oncol 39: 673–682. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altieri P, Barisione C, Lazzarini E, Garuti A, Bezante GP, Canepa M et al. (2016). Testosterone antagonizes doxorubicin‐induced senescence of cardiomyocytes. J Am Heart Assoc 5 .pii: e002383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azimzadeh O, Sievert W, Sarioglu H, Merl‐Pham J, Yentrapalli R, Bakshi MV et al. (2015). Integrative proteomics and targeted transcriptomics analyses in cardiac endothelial cells unravel mechanisms of long‐term radiation‐induced vascular dysfunction. J Proteome Res 14: 1203–1219. [DOI] [PubMed] [Google Scholar]
- Ban K, Noyan‐Ashraf MH, Hoefer J, Bolz SS, Drucker DJ, Husain M (2008). Cardioprotective and vasodilatory actions of glucagon‐like peptide 1 receptor are mediated through both glucagon‐like peptide 1 receptor‐dependent and ‐independent pathways. Circulation 117: 2340–2350. [DOI] [PubMed] [Google Scholar]
- Bardin N, Moal V, Anfosso F, Daniel L, Brunet P, Sampol J et al. (2003). Soluble CD146, a novel endothelial marker, is increased in physiopathological settings linked to endothelial junctional alteration. Thromb Haemost 90: 915–920. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhou M (2010). Determination of eniluracil and 5‐fluorouracil in human plasma by LC–MS/MS. Bioanalysis 2: 2011–2017. [DOI] [PubMed] [Google Scholar]
- Chien CT, Fan SC, Lin SC, Kuo CC, Yang CH, Yu TY et al. (2014). Glucagon‐like peptide‐1 receptor agonist activation ameliorates venous thrombosis‐induced arteriovenous fistula failure in chronic kidney disease. Thromb Haemost 112: 1051–1064. [DOI] [PubMed] [Google Scholar]
- Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B et al. (2010). Colorectal cancer. Lancet 375: 1030–1047. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cwikiel M, Eskilsson J, Albertsson M, Stavenow L (1996). The influence of 5‐fluorouracil and methotrexate on vascular endothelium. An experimental study using endothelial cells in the culture. Ann Oncol 7: 731–737. [DOI] [PubMed] [Google Scholar]
- De Falco E, Carnevale R, Pagano F, Chimenti I, Fianchini L, Bordin A et al. (2016). Role of NOX2 in mediating doxorubicin‐induced senescence in human endothelial progenitor cells. Mech Ageing Dev pii: S0047‐6374(16)30065‐3. [DOI] [PubMed] [Google Scholar]
- Erdogdu O, Nathanson D, Sjöholm A, Nyström T, Zhang Q (2010). Exendin‐4 stimulates proliferation of human coronary artery endothelial cells through eNOS‐, PKA‐ and PI3K/Akt‐dependent pathways and requires GLP‐1 receptor. Mol Cell Endocrinol 325: 26–35. [DOI] [PubMed] [Google Scholar]
- Favaro E, Granata R, Miceli I, Baragli A, Settanni F, Cavallo Perin P et al. (2012). The ghrelin gene products and exendin‐4 promote survival of human pancreatic islet endothelial cells in hyperglycaemic conditions, through phosphoinositide 3‐kinase/Akt, extracellular signal‐related kinase (ERK)1/2 and cAMP/protein kinase A (PKA) signalling pathways. Diabetologia 55: 1058–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focaccetti C, Bruno A, Magnani E, Bartolini D, Principi E, Dallaglio K et al. (2015). Effects of 5‐fluorouracil on morphology, cell cycle, proliferation, apoptosis, autophagy and ROS production in endothelial cells and cardiomyocytes. PLoS One 10: e0115686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox SB, Moghaddam A, Westwood M, Turley H, Bicknell R, Gatter KC et al. (1995). Platelet‐derived endothelial cell growth factor/thymidine phosphorylase expression in normal tissues: an immunohistochemical study. J Pathol 176: 183–190. [DOI] [PubMed] [Google Scholar]
- Freund A, Orjalo AV, Desprez PY, Campisi J (2010). Inflammatory networks during cellular senescence: causes and consequences. Trends Mol Med 16: 238–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Ushio S, Ozawa N, Masuguchi K, Kawashiri T, Oishi R et al. (2015). Exenatide facilitates recovery from oxaliplatin‐induced peripheral neuropathy in rats. PLoS One 10: e0141921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith YB, Roistacher N, Baum MS (2008). Capecitabine‐induced coronary vasospasm. J Clin Oncol 26: 3802–3804. [DOI] [PubMed] [Google Scholar]
- Hodjat M, Haller H, Dumler I, Kiyan Y (2013). Urokinase receptor mediates doxorubicin‐induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. J Vasc Res 50: 109–123. [DOI] [PubMed] [Google Scholar]
- Ignatescu MC, Gharehbaghi‐Schnell E, Hassan A, Rezaie‐Majd S, Korschineck I, Schleef RR et al. (1999). Expression of the angiogenic protein, platelet‐derived endothelial cell growth factor, in coronary atherosclerotic plaques: in vivo correlation of lesional microvessel density and constrictive vascular remodeling. Arterioscler Thromb Vasc Biol 19: 2340–2347. [DOI] [PubMed] [Google Scholar]
- Kang HM, Sohn I, Jung J, Jeong JW, Park C (2015). Exendin‐4 protects hindlimb ischemic injury by inducing angiogenesis. Biochem Biophys Res Commun 465: 758–763. [DOI] [PubMed] [Google Scholar]
- Lanza GA, Careri G, Crea F (2011). Mechanisms of coronary artery spasm. Circulation 124: 1774–1782. [DOI] [PubMed] [Google Scholar]
- Layoun ME, Wickramasinghe CD, Peralta MV, Yang EH (2016). Fluoropyrimidine‐induced cardiotoxicity: manifestations, mechanisms, and management. Curr Oncol Rep 18: 35. [DOI] [PubMed] [Google Scholar]
- Lazzarini E, Balbi C, Altieri P, Pfeffer U, Gambini E, Canepa M et al. (2016). The human amniotic fluid stem cell secretome effectively counteracts doxorubicin‐induced cardiotoxicity. Sci Rep 6: 29994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara J, Sugiyama S, Sugamura K, Nakamura T, Fujiwara Y, Akiyama E et al. (2012). A dipeptidyl peptidase‐4 inhibitor, des‐fluoro‐sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E‐deficient mice. J Am Coll Cardiol 59: 265–276. [DOI] [PubMed] [Google Scholar]
- Mattagajasingh I, Kim C‐S, Naqvi A, Yamamori T, Hoffman TA, Jung S‐B et al. (2007). SIRT1 promotes endothelium‐dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A 104: 14855–14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L et al. (2005). Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310: 314–317. [DOI] [PubMed] [Google Scholar]
- Oeseburg H, de Boer RA, Buikema H, van der Harst P, van Gilst WH, Silljé HH (2010). Glucagon‐like peptide 1 prevents reactive oxygen species‐induced endothelial cell senescence through the activation of protein kinase A. Arterioscler Thromb Vasc Biol 30: 1407–1414. [DOI] [PubMed] [Google Scholar]
- Ota H, Eto M, Kano MR, Kahyo T, Setou M, Ogawa S et al. (2010). Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler Thromb Vasc Biol 30: 2205–2211. [DOI] [PubMed] [Google Scholar]
- Park H, Kim CH, Jeong JH, Park M, Kim KS (2016). GDF15 contributes to radiation‐induced senescence through the ROS‐mediated p16 pathway in human endothelial cells. Oncotarget 7: 9634–9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk A, Vaage‐Nilsen M, Vistisen K, Nielsen DL (2013). Cardiotoxicity in cancer patients treated with 5‐fluorouracil or capecitabine: a systematic review of incidence, manifestations and predisposing factors. Cancer Treat Rev 39: 974–984. [DOI] [PubMed] [Google Scholar]
- Polk A, Vistisen K, Vaage‐Nilsen M, Nielsen DL (2014). A systematic review of the pathophysiology of 5‐fluorouracil‐induced cardiotoxicity. BMC Pharmacol Toxicol 15: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recio‐Mayoral A, Rimoldi OE, Camici PG, Kaski JC (2013). Inflammation and microvascular dysfunction in cardiac syndrome X patients without conventional risk factors for coronary artery disease. JACC Cardiovasc Imaging 6: 660–667. [DOI] [PubMed] [Google Scholar]
- Schüller J, Cassidy J, Dumont E, Roos B, Durston S, Banken L et al. (2000). Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol 45: 291–297. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al (2016) The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44:D1054‐1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spallarossa P, Altieri P, Aloi C, Garibaldi S, Barisione C, Ghigliotti G et al. (2009). Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am J Physiol Heart Circ Physiol 297: H2169–H2218. [DOI] [PubMed] [Google Scholar]
- Spallarossa P, Altieri P, Barisione C, Passalacqua M, Aloi C, Fugazza G et al. (2010). p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin‐treated endothelial progenitor cells. PLoS One 5: e15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate M, Chong A, Robinson E, Green BD, Grieve DJ (2015). Selective targeting of glucagon‐like peptide‐1 signalling as a novel therapeutic approach for cardiovascular disease in diabetes. Br J Pharmacol 172: 721–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tato‐Costa J, Casimiro S, Pacheco T, Pires R, Fernandes A, Alho I et al. (2016). Therapy‐induced cellular senescence induces epithelial‐to‐mesenchymal transition and increases invasiveness in rectal cancer. Clin Colorectal Cancer 15: 170–178. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Zalcberg JR (1998). 5‐fluorouracil: a pharmacological paradigm in the use of cytotoxics. Clin Exp Pharmacol Physiol 25: 887–895. [DOI] [PubMed] [Google Scholar]
- Timmers L, Henriques JP, de Kleijn DP, Devries JH, Kemperman H, Steendijk P et al. (2009). Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol 53: 501–510. [DOI] [PubMed] [Google Scholar]
- van der Loo B, Fenton MJ, Erusalimsky JD (1998). Cytochemical detection of a senescence‐associated beta‐galactosidase in endothelial and smooth muscle cells from human and rabbit blood vessels. Exp Cell Res 241: 309–315. [DOI] [PubMed] [Google Scholar]
- Walko CM, Lindley C (2005). Capecitabine: a review. Clin Ther 27: 23–44. [DOI] [PubMed] [Google Scholar]
- Woo JS, Kim W, Ha SJ, Kim JB, Kim SJ, Kim WS et al. (2013). Cardioprotective effects of exenatide in patients with ST‐segment‐elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol 33: 2252–2260. [DOI] [PubMed] [Google Scholar]
- Yin H, Pickering JG (2016). Cellular senescence and vascular disease: novel routes to better understanding and therapy. Can J Cardiol 32: 612–623. [DOI] [PubMed] [Google Scholar]
- Yokoi T, Fukuo K, Yasuda O, Hotta M, Miyazaki J, Takemura Y et al. (2006). Apoptosis signal‐regulating kinase 1 mediates cellular senescence induced by high glucose in endothelial cells. Diabetes 55: 1660–1665. [DOI] [PubMed] [Google Scholar]
- Zhu H, Zhang Y, Shi Z, Lu D, Li T, Ding Y et al. (2016). The neuroprotection of liraglutide against ischaemia‐induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci Rep 6: 26859. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers used in the study.
Figure S1 Senescence of EA.hy926 cells in response to increasing concentrations of 5FU. EA.hy926 cells were incubated for 4 h with the indicated concentrations of 5FU. After this treatment, senescence was assessed as percentage of SA β‐gal positive cells. No apoptosis, as evaluated by immunocytochemistry using a rabbit monoclonal anti‐cleaved caspase‐3 antibody (clone 5A1E, Cell Signalling Technologies), was observed. The experiment was repeated 5 times.
Figure S2 Effect of the signalling inhibitors used for functional experiments on senescence of EA.hy926 cells. EA.hy926 cells were treated with the following molecules (see Methods for times and concentrations): SB203580 (inhibitor of p38); SP600125 (inhibitor of JNK); PD98059 (inhibitor of ERK1/2); LY‐294002 (inhibitor of phosphatidylinositol‐3 kinase); L‐NAME (inhibitor of nitric oxide synthase); exendin (9–39) (antagonist of GLP‐1 receptor); or H‐89 (inhibitor of PKA). Senescence was evaluated by staining for SA β‐gal and compared to the one found in untreated cells (CTR). The upper part of the Figure shows representative pictures of SA β‐gal staining, whilst the percentage of positive cells (mean ± SEM of 5 independent replicates) is presented in the graph in the lower part.
Figure S3 Oxidative stress in EA.hy926 cells treated with 5FU and/or GLP‐1. Mean fluorescence intensity (MFI) of dichlorofluorescein in EA.hy926 cells after no treatment (CTR) or incubation with 5FU, 5FU preceded by GLP‐1, or GLP‐1 alone. N = 5.
Figure S4 GLP‐1R protein levels in EA.hy926 cells. Representative western blot and densitometry analysis of the expression of GLP‐1R protein in EA.hy926 cells after no treatment (CTR) or exposure to 5FU.
Figure S5 Effect of the signalling inhibitors used for functional experiments on eNOS and SIRT‐1 expression by EA.hy926 cells. Densitometry analysis and representative western blots for eNOS and SIRT‐1 in EA.hy926 after no treatment (CTR) or incubation with the following molecules (see Methods for times and concentrations): SB203580 (inhibitor of p38); SP600125 (inhibitor of JNK); PD98059 (inhibitor of ERK1/2); LY‐294002 (inhibitor of phosphatidylinositol‐3 kinase); L‐NAME (inhibitor of nitric oxide synthase); exendin (9–39) (antagonist of GLP‐1 receptor); or H‐89 (inhibitor of PKA). The optical density of the eNOS and SIRT‐1 bands was normalized for the one of actin. Data are mean ± SEM of 5 independent experiments.