

Abstract
Cardiotoxicity is a severe side effect of drugs that induce structural or electrophysiological changes in heart muscle cells. As a result, the heart undergoes failure and potentially lethal arrhythmias. It is still a major reason for drug failure in preclinical and clinical phases of drug discovery. Current methods for predicting cardiotoxicity are based on guidelines that combine electrophysiological analysis of cell lines expressing ion channels ectopically in vitro with animal models and clinical trials. Although no new cases of drugs linked to lethal arrhythmias have been reported since the introduction of these guidelines in 2005, their limited predictive power likely means that potentially valuable drugs may not reach clinical practice. Human pluripotent stem cell‐derived cardiomyocytes (hPSC‐CMs) are now emerging as potentially more predictive alternatives, particularly for the early phases of preclinical research. However, these cells are phenotypically immature and culture and assay methods not standardized, which could be a hurdle to the development of predictive computational models and their implementation into the drug discovery pipeline, in contrast to the ambitions of the comprehensive pro‐arrhythmia in vitro assay (CiPA) initiative. Here, we review present and future preclinical cardiotoxicity screening and suggest possible hPSC‐CM‐based strategies that may help to move the field forward. Coordinated efforts by basic scientists, companies and hPSC banks to standardize experimental conditions for generating reliable and reproducible safety indices will be helpful not only for cardiotoxicity prediction but also for precision medicine.
Linked Articles
This article is part of a themed section on New Insights into Cardiotoxicity Caused by Chemotherapeutic Agents. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v174.21/issuetoc
Abbreviations
- 2D
two dimensional
- 3D
three dimensional
- AP
action potential
- APD
AP duration
- APD90
APD at 90% of the repolarisation phase
- BVR
beat‐to‐beat variability of the repolarisation duration
- CiPA
comprehensive in vitro proarrhythmia assay
- CMs
cardiomyocytes
- ECG
Electrocardiogram
- EMA
European Medicines Agency
- FDA
US Food and Drug Administration
- hERG
human ether‐à‐go‐go‐related gene
- hESC‐CMs
human embryonic stem‐cell derived CMs
- hESC
human embryonic stem cells
- hiPSC‐CMs
human induced pluripotent stem‐cell derived CMs
- hiPSC
human induced pluripotent stem cells
- hPSC‐CMs
human pluripotent stem‐cell derived CMs
- hPSC
human pluripotent stem cells
- ICaL
L‐type Ca2 + current
- ICH
International Council on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use
- IK,ATP
ATP‐sensitive K+ current
- IK,slow1
Component 1 of the ultra‐rapid delayed rectifyer K+ current (4‐Aminopyridine sensitive)
- IK,slow2
Component 2 of the ultra‐rapid delayed rectifyer K+ current
- IK1
Inward‐rectifier K+ current
- IKr
Rapid component of the delayed‐rectifier K+ current
- IKs
Slow component of the delayed‐rectifier K+ current
- ISS
Steady‐state K+ current
- Ito
Transient‐outward K+ current
- MEA
multi electrode array
- PMDA
Japanese Pharmaceuticals and Medical Devices Agency
- TdP
Torsades de pointes
- Vm
membrane potential
Tables of Links
| TARGETS | |
|---|---|
| Voltage‐gated ion channels a | Transporters b |
| Cav1.2 | NCX |
| Kir2.1‐2.3 | |
| Kir6.2 | |
| Kv4.3 | |
| Kv7.1 | |
| Kv11.1 (hERG) |
| LIGANDS |
|---|
| Astemizole |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
Electrophysiological or structural damage of the myocardium as an unintentional side effect of pharmacological treatment is referred to as cardiotoxicity. The resulting contractile and electrical dysfunction often leads to abnormal heartbeat and life‐threatening arrhythmias (Ewer and Ewer, 2015; Gintant et al., 2016). Arrhythmia is currently an immense burden on public health, with millions of people at risk of heart malfunction because of the cardiotoxic effects of drugs (Rubinstein and Camm, 2002; Vejpongsa and Yeh, 2014; Viskin et al., 2015). Individuals with an underlying but undiagnosed genetic risk may be especially vulnerable because their need for safer alternatives already on the market may be unrecognized (Svanström et al., 2014; Schwartz and Woosley, 2016).
Cardiotoxicity is an important hurdle to drug discovery, with many compounds discarded during drug development because of potential toxic effects on the heart (Laverty et al., 2011; Hay et al., 2014) and difficulties in predicting how the human heart will respond. Cardiotoxicity is still a predominant cause of preclinical and clinical drug failure (McNaughton et al., 2014; Onakpoya et al., 2016; Siramshetty et al., 2016). In general, it is most conveniently categorized as (i) physical damage to cardiomyocytes (CMs) or (ii) electrical disruption, depending on the molecular target or function impaired by the drug.
Cardiotoxicity caused by physical damage to cardiomyocytes
Structural properties of CMs can be profoundly disrupted by pharmacological treatments. Physical damage is primarily associated with drugs specifically designed to be cytotoxic, such as chemotherapeutic agents in cancer treatment (Cross et al., 2015; Ewer and Ewer, 2015). These drugs induce mitochondrial damage (Varga et al., 2015), increase oxidative stress (Cross et al., 2015), activate DNA damage response pathways (Mercurio et al., 2016) and increase apoptosis (Arola et al., 2000). The net outcome for the heart may be many fewer CMs, with a residual proportion being vital but unhealthy. As a result, the contractile force of cardiac muscle is reduced over time (Ewer and Ewer, 2015). In turn, this reduces the ejection fraction from the left ventricle, and heart failure ensues (Cardinale et al., 2015). Indirect consequences of this are severe secondary electrical abnormalities emerging after medium‐ to long‐term drug exposure (Albini et al., 2010); the heart becomes arrhythmic and unable to cope with the physiological demand for oxygen and nutrients. Identifying these drug risks in humans has been challenging because of the paucity of predictive experimental models, but new approaches based on human pluripotent stem cell‐derived CMs (hPSC‐CMs) are revealing the ability to replicate cardiotoxicity as evident in the clinical context (Klimas et al., 2016), most recently in precision or personalized approaches to chemotherapeutic agents (Maillet et al., 2016), like the anthracyclines used for breast cancer treatment (Bellin and Mummery, 2016; Burridge et al., 2016).
Cardiotoxicity caused by electrical disruption
A second form of cardiotoxicity, commonly evident in the early phases of drug discovery, is the acute and direct, detrimental effect of drugs on the electrophysiological properties of CMs (Cook et al., 2014). Electrical homeostasis is crucial for coordinating synchronous and functional contraction of the heart, and its disruption significantly reduces cardiac output. In ventricular CMs, the cardiac action potential (AP), a transient oscillation of the electrical membrane potential (Vm), is initiated by an electrical stimulus sufficient to overcome the threshold of voltage‐gated Na+ channels, which open and rapidly depolarize Vm (Figure 1). Subsequently, tuned balance of the repolarizing currents [i.e. the transient outward current (Ito), rapid (IKr) and slow (IKs) components of the delayed‐rectifier K+ current, the inward‐rectifier K+ current (IK1) and the ATP‐sensitive K+ current (IK,ATP)] slowly counteract the L‐type Ca2 + current (ICaL) until they prevail, terminating the AP. Anything that modifies the frequency or the contour of the AP dictates arrhythmogenic risk. These properties are also precisely targeted by antiarrhythmic agents.
Figure 1.
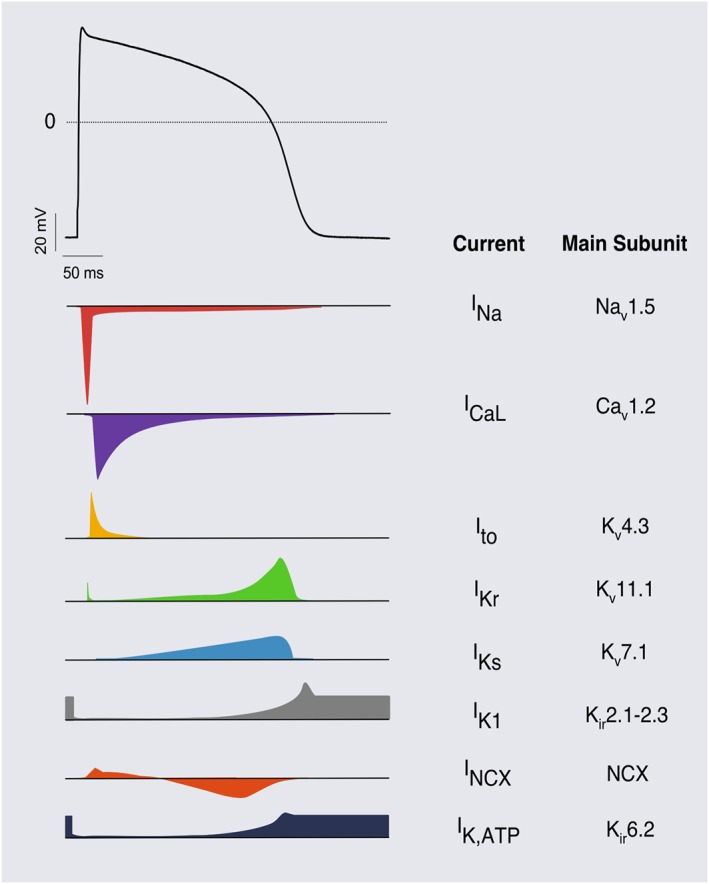
Typical ventricular action potential. Scheme of the relative contribution of ion currents to the cardiac AP. Currents and their respective α‐subunits are indicated on the right.
Some of these channels, however, have been described as being particularly prone to block by some compounds, and this can initiate arrhythmias. It is a recurring condition for the Kv11.1 channel [encoded by the human ether‐à‐go‐go‐related gene (hERG) also called KCNH2], which, together with its β‐subunit MinK‐related peptide 1, is responsible for IKr. Key aromatic residues in the S6 helix (Tyr652, Phe656) and the pore helix (Thr623, Ser624, Val625) of the Kv11.1 channel have been identified as the detrimental target of a broad spectrum of compounds (Perry, 2005) that impair K+ ion flux through the pore and therefore reduce IKr. This generates a prolongation of the QT interval of the electrocardiogram (ECG), with the potential occurrence of a polymorphic form of ventricular tachycardia known as torsades de pointes (TdP), which can cause sudden death (Sanguinetti and Tristani‐Firouzi, 2006).
Life‐threatening arrhythmic events may arise when any drug (or its metabolites) is (i) itself pro‐arrhythmic in all individuals in any population; (ii) generally safe in most of the population but cardiotoxic on specific genetic backgrounds or in the context of pre‐existing conditions; and (iii) generally safe but cardiotoxic in combination with other drugs, perhaps on specific genetic backgrounds. A major goal in drug discovery is then to design drugs with low chances of interactions with ion channels other than the ones targeted and, most importantly, to be able to predict in which of the three categories above that each potential new drug belongs. This would allow adverse drug effects to be identified in early preclinical phases of development, before entering (expensive) phase III clinical trials and introducing the drug to market when it may be prescribed unknowingly to patients at risk. The lack of unequivocal predictive scores has resulted in a series of tests that can only provide an indication of clinical safety when combined together (ICH, 2005a). This is illustrated by the sharp reduction in drug withdrawal over the last decade because of arrhythmogenic events, largely attributable to the widespread introduction of compulsory hERG channel assays (Stockbridge et al., 2012). What sometimes goes almost unnoticed though is the concomitant fall in the approval rate of new drugs despite the unprecedented increase in R&D investment in the last 10 years (Kola and Landis, 2004; Bunnage, 2011; Hay et al., 2014; Waring et al., 2015). This indicates that current methods are still not meeting needs, perhaps providing negative indications for drugs that could be of clinical benefit. Novel and more comprehensive approaches specifically addressing risk in humans should be pursued, both in the early preclinical phases as well as later as drugs move towards human studies (Woosley and Romero, 2013), most particularly for drugs that cannot, or must not, be tested on healthy volunteers because of narrow therapeutic windows.
In this review, we describe how hPSC‐CMs have been used for cardiotoxicity detection, how they compare with current alternative technologies, and whether they might eventually be introduced as effective in vitro predictors of cardiotoxicity in vivo. We also discuss how they might contribute to solving the current stagnation in drug approval.
Cardiotoxicity screening methodologies
During the 1990s, the main drug regulatory agencies, the European Medicines Agency (EMA), the US Food and Drug Administration (FDA) and the Japanese Pharmaceuticals and Medical Devices Agency (PMDA) started investigating the interactions between approved drugs and Kv11.1 (hERG) channels following several cases of lethal arrhythmias associated with commonly prescribed drugs, which was later correlated with an unusually high affinity for Kv11.1 (Friedrichs et al., 2005; Shah, 2005). Since 2005, it has become mandatory for all new drugs seeking regulatory approval to demonstrate that the IC50 of the candidate drug for Kv11.1 is as distant as possible from the target of interest [ICH‐S7B, (ICH, 2005b)]. A 30‐fold separation margin was found to be highly predictive for hERG block in preclinical tests (Redfern et al., 2003); furthermore, clinical studies on healthy volunteers [ICH‐E14, Thorough QT/QTc Study (ICH, 2005a; Fermini et al., 2015)] must follow in vitro hERG tests for new drugs, and also be carried out for any other drugs that have undergone substantial alterations in formulation, administration route or target population (Shah, 2005).
The most convenient way to assess Kv11.1 interaction in vitro is the hERG blockade assay (Hancox et al., 2008). Immortalized mammalian cell lines such as HEK or CHO are genetically engineered to express Kv11.1 channels ectopically [or, better, combinations of selected ion channels as recently proposed in the MICE approach (Kramer et al., 2013)]. These assays are extensively used in the preclinical characterization of drugs because of low cost and flexibility. Traditional low‐throughput techniques for measuring effects on these channels such as the manual patch clamp, although very sophisticated and accurate, have been complemented in recent years by reliable and more affordable high‐throughput technologies, which allow simultaneous automated screening of many compounds and ion channels. A recent alternative, although not yet formally approved as a replacement for the standard hERG blockade assay, is the use of [3H]‐dofetilide displacement from Kv11.1 channels assessed as radioactive decay over time when the [3H]‐dofetilide is displaced by higher affinity, non‐radioactive compounds competing for its same binding site (Diaz et al., 2004; Yu et al., 2015). Although this increases the throughput over classical assays, it falls short in capturing the functional and biophysical aspects of hERG blocks, in particular for those compounds that do not compete for the dofetilide binding site on Kv11.1 channels.
However, although Kv11.1 block is a discrete indicator of arrhythmogenicity, the former does not imply the latter, as demonstrated for verapamil (Liang et al., 2013) and ranolazine (Hancox et al., 2008), with a significant chance of generating false‐positive or false‐negative outcomes. Moreover, no information is obtained on the contractile behaviour or temporal dispersion of repolarization; the latter, with a crucial role in the genesis of lethal arrhythmia (Townsend and Brown, 2013), cannot be derived from simplified in vitro models, which further limits the predictivity of these approaches.
The spectrum of ion channels and contractile proteins required for reliable pro‐arrhythmic prediction scores should reflect that of adult CMs. Animal models recapitulate human cardiac physiology to a certain approximation so that they are widely used for arrhythmia predictivity. Small animal species like mice can also be genetically engineered to generate tailored models of either monogenic or complex diseases. Although some differences between drug effects in vivo and ex vivo have been reported (Bentzen et al., 2011), isolated hearts connected to Langendorff perfusion devices have been valuable for studying drug effects in relatively controlled, but metabolically isolated conditions. This has often been carried out in combination with Ca2 +‐ and voltage‐sensitive dyes, to analyse electrical dispersion and propagation (Akar and Akar, 2006), generate conduction maps (Yu et al., 2016) and Ca2 +‐handling profiles (Herron et al., 2012).
Isolated CMs from medium/large animals, most particularly rabbits/dogs, are presently regarded as the gold standard for the investigation of pharmacological targets and for the prediction of the pro‐arrhythmic potential of novel compounds. However, there are substantial and not negligible inter‐species differences, in particular in the genetics of transgenic models because the human genome differs from that of widely used animal species; correctly mimicking the effects of genetic mutations is only performed routinely in mice, which differ substantially from humans in electrophysiology and Ca2 +‐handling. A more rapid heartbeat (500 versus 60 beats min‐1 in humans), the predominant role of potassium currents [transient‐outward (Ito), ultra‐rapid components 1 (IK,slow1) and 2 (IK,slow2) of the delayed‐rectifier K+ current, and steady state (ISS) K+ currents], the absence of the plateau phase generated by the L‐type Ca2 + current and a smaller contribution of the Na+‐Ca2 + exchanger in the removal of Ca2 + from the cytosol, shape the AP in CMs from small rodents, limiting their arrhythmogenic predictivity, with pharmacological responses and drug sensitivities often far from those of human CMs (Nerbonne, 2004; Martignoni et al., 2006; Ahrens‐Nicklas and Christini, 2009; O'Hara and Rudy, 2012; Himmel, 2013). Large animal models are then required as endpoints in preclinical phases of research (Koeppel et al., 2012), but they come at high cost, cannot easily be genetically modified and they still show the inter‐species differences from humans. Although extremely useful and not replaceable in the short term, these models are nevertheless low throughput and do not support large‐scale drug–drug interaction screening (Milan and MacRae, 2005).
Finally, primary human cardiac tissue can be obtained from biopsies collected during heart surgery. However, this is limited in quantity and is often only available from certain patient groups, for example, after sudden death from non‐cardiac causes, so that they cannot be considered as representative of the general population. In addition, because samples cannot be collected repeatedly from the same source, their use in drug screening is limited. Furthermore, adult CMs de‐differentiate in prolonged culture, which is also a major drawback (Mitcheson et al., 1998).
Human pluripotent stem cell‐derived cardiomyocytes
An alternative source of functional human CMs without the limitations of collecting primary heart tissue is to derive them from human embryonic‐ (hESC) or induced pluripotent (hiPSC) stem cells. Protocols are available to direct cell differentiation towards many somatic cell lineages, including CMs (Mummery et al., 2012; Schwach and Passier, 2016). Chemically defined media supplemented with developmentally relevant growth factors [or small molecules that activate similar pathways, (Burridge et al., 2014; Chen et al., 2014; van den Berg et al., 2016)] are now commercially available or sold as complete kits so that deriving CMs from pluripotent stem cells (as hESC‐CM or hiPSC‐CM) is now feasible for most laboratories. Because legal and ethical issues restrict the use of hESC in some countries, hiPSC are preferred by most regulatory authorities seeking to establish reliable cardiotoxicity assays. Healthy and diseased hiPSC lines are increasingly available through dedicated cell banks [e.g. NIH (USA), StemBancc (EU), Human Induced Pluripotent Stem Cells Initiative (UK), Wellcome Trust (UK), New York Stem Cell Foundation (USA), and California Institute of Regenerative Medicine (USA)], and hiPSC‐CMs are also commercially available (e.g. from Cellular Dynamics International iCell® CMs, Axiogenesis Cor.4U®, Pluriomics Pluricyte®.).
Research over the last few years has increasingly documented the potential of hiPSC in replicating (genetic) cardiovascular disease phenotypes, monogenic cardiac channelopathies being among the most common class of diseases modelled (Bellin et al., 2012; Šarić et al., 2014). In addition, hiPSC‐CMs are also being used as model systems to test the effect of both known and novel drugs on cardiac channelopathies (Matsa et al., 2011; Zhang et al., 2012; Bellin et al., 2013; Di Pasquale et al., 2013; Zhang et al., 2014), with suppression of pathological pro‐arrhythmic potential and restoration of altered AP duration (APD) being two main endpoints. Clinical evidence has shown that pharmacological treatments can be of more benefit when properly calibrated in a disease‐specific or even mutation‐specific manner (Nebert and Vesell, 2006; Cavallari, 2012; Amin and Wilde, 2016; Itoh et al., 2016; Martiniano et al., 2016). This has been particularly evident for monogenic diseases targeting the cardiac electrical system (Priori, 1998; Tan et al., 2006), because genetic modifiers critically determine the clinical phenotype (Duchatelet et al., 2013; de Villiers et al., 2014). For these reasons, a renewable source of healthy human‐ and patient‐derived CMs represented by hiPSC have potential as valuable tools for measuring pathological phenotypes, mechanisms underlying disease and pharmacological responses.
More recently, translational crosstalk between patients and hiPSC‐CMs has shown how closely they can mimic clinical drug responses. Examples include long‐QT syndrome (Terrenoire et al., 2013; Malan et al., 2016), catecholaminergic polymorphic ventricular tachycardia (Penttinen et al., 2015) and heart failure with preserved ejection fraction (Raphael et al., 2016). The cardiotoxic effects of anthracyclines in cardio‐oncology (Burridge et al., 2016) have also been recently validated (Avior et al., 2016).
Assays and readouts
At appropriate seeding densities, hPSC‐CMs can form a functional cardiac syncytium. Electrical signals resembling the QT interval of the ECG and defined field potentials can be recorded through extracellular‐ (Harris et al., 2013; Nozaki et al., 2014; Clements, 2016) or intracellular‐ (Fendyur and Spira, 2012; Spira and Hai, 2013) electrodes using multi electrode arrays (MEAs). Some medium‐throughput MEA platforms also allow contractions (displacement) to be measured through impedance (Li et al., 2016; Obergrussberger et al., 2016), generating highly reproducible results (Lu et al., 2015). In some cases, it can be valuable to electrically ‘pace’ CMs so that their beating rate (defined RR‐like interval because it reflects the analogue ECG parameter) is standardized and constant rather than irregular, as often occurs if beating is spontaneous. However, in contrast to isolated cells or neuronal networks, clusters of hPSC‐CMs seeded on MEA cannot efficiently be paced in all available devices, because the hardware is not suitable (Natarajan et al., 2011). Although methods for electrical stimulation have been implemented in some custom‐made MEAs (Kaneko et al., 2012) and impedance‐based systems (Xi et al., 2011), these are not yet widely used. The QT interval duration in humans and in hPSC‐CMs directly depends on the RR interval, such that heart rate correction methods based on Bazett's or Fridericia's formulae (Johnson and Ackerman, 2009; Kaneko et al., 2012) have been developed to minimize this dependency and allow the comparison of QT intervals from samples/patients with different beat rates. Although these corrections are valid for patients (i.e. when frequencies fall in the clinical range), the absence of uniform QT‐RR relationships in hPSC‐CMs prevents calibration of such methodologies in vitro on a large scale and could cause biased interpretation of corrected QT intervals (Sala et al., 2016). A standardized stimulation protocol with crescent frequencies carried out on multiple cell lines would be useful for this purpose and might address QT correction issues that have been a matter of dispute, eventually leading to more reliable cell line‐specific QT correction methods for hPSC‐CM disease modelling and pharmacology (Woosley and Romero, 2013).
Furthermore, because QT‐like and RR‐like intervals on MEAs share certain similarities to ECG signals, it could be feasible to apply some of the clinical pro‐arrhythmic evaluation strategies to MEA; QT variability index (Berger, 2003) or beat‐to‐beat variability of the repolarisation duration [BVR (Pueyo et al., 2016; Stams et al., 2016)] can be informative, especially considering the pivotal role of the Kv11.1 channel in shaping the repolarisation stability (Altomare et al., 2015). Data analysis algorithms could allow automatic quantification of both short‐ and long‐term components of the repolarisation variability in a selected series of beats (Sala et al., 2016). Its high dependence on the beating frequency could also allow the identification of key regulators of the RR interval variability in hPSC‐CMs. Representative examples of the application of BVR analysis to hPSC‐CMs are shown in Figure 2. Particular attention should be given to generating standard recording protocols for drug testing, because it has been demonstrated (for irreversible agonists/antagonists) that the volume of liquid in which a drug is dissolved may influence the outcome of the test (Cavero et al., 2016).
Figure 2.
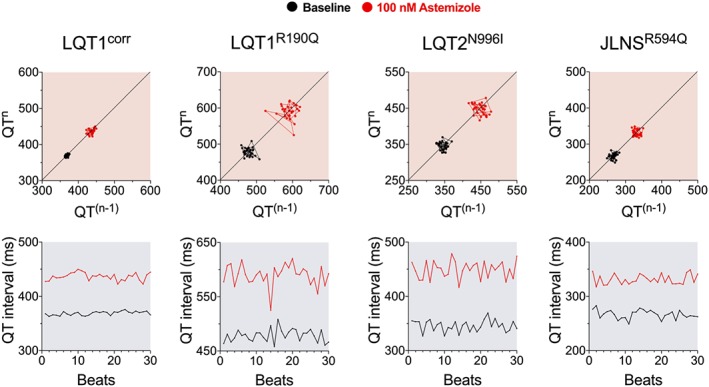
BVR analysis. Representative Poincaré plots (top) and QT interval sequences for 30 consecutive beats (bottom) for individual experiments at MEA, under control conditions (black) and after IKr block with 100 nM astemizole (red) in hiPSC‐CMs lines with long‐QT syndrome (LQT1R190Q, LQT2N996I, JLNSR594Q) and one hiPSC‐CMs control isogenic to the LQT1R190Q (LQT1corr).
Although straightforward to produce and maintain in culture, two‐dimensional (2D) layers of hPSC‐CMs on plastic or glass substrates do not entirely replicate the three‐dimensional (3D) micro‐environment of myocardium nor its physiological properties such as cyclic stretch, CM‐non‐CM or CM–CM interaction and communication, contractile force and paracrine signalling (Beauchamp et al., 2015). In addition, 2D monotypic cultures do not resemble the complex cellular composition of the adult heart, where CMs represent only one third of the total number of cells: they are normally incorporated within networks of smooth muscle cells/pericytes, endothelial cells and fibroblasts (Tirziu et al., 2010) and covered by epicardium on the outside and endocardium on the inside layer. The generation of cardiac microtissues (Beauchamp et al., 2015), engineered heart tissues (Zimmermann and Schneiderbanger, 2002; Turnbull et al., 2014) or 3D vascularized cardiac layers (Mosadegh et al., 2014) have been reported. These multidimensional and heterogeneous environments promote CM organization and maturity and will thus probably contribute, in the future, to enhancing the predictivity and reliability of these platforms for drug screening when compared with 2D cultures (Eder et al., 2016). Using edge‐detection contractility, Ravenscroft et al. observed that in vitro measurements were more likely to replicate in vivo data when CMs were cultured with endothelial cells and cardiac fibroblasts, suggesting a contribution of non‐CMs to drug responses (Ravenscroft et al., 2016).
The immature phenotype, low levels of extracellular matrix proteins and the membrane structure of hPSC‐CMs make them considerably more tolerant of enzymatic dissociation for FACS analyses or automated cell manipulation than adult CMs (Bhattacharya et al., 2014; Scheel et al., 2014). Ion currents and AP can be measured with a sufficient level of precision with these systems that the chances of generating large‐scale, standardized methods for measuring cardiotoxicity are increased (Scheel et al., 2014). However, the success of these approaches will, nevertheless, be determined by progress in improving the maturation status of hPSC‐CMs, that is, the more hPSC‐CMs resemble adult CMs, the less they will tolerate invasive techniques. For this reason, less invasive alternatives are being already being developed in parallel, with the interest in voltage‐sensitive dyes (Burridge et al., 2011) and optogenetics (Park et al., 2014; Chang Liao et al., 2015; Song et al., 2015) significantly rising (Dempsey et al., 2016). At present, their widespread implementation in drug screening is limited by the slow kinetics and the relatively low signal/noise ratio of voltage sensors, with data still requiring proper validation by low‐throughput patch clamp analyses. Incorporation of these new methods in ICH guidelines would be premature and unlikely short term. For chemical and genetically encoded Ca2 +‐sensitive dyes, the situation is easier, mainly due to the slower kinetics of Ca2 + fluxes. These probes have already been widely used for high‐throughput screening in both disease modelling and drug development with encouraging results (Kaestner et al., 2014; Huebsch et al., 2015; Lu et al., 2015; Rast et al., 2015; Dempsey et al., 2016; Klimas et al., 2016). Ca2 +‐handling also affects cell contractility, which can also be measured, as recent examples with sophisticated optical mapping systems show (Herron et al., 2012; Hayakawa et al., 2014; Maddah et al., 2015). The combination of engineering and biology has allowed the precise quantification of contractile force in hPSC‐CMs (Ribeiro et al., 2015a), although it has been demonstrated that substrate stiffness plays a major role in the quantification of absolute force values (Feinberg et al., 2012; Ribeiro et al., 2015b). More recently, integrated technologies capable of simultaneously quantifying multiple parameters are being implemented with the view to providing reliable, high‐throughput tools for early preclinical stages of drug development (Hochbaum et al., 2014; Kijlstra et al., 2015; Rast et al., 2015; Song et al., 2015; Dempsey et al., 2016; Klimas et al., 2016).
In silico approaches
In silico approaches are also proving valuable in predicting the pro‐arrhythmic potential of drugs. Computational models for ion currents are being generated based on existing electrophysiological data measured in adult animal/human CMs as well as hPSC‐CMs and then integrated into complex single‐ or multi‐cellular models (O'Hara et al., 2011; Paci et al., 2013). It is expected that, as computational power increases, the accuracy and complexity of such models will rise. However, the heterogeneity of hiPSC‐CM phenotypes may limit the predictive power of current models (Paci et al., 2013, 2015). For example, different differentiation protocols lead to different ventricular phenotypes (Figure 3), integrating reprogramming methods may disrupt genes of interest for the phenotype under study, and the kinetics of hiPSC‐CM differentiation may differ between individual lines. This particularly impacts IKs and IK1, whose contribution is essential for a correct prediction of drug effects. Only few attempts have been made to promote phenotypic uniformity in hiPSC‐CMs (Hwang et al., 2015a; Zhu et al., 2016), and we believe that the reproducibility of outcomes would benefit from standardized conditions to provide better input for computational models. On the other hand, as highlighted by Rodriguez et al., the cross‐institutional limitations of accessing in vitro propriety data profoundly slows the development of comprehensive computational models (Rodríguez et al., 2015).
Figure 3.
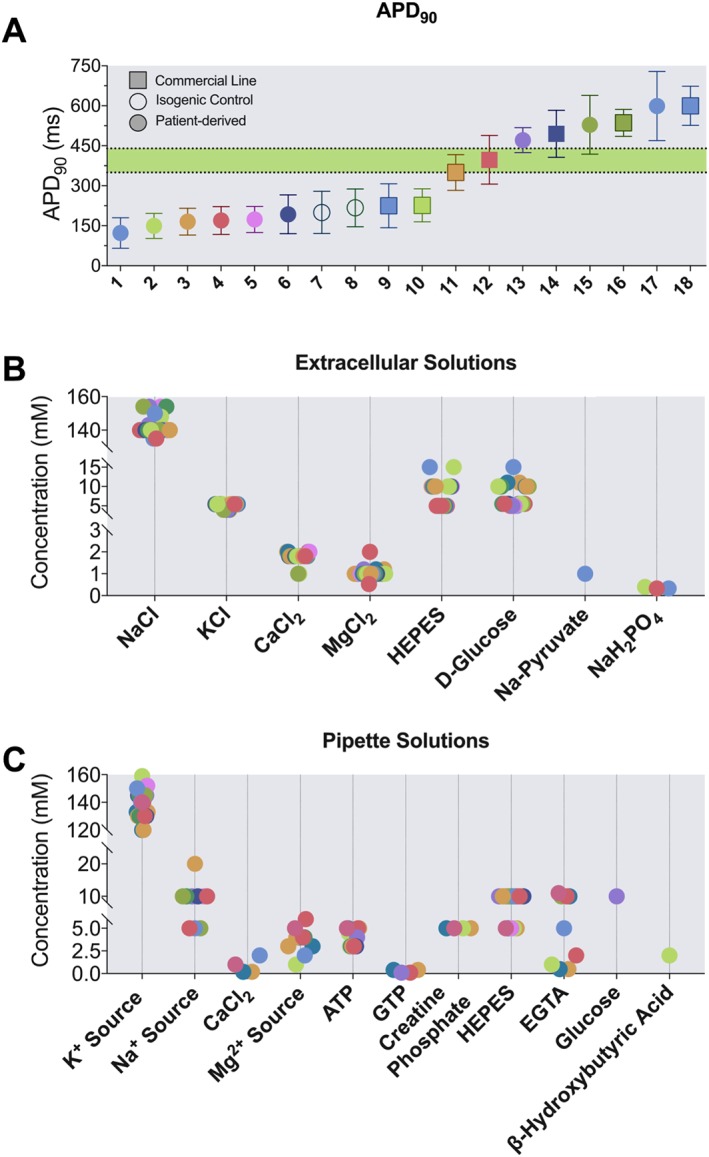
Comparison of wild‐type hiPSC‐CMs in the literature. (A) APD90 values measured with patch clamp in paced (1 Hz) hiPSC‐CMs at physiological temperature (~37°C); numbers 1–18 indicate wild‐type lines as specified below. The green area defines the normal QT interval range for humans. Data are shown as mean ± SD: 1, (Malan et al., 2016); 2, (Rocchetti/Sala et al., unpublished); 3, (Zhang et al., 2014); 4, (Zhang et al., 2014); 5, (Davis et al., 2012); 6, (Rocchetti/Sala et al, unpublished); 7, (Bellin et al., 2013); 8, (Sala et al., 2016); 9, (Bizy et al., 2013); 10, (Ma et al., 2013); 11, (Ma et al., 2015); 12, (Ma et al., 2015); 13, (Gibson et al., 2014a); 14, (Itzhaki et al., 2011); 15, (Gibson et al., 2014c); 16, (Lu et al., 2014); 17, (Gibson et al., 2014b); 18, (Mehta et al., 2014). (B) and (C) Composition of extracellular buffers (B) and pipette solutions (C) for current clamp experiments. K+ source indicates the sum of KCl and/or K‐aspartate and/or K‐glutamate. Na+ source indicates the sum of NaCl and/or the conjugated Na‐salts to ATP, GTP or creatine phosphate. Mg2 + source indicates the sum of MgCl2 and/or the conjugated Na‐salts of ATP, GTP or creatine phosphate.
The result is that current mathematical AP models of hPSC‐CMs (Paci et al., 2013, 2015, 2012) still represent a fraction of the heterogeneity that has been experimentally observed in literature, although their usefulness and predictivity are beyond doubt (Figure 3). In this context, the coordination and promotion of standard methodologies for phenotypic characterization of differentiated cells may come from cell banks.
Future perspectives
The comprehensive in vitro pro‐arrhythmia assay (CiPA) initiative proposes integration of different approaches to quantify the proarrhythmic potential of drugs at three levels: (i) heterologous system, with the expression of a representative spectrum of cardiac ion channels; (ii) in silico mathematical models of CMs; and (iii) confirmation of the data from integrated human cellular studies in advanced systems like hiPSC‐CMs.
In the coming years, the FDA plans to update the current guidelines, ICH‐S7B (ICH, 2005b) and ICH‐E14 (ICH, 2005a) for preclinical and clinical evaluation of drug arrhythmogenicity, and it is expected that ICH‐S7B will integrate hiPSC‐CMs as a platform for ‘personalized’ drug testing (van der Heyden and Jonsson, 2012; Chen et al., 2016). A very detailed overview on the current status of the CiPA initiative has recently been provided by Cavero et al., who listed in detail many of the technical and regulatory limitations that must be addressed to provide a robust set of protocols that would be needed before implementation of these assays in a safety pharmacology pipeline (Cavero et al., 2016). Although significant progress has been made in this context, in particular with the standardization of patch clamp protocols to evoke IKr (although only in the manual configuration) and the proposal of an in vitro TdPrisk score (Crumb et al., 2016), a complete list of unresolved issues and areas needing more discussion, including agreement among researchers, agencies and companies on how precisely to move forward, is at present lacking. This to some extent undermines full implementation in the short term.
Limitations in applicability of hiPSC‐CM to large‐scale drug testing
Phenotypic immaturity
As mentioned earlier, one major limitation of hPSC‐CMs is their phenotypic immaturity compared with adult CMs. Typical fetal features that they display include automaticity of beating, depolarized diastolic Vm, low ion channel expression, delayed excitation‐contraction coupling and low contractile force (Veerman et al., 2015). Defined culture conditions (Burridge et al., 2014; Ribeiro et al., 2015a, 2015b) can be used to improve the maturation of hPSC‐CMs to reveal hidden disease phenotypes (Birket et al., 2015) or to drive differentiation to chamber‐specific cell populations (Devalla et al., 2015), and drug testing might benefit from their standardization. Exogenous stimuli, such as adjusted pacing frequency (Chan et al., 2013), 3D‐microenvironments (Zhao et al., 1999; Nunes et al., 2013; Hirt et al., 2014; Eder et al., 2016) and heterotypic cell co‐culture (Robertson et al., 2013) may all contribute to cell maturation, increasing the expression of ion channels and improving functional properties. Recent advances in electrophysiology (Meijer van Putten et al., 2015) and cell biology (Vaidyanathan et al., 2016) allow the introduction of exogenous IK1 conductance in hiPSC‐CMs, although neither single nor combined approaches were successful in mimicking adult maturation state entirely, in particular of Ca2 +‐handling, signalling and compartmentalization, mainly due to their lack of T‐tubules (Kane and Terracciano, 2015). Thus, caution is needed when attempting to translate results using hiPSC‐CMs to more complex systems. Furthermore, the integration of hiPSC‐CMs into drug screening platforms would preferably be based on defined (xeno‐free) reagents to implement current Good Manufacturing Practice guidelines and would extend to new coatings for substrates that are resistant to drug adsorption and not yet broadly implemented (Rodin et al., 2014; Leha et al., 2016).
Phenotypic variability
High line‐to‐line variability (although only modest clonal variability in cells from one line) is still a widespread problem that requires joint efforts to solve, although recent, robust, methodologies have contributed to mitigating the issues (Burridge et al., 2011; Hartjes et al., 2014; Denning et al., 2016). The reproducibility of the results in terms of the absolute values of parameters measured may indeed be due to limited standardization of experimental conditions: few studies carry out head‐to‐head comparisons of cells from different hiPSC lines derived under identical conditions. Fully automated, high‐throughput procedures to reprogramme hiPSC from somatic cells have been developed (Paull et al., 2015) with the aim of limiting variability. Pharmacogenomic analysis of these cell populations may unveil the key sources of variability. A partial solution could be obtained by pooling data from large cohorts of patients to reveal unusual patterns of pharmacological responses associated with specific genotypes, contributing to insights into precision medicine (Collins and Varmus, 2015), although this is still far from the goals of personalized medicine (Food and Drug Administration, 2013), (Cuchiara et al., 2015). It would then be feasible to capture key aspects of variability in vitro during preclinical phases of research, before the last – extremely expensive – phases of drug testing. This may reduce the failure rate of current pro‐arrhythmic assays by unmasking unexpected drug sensitivities and cardiotoxic effects in the presence of specific triggers.
Individual genetic backgrounds, or DNA epigenetic status (Burridge et al., 2015), appear to influence the differentiation potential (Miyamoto et al., 2015) of hPSC and the cell phenotype after differentiation. A high phenotypic variability has been observed among different lines described as ‘healthy controls’ (sometimes age and gender matched) and used as a reference in multiple studies (Figure 3). In a study on four isogenic lines from healthy donors (Kyttälä et al., 2016), up to 158 genes were found to be differently expressed among the lines; some were imprinted, evidence for a role of epigenetic memory in determining the phenotype and differentiation potential of hPSC, possibly by affecting their response to growth factors (Pekkanen‐Mattila et al., 2009). A consequence may then include differential responses to drugs. Jones et al. proposed Ca2 + synchronization in aggregates of hiPSC as a reliable parameter to identify sources of variability related to culture conditions and maturation status of hiPSC‐CMs (Jones et al., 2015).
Even though Ca2 +‐handling features appear conserved in independent hiPSC‐CM cultures from different research groups (Hwang et al., 2015a, 2015b; Kane and Terracciano, 2015), heterogeneity has been widely observed in other electrophysiological properties such as the APD (Figure 3), upstroke velocity and current densities (Hoekstra et al., 2012). Inconsistent results on the mechanism of hiPSC‐CM automaticity have also been noted: Kim et al. reported a Ca2 + clock mechanism, consistent with ivabradine having no effect on beating frequency (Kim et al., 2015). In contrast, Bedut et al. demonstrated reduced beating rates in hiPSC‐CMs at lower doses of ivabradine (1‐2 μM vs 3‐9 μM) (Bedut et al., 2016). Furthermore, when drug concentrations were compared among different hPSC‐CM lines, techniques or studies, the differences appear to be significant and functionally relevant (Kuusela et al., 2016). Broader studies are still required to pinpoint the contribution of donor genetic background and subsequent epigenetic modifications on both cardiac phenotype and drug responses. This could be provided by recent advances in genome‐editing techniques, with CRISPR/Cas9 being the current leading method (Sander and Joung, 2014). This approach allows rapid and precise modification of the hPSC genome so that generating isogenically matched controls for (genetically) diseased hiPSC lines is feasible for many labs, reducing one source of variability. This type of gene repair service is also available commercially. Creating hiPSC biobanks that include not only diseased hiPSC lines as mentioned earlier but also genetically matched controls, with multiple clones of each, is an ambitious goal but could be of major benefit to the field particularly if accompanied by whole genome sequence data. A more modest goal could be the generation of smaller panels (10–50 lines) of bona fide wild‐type hiPSC lines, representative of the gender and ethnic diversity in specific populations, which might be extremely useful to broaden comparisons from an individual perspective with major genotype classes. These small panels should be extensively characterized, by multiple laboratories and univocally validated with standard procedures under agreed conditions. Updates could be provided by the community of researchers using the panel, so that over time ‘golden standards’ could be created among hiPSC lines, much as already available for hESC (Figure 4).
Figure 4.
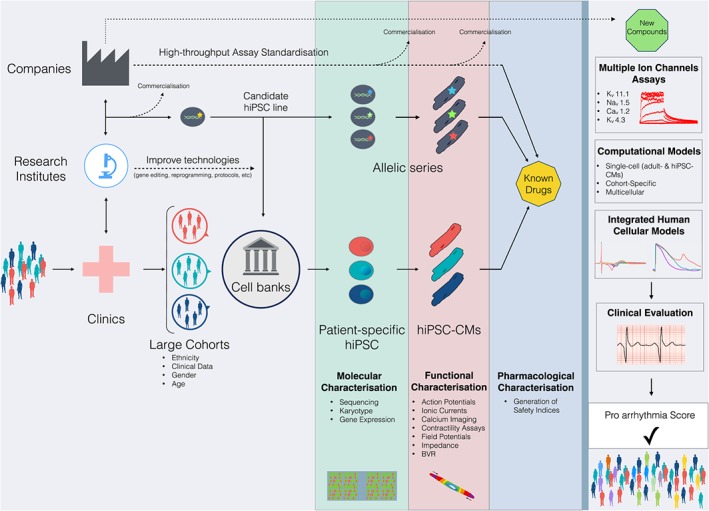
Anticipated integration of hPSC‐CMs in cardiotoxicity. Samples are collected from large cohorts of patients in clinics along with patient‐relevant clinical parameters. Cell banks will reprogramme somatic cells to patient‐specific hiPSC using standardized methodologies. The same samples can be used to produce control and/or mutated hiPSC candidate lines in which mutations are rescued or introduced with gene editing technologies. Molecular characterisation of undifferentiated hiPSC and functional characterisations of hiPSC‐CMs will outline the pharmacological response to known drugs to generate reliable safety indices. Novel drugs will then be tested based on the CiPA guidelines with integrated human cellular models as a predominant preclinical experimental component. The aim is to generate pro‐arrhythmic scores applicable to the general population.
Alternatively, very small subsets could be created with whole genome sequence data and validated high differentiation efficiency to all three germ layers. These should be made widely (commercially) available to allow the generation of allelic series for different mutations on the same genetic background (Musunuru, 2015). Commercializing disease‐specific hiPSC lines with appropriate informed consents from donors for drug screening process is likely to have great value for pharmaceutical companies, because mutations and polymorphisms have been implicated as major contributors to drug sensitivity (Roden and George, 2002; Tomalik‐Scharte et al., 2007).
A robust, highly standardized example was recently described by Kitaguchi et al. (2016). With just minor discrepancies, they demonstrated, in a mixed private–public multicentre study, that commercial lines can offer reliable and consistent results, suggesting that one way to pursue a prompt integration of hiPSC‐CMs in safety pharmacology could be through biotech companies; although information on protocols and reagents used (e.g. electrolyte molarity and protein supplements) may not be provided presenting difficulties in comparing and publishing results (Figure 3).
Conclusions
The growing appreciation of the value of hiPSC as a resource in drug discovery and safety pharmacology, not only for the heart but also for the brain, kidney and liver, has also driven the establishment of novel repositories for raw data from genomics, proteomics and metabolomic studies. Some cell banks, with the Human Induced Pluripotent Stem Cells Initiative (HipSci) at the Wellcome Trust/Sanger Institute (UK) leading this approach, make genomics and FACS data publicly available for many hiPSC lines. However, at present, the data available are only phenotypic/molecular characterisations of undifferentiated cells. To the best of our knowledge, these lines have not been compared directly for their differentiation capabilities, for example, towards the cardiac lineage or the electrophysiological properties of derivative hiPSC‐CMs in standard conditions. This would be extremely useful before their use in safety pharmacology (Figure 4).
Phenotypic data derived from large cohorts of patient‐specific hiPSC‐CMs can be categorized by variations such as ethnicity, gender, age, genotype, pre‐existing (clinical) conditions and much more, as we suggest in Figure 5. Furthermore, FDA/EMA/PMDA safety profile data for active concentrations of prescription drugs can also serve as a reference for comparing hiPSC‐CM data. A remarkable example has been recently presented by Burridge et al. (2016), in which in vitro doxorubicin‐induced cardiotoxicity investigated in hiPSC‐CMs replicated the clinical condition observed in a very small cohort of patients (Bellin and Mummery, 2016). It will be interesting to extend this type of approach to larger cohorts to reproduce different severities in drug responses or even temporal onset of the disease (early/late toxicity). Sufficiently large cohorts may make it possible to develop broadly applicable safety indices. This should also be encouraged in the context of cardiotoxicity evaluation.
Figure 5.
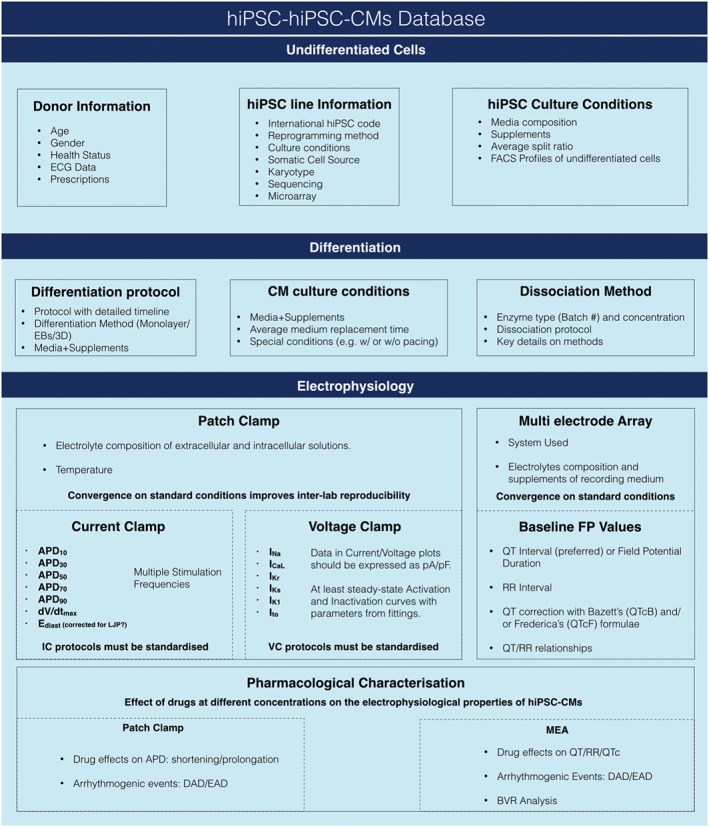
Useful parameters for hiPSC and hiPSC‐CMs banking. Data information on hiPSC lines stored in dedicated banks (top). Details of differentiation protocols and culture conditions (centre). Parameters for a comprehensive electrophysiological characterisation of hiPSC‐CMs with patch clamp and MEA. EB, embryoid bodies; MEA, multi electrode array; LJP, liquid junction potential; IC, current clamp; VC, voltage clamp; FP, field potential; QTc, corrected QT interval; DAD, delayed after depolarizations; EAD, early after depolarizations.
Drug repurposing and ‘forgotten drugs’
Drug repurposing strategies will benefit from the introduction of more predictive and sensitive in vitro models, in particular those simulating specific traits of diseases of patients' cohorts (McNeish et al., 2015). More refined technologies will also pave the way to rediscovering ‘forgotten drugs’, drugs that are no longer in production because they are no longer covered by patents or were not a commercial success for the purposes for which they were designed or were considered insufficiently safe. Having them rehabilitated in part through hiPSC technology would require (i) the predictive value of hiPSC‐CMs to be confirmed; (ii) evidence that the costs of drug development justify the potential revenue and (iii) that sufficient numbers of patients are eligible for treatment in the later clinical phases of development.
We anticipate that the integration of hiPSC‐CMs in early, preclinical stages of drug development is imminent and will be of benefit in: (i) the early detection of cardiotoxic drugs in safety pharmacology assays, which will help reduce the withdrawal of drugs already introduced to the market; (ii) the refinement of drug safety assays, as they are based on human cell models at early stages, which may improve the yield of drug discovery activities by selectively discarding potentially harmful compounds in ‘quick win, fail fast’ approaches (Paul et al., 2010); and (iii) reducing the number of animal used for drug testing.
The prerequisite, however, is process standardization within the scientific communities of academia and pharma. Joint efforts from research institutes, cell banks, biotech companies and public institutions comparing different lines in identical experimental conditions will help make hiPSC‐CMs an intrinsic part of the drug testing process.
Conflict of interest
C.L.M. is co‐founder of Pluriomics bv.
Acknowledgements
Work in the lab of C.L.M. is supported by CVON‐HUSTCARE (LS), the European Research Council (ERC‐AdG STEMCARDIOVASC) (MB), CrackIt InPulse (NC3R/GSK) and H2020 TECHNOBEAT. We greatly thank Prof. Rocchetti M. from the University of Milano ‐ Bicocca, Italy, for kindly sharing some of the data necessary for Figure 3.
Sala, L. , Bellin, M. , and Mummery, C. L. (2017) Integrating cardiomyocytes from human pluripotent stem cells in safety pharmacology: has the time come?. British Journal of Pharmacology, 174: 3749–3765. doi: 10.1111/bph.13577.
References
- Ahrens‐Nicklas RC, Christini DJ (2009). Anthropomorphizing the mouse cardiac action potential via a novel dynamic clamp method. Biophys J 97: 2684–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akar JG, Akar FG (2006). Mapping arrhythmias in the failing heart: from Langendorff to patient. J Electrocardiol 39: S19–S23. [DOI] [PubMed] [Google Scholar]
- Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM (2010). Cardiotoxicity of anticancer drugs: the need for cardio‐oncology and cardio‐oncological prevention. J Natl Cancer Inst 102: 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altomare C, Bartolucci C, Sala L, Bernardi J, Mostacciuolo G, Rocchetti M et al. (2015). IKr impact on repolarization and its variability assessed by dynamic clamp. Circ Arrhythm Electrophysiol 8: 1265–1275. [DOI] [PubMed] [Google Scholar]
- Amin AS, Wilde AAM (2016). Genetic screening in acquired long QT syndrome? Caution: proceed carefully. Eur Heart J 37: 1465–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arola OJ, Saraste A, Pulkki K, Kallajoki M, Parvinen M, Voipio‐Pulkki LM (2000). Acute doxorubicin cardiotoxicity involves cardiomyocyte apoptosis. Cancer Res 60: 1789–1792. [PubMed] [Google Scholar]
- Avior Y, Sagi I, Benvenisty N (2016). Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol 17: 170–182. [DOI] [PubMed] [Google Scholar]
- Beauchamp P, Moritz W, Kelm JM, Ullrich ND, Agarkova I, Anson BD et al. (2015). Development and characterization of a scaffold‐free 3D spheroid model of induced pluripotent stem cell‐derived human cardiomyocytes. Tissue Eng Part C Methods 21: 852–861. [DOI] [PubMed] [Google Scholar]
- Bedut S, Seminatore Nole C, Lamamy V, Caignard S, Boutin JA, Nosjean O et al. (2016). High‐throughput drug profiling with voltage‐ and calcium‐sensitive fluorescent probes in human iPSC‐derived cardiomyocytes. Am J Physiol Heart Circ Physiol 311: H44–H53. [DOI] [PubMed] [Google Scholar]
- Bellin M, Mummery CL (2016). Stem cells: the cancer's gone, but did chemotherapy damage your heart? Nat Rev Cardiol 13: 383–384. [DOI] [PubMed] [Google Scholar]
- Bellin M, Casini S, Davis RP, D'Aniello C, Haas J, Ward‐van Oostwaard D et al. (2013). Isogenic human pluripotent stem cell pairs reveal the role of a KCNH2 mutation in long‐QT syndrome. EMBO J 32: 3161–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellin M, Marchetto MC, Gage FH, Mummery CL (2012). Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol 13: 713–726. [DOI] [PubMed] [Google Scholar]
- Bentzen BH, Bahrke S, Wu K, Larsen AP, Odening KE, Franke G et al. (2011). Pharmacological activation of Kv11.1 in transgenic long QT‐1 rabbits. J Cardiovasc Pharmacol 57: 223–230. [DOI] [PubMed] [Google Scholar]
- Berger RD (2003). QT variability. J Electrocardiol 36 (Suppl.): 83–87. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Burridge PW, Kropp EM, Chuppa SL, Kwok W‐M, Wu JC et al. (2014). High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J Vis Exp : 52010. doi:10.3791/52010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birket MJ, Ribeiro MC, Kosmidis G, Ward D, Leitoguinho AR, van de Pol V et al. (2015). Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC‐cardiomyocyte function. Cell Rep 13: 733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizy A, Guerrero‐Serna G, Hu B, Ponce‐Balbuena D, Willis BC, Zarzoso M et al. (2013). Myosin light chain 2‐based selection of human iPSC‐derived early ventricular cardiac myocytes. Stem Cell Res 11: 1335–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnage ME (2011). Getting pharmaceutical R&D back on target. Nat Chem Biol 7: 335–339. [DOI] [PubMed] [Google Scholar]
- Burridge PW, Li YF, Matsa E, Wu H, Ong S‐G, Sharma A et al. (2016). Human induced pluripotent stem cell‐derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin‐induced cardiotoxicity. Nat Med 22: 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD et al. (2014). Chemically defined generation of human cardiomyocytes. Nat Methods 11: 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Sharma A, Wu JC (2015). Genetic and epigenetic regulation of human cardiac reprogramming and differentiation in regenerative medicine. Annu Rev Genet 49: 461–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Thompson S, Millrod MA, Weinberg S, Yuan X, Peters A et al. (2011). A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One 6: e18293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F et al. (2015). Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 131: 1981–1988. [DOI] [PubMed] [Google Scholar]
- Cavallari LH (2012). Tailoring drug therapy based on genotype. J Pharm Pract 25: 413–416. [DOI] [PubMed] [Google Scholar]
- Cavero I, Guillon J‐M, Ballet V, Clements M, Gerbeau J‐F, Holzgrefe H (2016). Comprehensive in vitro proarrhythmia assay (CiPA): pending issues for successful validation and implementation. J Pharmacol Toxicol Methods . doi:10.1016/j.vascn.2016.05.012. [DOI] [PubMed] [Google Scholar]
- Chan Y‐C, Ting S, Lee Y‐K, Ng K‐M, Zhang J, Chen Z et al. (2013). Electrical stimulation promotes maturation of cardiomyocytes derived from human embryonic stem cells. J Cardiovasc Transl Res 6: 989–999. [DOI] [PubMed] [Google Scholar]
- Chang Liao M‐L, de Boer TP, Mutoh H, Raad N, Richter C, Wagner E et al. (2015). Sensing cardiac electrical activity with a cardiac myocyte – targeted optogenetic voltage indicator. Circ Res 117: 401–412. [DOI] [PubMed] [Google Scholar]
- Chen A, Ting S, Seow J, Reuveny S, Oh S (2014). Considerations in designing systems for large scale production of human cardiomyocytes from pluripotent stem cells. Stem Cell Res Ther 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen IY, Matsa E, Wu JC (2016). Induced pluripotent stem cells: at the heart of cardiovascular precision medicine. Nat Rev Cardiol 13: 333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements M (2016). Multielectrode array (mea) assay for profiling electrophysiological drug effects in human stem cell‐derived cardiomyocytes. Curr Protoc Toxicol 68: 22.4.1–22.4.32. [DOI] [PubMed] [Google Scholar]
- Collins FS, Varmus H (2015). A new initiative on precision medicine. N Engl J Med 372: 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G et al. (2014). Lessons learned from the fate of AstraZeneca's drug pipeline: a five‐dimensional framework. Nat Rev Drug Discov 13: 419–431. [DOI] [PubMed] [Google Scholar]
- Cross MJ, Berridge BR, Clements PJM, Cove‐Smith L, Force TL, Hoffmann P et al. (2015). Physiological, pharmacological and toxicological considerations of drug‐induced structural cardiac injury. Br J Pharmacol 172: 957–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crumb WJ, Vicente J, Johannesen L, Strauss DG (2016). An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J Pharmacol Toxicol Methods . doi:10.1016/j.vascn.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Cuchiara ML, Olive JK, Matthews K (2015). Regulating the therapeutic translation of regenerative medicine. Expert Opin Biol Ther 15: 1387–1390. [DOI] [PubMed] [Google Scholar]
- Davis RP, Casini S, van den Berg CW, Hoekstra M, Remme CA, Dambrot C et al. (2012). Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation 125: 3079–3091. [DOI] [PubMed] [Google Scholar]
- de Villiers CP, van der Merwe L, Crotti L, Goosen A, George AL, Schwartz PJ et al. (2014). AKAP9 is a genetic modifier of congenital long‐QT syndrome type 1. Circ Cardiovasc Genet 7: 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey GT, Chaudhary KW, Atwater N, Nguyen C, Brown BS, McNeish JD et al. (2016). Cardiotoxicity screening with simultaneous optogenetic pacing, voltage imaging and calcium imaging. J Pharmacol Toxicol Methods . doi:10.1016/j.vascn.2016.05.003. [DOI] [PubMed] [Google Scholar]
- Denning C, Borgdorff V, Crutchley J, Firth KSA, George V, Kalra S et al. (2016). Cardiomyocytes from human pluripotent stem cells: from laboratory curiosity to industrial biomedical platform. Biochim Biophys Acta 1863: 1728–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devalla HD, Schwach V, Ford JW, Milnes JT, El‐Haou S, Jackson C et al. (2015). Atrial‐like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial‐selective pharmacology. EMBO Mol Med 7: 394–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pasquale E, Lodola F, Miragoli M, Denegri M, Avelino‐Cruz JE, Buonocore M et al. (2013). CaMKII inhibition rectifies arrhythmic phenotype in a patient‐specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis 4: e843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz GJ, Daniell K, Leitza ST, Martin RL, Su Z, McDermott JS et al. (2004). The [3H]dofetilide binding assay is a predictive screening tool for hERG blockade and proarrhythmia: comparison of intact cell and membrane preparations and effects of altering [K+]o. J Pharmacol Toxicol Methods 50: 187–199. [DOI] [PubMed] [Google Scholar]
- Duchatelet S, Crotti L, Peat RA, Denjoy I, Itoh H, Berthet M et al. (2013). Identification of a KCNQ1 polymorphism acting as a protective modifier against arrhythmic risk in long‐QT syndrome. Circ Cardiovasc Genet 6: 354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder A, Vollert I, Hansen A, Eschenhagen T (2016). Human engineered heart tissue as a model system for drug testing. Adv Drug Deliv Rev 96: 214–224. [DOI] [PubMed] [Google Scholar]
- Ewer MS, Ewer SM (2015). Cardiotoxicity of anticancer treatments. Nat Rev Cardiol 12: 547–558. [DOI] [PubMed] [Google Scholar]
- Feinberg AW, Alford PW, Jin H, Ripplinger CM, Werdich AA, Sheehy SP et al. (2012). Controlling the contractile strength of engineered cardiac muscle by hierarchal tissue architecture. Biomaterials 33: 5732–5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendyur A, Spira ME (2012). Toward on‐chip, in‐cell recordings from cultured cardiomyocytes by arrays of gold mushroom‐shaped microelectrodes. Front Neuroeng 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermini B, Hancox JC, Abi‐Gerges N, Bridgland‐Taylor M, Chaudhary KW, Colatsky T et al. (2015). A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J Biomol Screen 21: 1–11. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration (2013). Paving the Way for Personalized Medicine: 1–62.
- Friedrichs GS, Patmore L, Bass A (2005). Non‐clinical evaluation of ventricular repolarization (ICH S7B): results of an interim survey of international pharmaceutical companies. J Pharmacol Toxicol Methods 52: 6–11. [DOI] [PubMed] [Google Scholar]
- Gibson JK, Yue Y, Bronson J, Palmer C, Numann R (2014a). Human stem cell‐derived cardiomyocytes detect drug‐mediated changes in action potentials and ion currents. J Pharmacol Toxicol Methods 70: 255–267. [DOI] [PubMed] [Google Scholar]
- Gibson K, Yue Y, Li P (2014b), Evaluation of drug mediated changes in action potentials recorded from adult human stem cell‐derived Cor.4 U® cardiomyocytes. [DOI] [PubMed]
- Gibson K, Yue Y, Li P (2014c). Evaluation of drug mediated changes in action potentials recorded from adult human stem cell‐derived iCell® cardiomyocytes. [DOI] [PubMed]
- Gintant G, Sager PT, Stockbridge N (2016). Evolution of strategies to improve preclinical cardiac safety testing. Nat Rev Drug Discov 15: 457–471. [DOI] [PubMed] [Google Scholar]
- Hancox JC, McPate MJ, El Harchi A, Zhang YH (2008). The hERG potassium channel and hERG screening for drug‐induced torsades de pointes. Pharmacol Ther 119: 118–132. [DOI] [PubMed] [Google Scholar]
- Harris K, Aylott M, Cui Y, Louttit JB, McMahon NC, Sridhar A (2013). Comparison of electrophysiological data from human‐induced pluripotent stem cell‐derived cardiomyocytes to functional preclinical safety assays. Toxicol Sci 134: 412–426. [DOI] [PubMed] [Google Scholar]
- Hartjes KA, Li X, Martinez‐Fernandez A, Roemmich AJ, Larsen BT, Terzic A et al. (2014). Selection via pluripotency‐related transcriptional screen minimizes the influence of somatic origin on iPSC differentiation propensity. Stem Cells 32: 2350–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J (2014). Clinical development success rates for investigational drugs. Nat Biotechnol 32: 40–51. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Kunihiro T, Ando T, Kobayashi S, Matsui E, Yada H et al. (2014). Image‐based evaluation of contraction–relaxation kinetics of human‐induced pluripotent stem cell‐derived cardiomyocytes: correlation and complementarity with extracellular electrophysiology. J Mol Cell Cardiol 77: 178–191. [DOI] [PubMed] [Google Scholar]
- Herron TJ, Lee P, Jalife J (2012). Optical imaging of voltage and calcium in cardiac cells & tissues. Circ Res 110: 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel HM (2013). Drug‐induced functional cardiotoxicity screening in stem cell‐derived human and mouse cardiomyocytes: effects of reference compounds. J Pharmacol Toxicol Methods 68: 97–111. [DOI] [PubMed] [Google Scholar]
- Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Börnchen C, Müller C et al. (2014). Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol 74: 151–161. [DOI] [PubMed] [Google Scholar]
- Hochbaum DR, Zhao Y, Farhi SL, Klapoetke N, Werley CA, Kapoor V et al. (2014). All‐optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat Methods 11: 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra M, Mummery CL, Wilde AAM, Bezzina CR, Verkerk AO (2012). Induced pluripotent stem cell derived cardiomyocytes as models for cardiac arrhythmias. Front Physiol 3: 346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebsch N, Loskill P, Mandegar MA, Marks NC, Sheehan AS, Ma Z et al. (2015). Automated video‐based analysis of contractility and calcium flux in human‐induced pluripotent stem cell‐derived cardiomyocytes cultured over different spatial scales. Tissue Eng Part C Methods 21: 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HS, Kryshtal DO, Feaster TK, Sanchez‐Freire V, Zhang J, Kamp TJ et al. (2015a). Comparable calcium handling of human iPSC‐derived cardiomyocytes generated by multiple laboratories. J Mol Cell Cardiol 85: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HS, Kryshtal DO, Feaster TK, Sanchez‐Freire V, Zhang J, Kamp TJ et al. (2015b). Human induced pluripotent stem cell (hiPSC) derived cardiomyocytes to understand and test cardiac calcium handling: a glass half full. J Mol Cell Cardiol 89: 379–380. [DOI] [PubMed] [Google Scholar]
- ICH (2005a). ICH E14 guidance for industry. Clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs (Center for Drug Evaluation and Research).
- ICH (2005b) The non‐clinical evaluation of the potential for delayed ventricular repolarization (Qt Interval Prolongation) by human pharmaceuticals. S7B Guideline. [PubMed]
- Itoh H, Crotti L, Aiba T, Spazzolini C, Denjoy I, Fressart V et al. (2016). The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur Heart J 37: 1456–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itzhaki I, Maizels L, Huber I, Zwi‐Dantsis L, Caspi O, Winterstern A et al. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471: 225–229. [DOI] [PubMed] [Google Scholar]
- Johnson JN, Ackerman MJ (2009). QTc: how long is too long? Br J Sports Med 43: 657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones AR, Edwards DH, Cummins MJ, Williams AJ, George CH (2015). A systemized approach to investigate Ca(2+) synchronization in clusters of human induced pluripotent stem‐cell derived cardiomyocytes. Front Cell Dev Biol 3: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner L, Scholz A, Tian Q, Ruppenthal S, Tabellion W, Wiesen K et al. (2014). Genetically encoded Ca2+ indicators in cardiac myocytes. Circ Res 114: 1623–1639. [DOI] [PubMed] [Google Scholar]
- Kane C, Terracciano CM (2015). Induced pluripotent stem cell‐derived cardiac myocytes to understand and test calcium handling: pie in the sky? J Mol Cell Cardiol 89: 376–378. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Nomura F, Hattori A (2012). Improvement of electrical stimulation protocol for simultaneous measurement of extracellular potential with on‐chip multi‐electrode array system. Jpn J of Appl Phys 51: 06FK02. [Google Scholar]
- Kijlstra JD, Hu D, Mittal N, Kausel E, van der Meer P, Garakani A et al. (2015). Integrated analysis of contractile kinetics, force generation, and electrical activity in single human stem cell‐derived cardiomyocytes. Stem Cell Rep 5: 1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JJ, Yang L, Lin B, Zhu X, Sun B, Kaplan AD et al. (2015). Mechanism of automaticity in cardiomyocytes derived from human induced pluripotent stem cells. J Mol Cell Cardiol 81C: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitaguchi T, Moriyama Y, Taniguchi T, Ojima A, Ando H, Uda T et al. (2016). CSAHi study: evaluation of multi‐electrode array in combination with human iPS cell‐derived cardiomyocytes to predict drug‐induced QT prolongation and arrhythmia – effects of 7 reference compounds at 10 facilities. J Pharmacol Toxicol Methods 78: 93–102. [DOI] [PubMed] [Google Scholar]
- Klimas A, Ambrosi CM, Yu J, Williams JC, Bien H, Entcheva E (2016). OptoDyCE as an automated system for high‐throughput all‐optical dynamic cardiac electrophysiology. Nat Commun 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppel F, Labarre D, Zitoun P (2012). Quickly finding a needle in a haystack: a new automated cardiac arrhythmia detection software for preclinical studies. J Pharmacol Toxicol Methods 66: 92–97. [DOI] [PubMed] [Google Scholar]
- Kola I, Landis J (2004). Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3: 711–716. [DOI] [PubMed] [Google Scholar]
- Kramer J, Obejero‐Paz CA, Myatt G, Kuryshev YA, Bruening‐Wright A, Verducci JS et al. (2013). MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci Rep 3: 2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuusela J, Kujala VJ, Kiviaho A, Ojala M, Swan H, Kontula K et al. (2016). Effects of cardioactive drugs on human induced pluripotent stem cell derived long QT syndrome cardiomyocytes. Springerplus 5: 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttälä A, Moraghebi R, Valensisi C, Kettunen J, Andrus C, Pasumarthy KK et al. (2016). Genetic variability overrides the impact of parental cell type and determines iPSC differentiation potential. Stem Cell Rep 6: 200–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laverty HG, Benson C, Cartwright EJ, Cross MJ, Garland C, Hammond T et al. (2011). How can we improve our understanding of cardiovascular safety liabilities to develop safer medicines? Br J Pharmacol 163: 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leha A, Moens N, Meleckyte R, Culley OJ, Gervasio MK, Kerz M et al. (2016). A high‐content platform to characterise human induced pluripotent stem cell lines. Methods 96: 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang R, Zhao B, Lossin C, Cao Z (2016). Cardiotoxicity screening: a review of rapid‐throughput in vitro approaches. Arch Toxicol 90: 1803–1816. [DOI] [PubMed] [Google Scholar]
- Liang P, Lan F, Lee AS, Gong T, Sanchez‐Freire V, Wang Y et al. (2013). Drug screening using a library of human induced pluripotent stem cell‐derived cardiomyocytes reveals disease‐specific patterns of cardiotoxicity. Circulation 127: 1677–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu HR, Whittaker R, Price JH, Vega R, Pfeiffer ER, Cerignoli F et al. (2015). High throughput measurement of Ca++ dynamics in human stem cell‐derived cardiomyocytes by kinetic image cytometery: a cardiac risk assessment characterization using a large panel of cardioactive and inactive compounds. Toxicol Sci 148: 503–516. [DOI] [PubMed] [Google Scholar]
- Lu J, Wei H, Wu J, Jamil MFA, Tan ML, Adenan MI et al. (2014). Evaluation of the cardiotoxicity of mitragynine and its analogues using human induced pluripotent stem cell‐derived cardiomyocytes. PLoS One 9: e115648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Wei H, Lu J, Huang D, Liu Z, Loh LJ et al. (2015). Characterization of a novel KCNQ1 mutation for type 1 long QT syndrome and assessment of the therapeutic potential of a novel IKs activator using patient‐specific induced pluripotent stem cell‐derived cardiomyocytes. Stem Cell Res Ther 6: 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D, Wei H, Zhao Y, Lu J, Li G, Sahib NBE et al. (2013). Modeling type 3 long QT syndrome with cardiomyocytes derived from patient‐specific induced pluripotent stem cells. Int J Cardiol 168: 5277–5286. [DOI] [PubMed] [Google Scholar]
- Maddah M, Heidmann JD, Mandegar MA, Walker CD, Bolouki S, Conklin BR et al. (2015). A non‐invasive platform for functional characterization of stem‐cell‐derived cardiomyocytes with applications in cardiotoxicity testing. Stem Cell Rep 4: 621–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet A, Tan K, Chai X, Sadananda SN, Mehta A, Ooi J et al. (2016). Modeling doxorubicin‐induced cardiotoxicity in human pluripotent stem cell derived‐cardiomyocytes. Sci Rep 6: 25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan D, Zhang M, Stallmeyer B, Müller J, Fleischmann BK, Schulze‐Bahr E et al. (2016). Human iPS cell model of type 3 long QT syndrome recapitulates drug‐based phenotype correction. Basic Res Cardiol 111: 14–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignoni M, Groothuis GMM, de Kanter R (2006). Species differences between mouse, rat, dog, monkey and human CYP‐mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol 2: 875–894. [DOI] [PubMed] [Google Scholar]
- Martiniano SL, Sagel SD, Zemanick ET (2016). Cystic fibrosis: a model system for precision medicine. Curr Opin Pediatr 28: 312–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A et al. (2011). Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J 32: 952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton R, Huet G, Shakir S (2014). An investigation into drug products withdrawn from the EU market between 2002 and 2011 for safety reasons and the evidence used to support the decision‐making. BMJ Open 4: e004221–e004221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeish J, Gardner JP, Wainger BJ, Woolf CJ, Eggan K (2015). From dish to bedside: lessons learned while translating findings from a stem cell model of disease to a clinical trial. Cell Stem Cell 17: 8–10. [DOI] [PubMed] [Google Scholar]
- Mehta A, Sequiera GL, Ramachandra CJA, Sudibyo Y, Chung Y, Sheng J et al. (2014). Re‐trafficking of hERG reverses long QT syndrome 2 phenotype in human iPS‐derived cardiomyocytes. Cardiovasc Res 102: 497–506. [DOI] [PubMed] [Google Scholar]
- Meijer van Putten RME, Mengarelli I, Guan K, Zegers JG, van Ginneken ACG, Verkerk AO et al. (2015). Ion channelopathies in human induced pluripotent stem cell derived cardiomyocytes: a dynamic clamp study with virtual IK1. Front Physiol 6: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercurio V, Pirozzi F, Lazzarini E, Marone G, Rizzo P, Agnetti G et al. (2016). Models of heart failure based on the cardiotoxicity of anticancer drugs. J Card Fail 22: 449–458. [DOI] [PubMed] [Google Scholar]
- Milan DJ, MacRae CA (2005). Animal models for arrhythmias. Cardiovasc Res 67: 426–437. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Hancox JC, Levi AJ (1998). Cultured adult cardiac myocytes: future applications, culture methods, morphological and electrophysiological properties. Cardiovasc Res 39: 280–300. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Furusawa C, Kaneko K (2015). Pluripotency, differentiation, and reprogramming: a gene expression dynamics model with epigenetic feedback regulation. PLoS Comput Biol 11: e1004476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosadegh B, Dabiri BE, Lockett MR, Derda R, Campbell P, Parker KK et al. (2014). Three‐dimensional paper‐based model for cardiac ischemia. Adv Healthc Mater 3: 1036–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery CL, Zhang J, Ng ES, Elliott DA, Elefanty AG, Kamp TJ. (2012). Differentiation of human embryonic stem cells and induced pluripotent stem cells to cardiomyocytes: a methods overview. Circ Res 111: 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K (2015). Personalized genomes and cardiovascular disease. Cold Spring Harb Perspect Med 5: a014068–a014068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan A, Stancescu M, Dhir V, Armstrong C, Sommerhage F, Hickman JJ et al. (2011). Patterned cardiomyocytes on microelectrode arrays as a functional, high information content drug screening platform. Biomaterials 32: 4267–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Vesell ES (2006). Can personalized drug therapy be achieved? A closer look at pharmaco‐metabonomics. Trends Pharmacol Sci 27: 580–586. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM (2004). Studying cardiac arrhythmias in the mouse – a reasonable model for probing mechanisms? Trends Cardiovasc Med 14: 83–93. [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Honda Y, Tsujimoto S, Watanabe H, Kunimatsu T, Funabashi H (2014). Availability of human induced pluripotent stem cell‐derived cardiomyocytes in assessment of drug potential for QT prolongation. Toxicol Appl Pharmacol 278: 72–77. [DOI] [PubMed] [Google Scholar]
- Nunes SS, Miklas JW, Liu J, Aschar‐Sobbi R, Xiao Y, Zhang B et al. (2013). Biowire: a platform for maturation of human pluripotent stem cell‐derived cardiomyocytes. Nat Methods 10: 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obergrussberger A, Juhasz K, Thomas U, Stölzle‐Feix S, Becker N, Dörr L et al. (2016). Safety pharmacology studies using EFP and impedance. J Pharmacol Toxicol Methods . doi:10.1016/j.vascn.2016.04.006. [DOI] [PubMed] [Google Scholar]
- O'Hara T, Rudy Y (2012). Quantitative comparison of cardiac ventricular myocyte electrophysiology and response to drugs in human and nonhuman species. Am J Physiol Heart Circ Physiol 302: H1023–H1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara T, Virág L, Varró A, Rudy Y (2011). Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol 7: e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onakpoya IJ, Heneghan CJ, Aronson JK (2016). Worldwide withdrawal of medicinal products because of adverse drug reactions: a systematic review and analysis. Crit Rev Toxicol 46: 477–489. [DOI] [PubMed] [Google Scholar]
- Paci M, Hyttinen J, Aalto‐Setälä K, Severi S (2013). Computational models of ventricular‐ and atrial‐like human induced pluripotent stem cell derived cardiomyocytes. Ann Biomed Eng 41: 2334–2348. [DOI] [PubMed] [Google Scholar]
- Paci M, Hyttinen J, Rodriguez B, Severi S (2015). Human induced pluripotent stem cell‐derived versus adult cardiomyocytes: an in silico electrophysiological study on effects of ionic current block. Br J Pharmacol 172: 5147–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paci M, Sartiani L, Del Lungo M, Jaconi M, Mugelli A, Cerbai E et al. (2012). Mathematical modelling of the action potential of human embryonic stem cell derived cardiomyocytes. Biomed Eng Online 11: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SA, Lee S‐R, Tung L, Yue DT (2014). Optical mapping of optogenetically shaped cardiac action potentials. Sci Rep 4: 6125–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR et al. (2010). How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 9: 203–214. [DOI] [PubMed] [Google Scholar]
- Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C et al. (2015). Automated, high‐throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods 12: 885–892. [DOI] [PubMed] [Google Scholar]
- Pekkanen‐Mattila M, Kerkelä E, Tanskanen JMA, Pietilä M, Pelto‐Huikko M, Hyttinen J et al. (2009). Substantial variation in the cardiac differentiation of human embryonic stem cell lines derived and propagated under the same conditions – a comparison of multiple cell lines. Ann Med 41: 360–370. [DOI] [PubMed] [Google Scholar]
- Penttinen K, Swan H, Vanninen S, Paavola J, Lahtinen AM, Kontula K et al. (2015). Correction: antiarrhythmic effects of dantrolene in patients with catecholaminergic polymorphic ventricular tachycardia and replication of the responses using iPSC models. PLoS One 10: e0134746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M (2005). Drug binding interactions in the inner cavity of hERG channels: molecular insights from structure–activity relationships of clofilium and ibutilide analogs. Mol Pharmacol 69: 509–519. [DOI] [PubMed] [Google Scholar]
- Priori SG (1998). Gene specific therapy for cardiac disease: the case of long QT syndrome. Rev Port Cardiol 17 (Suppl 3): III27–III38. [PubMed] [Google Scholar]
- Pueyo E, Dangerfield CE, Britton OJ, Virág L, Kistamás K, Szentandrássy N et al. (2016). Experimentally‐based computational investigation into beat‐to‐beat variability in ventricular repolarization and its response to ionic current inhibition. PLoS One 11: e0151461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael R, Purushotham D, Gastonguay C, Chesnik MA, Kwok W‐M, Wu H‐E et al. (2016). Combining patient proteomics and in vitro cardiomyocyte phenotype testing to identify potential mediators of heart failure with preserved ejection fraction. J Transl Med 14: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rast G, Weber J, Disch C, Schuck E, Ittrich C, Guth BD (2015). An integrated platform for simultaneous multi‐well field potential recording and Fura‐2‐based calcium transient ratiometry in human induced pluripotent stem cell (hiPSC)‐derived cardiomyocytes. J Pharmacol Toxicol Methods 75: 91–100. [DOI] [PubMed] [Google Scholar]
- Ravenscroft SM, Pointon A, Williams AW, Cross MJ, Sidaway JE (2016). Cardiac non‐myocyte cells show enhanced pharmacological function suggestive of contractile maturity in stem cell derived cardiomyocyte microtissues. Toxicol Sci 152: 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern WS, Carlsson L, Davis AS, Lynch WG, Mackenzie I, Palethorpe S et al. (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 58: 32–45. [DOI] [PubMed] [Google Scholar]
- Ribeiro AJS, Ang Y‐S, Fu J‐D, Rivas RN, Mohamed TMA, Higgs GC et al. (2015a). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc Natl Acad Sci U S A 112: 12705–12710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MC, Tertoolen LG, Guadix JA, Bellin M, Kosmidis G, D'Aniello C et al. (2015b). Functional maturation of human pluripotent stem cell derived cardiomyocytes in vitro – correlation between contraction force and electrophysiology. Biomaterials 51: 138–150. [DOI] [PubMed] [Google Scholar]
- Robertson C, Tran DD, George SC (2013). Concise review: maturation phases of human pluripotent stem cell‐derived cardiomyocytes. Stem Cells 31: 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, George AL (2002). The genetic basis of variability in drug responses. Nat Rev Drug Discov 1: 37–44. [DOI] [PubMed] [Google Scholar]
- Rodin S, Antonsson L, Hovatta O, Tryggvason K (2014). Monolayer culturing and cloning of human pluripotent stem cells on laminin‐521–based matrices under xeno‐free and chemically defined conditions. Nat Protoc 9: 2354–2368. [DOI] [PubMed] [Google Scholar]
- Rodríguez B, Carusi A, Abi Gerges N, Ariga R, Britton O, Bub G et al (2015). Human‐based approaches to pharmacology and cardiology: an interdisciplinary and intersectorial workshop. Europace euv320. [DOI] [PMC free article] [PubMed]
- Rubinstein E, Camm J (2002). Cardiotoxicity of fluoroquinolones. J Antimicrob Chemother 49: 593–596. [DOI] [PubMed] [Google Scholar]
- Sala L, Yu Z, Ward‐van Oostwaard D, van Veldhoven JPD, Moretti A, Laugwitz K‐L et al. (2016). A new hERG allosteric modulator rescues genetic and drug‐induced long‐QT syndrome phenotypes in cardiomyocytes from isogenic pairs of patient induced pluripotent stem cells. EMBO Mol Med. doi: 10.15252/emmm.201606260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK (2014). CRISPR‐Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32: 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Tristani‐Firouzi M (2006). hERG potassium channels and cardiac arrhythmia. Nature 440: 463–469. [DOI] [PubMed] [Google Scholar]
- Šarić T, Halbach M, Khalil M, Er F (2014). Induced pluripotent stem cells as cardiac arrhythmic in vitro models and the impact for drug discovery. Expert Opin Drug Discovery 9: 55–76. [DOI] [PubMed] [Google Scholar]
- Scheel O, Frech S, Amuzescu B, Eisfeld J, Lin KH, Knott T (2014). Action potential characterization of human induced pluripotent stem cell‐derived cardiomyocytes using automated patch‐clamp technology. Assay Drug Dev Technol 12: 457–469. [DOI] [PubMed] [Google Scholar]
- Schwach V, Passier R (2016). Generation and purification of human stem cell‐derived cardiomyocytes. Differentiation 91: 126–138. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Woosley RL (2016). Predicting the unpredictable: drug‐induced QT prolongation and torsades de pointes. J Am Coll Cardiol 67: 1639–1650. [DOI] [PubMed] [Google Scholar]
- Shah RR (2005). Drugs, QTc interval prolongation and final ICH E14 guideline : an important milestone with challenges ahead. Drug Saf 28: 1009–1028. [DOI] [PubMed] [Google Scholar]
- Siramshetty VB, Nickel J, Omieczynski C, Gohlke B‐O, Drwal MN, Preissner R (2016). WITHDRAWN – a resource for withdrawn and discontinued drugs. Nucleic Acids Res 44: D1080–D1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Awari DW, Han EY, Uche‐Anya E, Park S‐HE, Yabe YA et al. (2015). Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cells Transl Med 4: 468–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SPH et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira ME, Hai A (2013). Multi‐electrode array technologies for neuroscience and cardiology. Nat Nanotechnol 8: 83–94. [DOI] [PubMed] [Google Scholar]
- Stams TR, Oosterhoff P, Heijdel A, Dunnink A, Beekman JD, van der Nagel R et al. (2016). Beat‐to‐beat variability in preload unmasks latent risk of Torsade de Pointes in anesthetized chronic atrioventricular block dogs. Circ J 80: 1336–1345. [DOI] [PubMed] [Google Scholar]
- Stockbridge N, Zhang J, Garnett C, Malik M (2012). Practice and challenges of thorough QT studies. J Electrocardiol 45: 582–587. [DOI] [PubMed] [Google Scholar]
- Svanström H, Pasternak B, Hviid A (2014). Use of clarithromycin and roxithromycin and risk of cardiac death: cohort study. BMJ 349: g4930–g4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HL, Bardai A, Shimizu W, Moss AJ, Schulze‐Bahr E, Noda T et al. (2006). Genotype‐specific onset of arrhythmias in congenital long‐QT syndrome: possible therapy implications. Circulation 114: 2096–2103. [DOI] [PubMed] [Google Scholar]
- Terrenoire C, Wang K, Tung KWC, Chung WK, Pass RH, Lu JT et al. (2013). Induced pluripotent stem cells used to reveal drug actions in a long QT syndrome family with complex genetics. J Gen Physiol 141: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirziu D, Giordano FJ, Simons M (2010). Cell communications in the heart. Circulation 122: 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomalik‐Scharte D, Lazar A, Fuhr U, Kirchheiner J (2007). The clinical role of genetic polymorphisms in drug‐metabolizing enzymes. Pharmacogenomics J 8: 4–15. [DOI] [PubMed] [Google Scholar]
- Townsend C, Brown BS (2013). Predicting drug‐induced QT prolongation and torsades de pointes: a review of preclinical endpoint measures. Curr Protoc Pharmacol Chapter 10: Unit 10.16. [DOI] [PubMed]
- Turnbull IC, Karakikes I, Serrao GW, Backeris P, Lee J‐J, Xie C et al. (2014). Advancing functional engineered cardiac tissues toward a preclinical model of human myocardium. FASEB J 28: 644–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidyanathan R, Markandeya YS, Kamp TJ, Makielski JC, Janaury CT, Eckhardt LL (2016). IK1‐enhanced human induced pluripotent stem cell‐derived cardiomyocytes: an improved cardiomyocyte model to investigate inherited arrhythmia syndromes. Am J Physiol Heart Circ Physiol 310: H1611–H1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg CW, Elliott DA, Braam SR, Mummery CL, Davis RP (2016). Differentiation of human pluripotent stem cells to cardiomyocytes under defined conditions. Methods Mol Biol 1353: 163–180. [DOI] [PubMed] [Google Scholar]
- van der Heyden MAG, Jonsson MKB (2012). Personalized medicine and the role of induced pluripotent stem cells. Cardiovasc Res 95: 395–396. [DOI] [PubMed] [Google Scholar]
- Varga ZV, Ferdinandy P, Liaudet L, Pacher P (2015). Drug‐induced mitochondrial dysfunction and cardiotoxicity. Am J Physiol Heart Circ Physiol 309: H1453–H1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerman CC, Kosmidis G, Mummery CL, Casini S, Verkerk AO, Bellin M (2015). Immaturity of human stem‐cell‐derived cardiomyocytes in culture: fatal flaw or soluble problem? Stem Cells Dev 24: 1035–1052. [DOI] [PubMed] [Google Scholar]
- Vejpongsa P, Yeh ETH (2014). Prevention of anthracycline‐induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol 64: 938–945. [DOI] [PubMed] [Google Scholar]
- Viskin S, Havakuk O, Schwaber MJ (2015). Pro‐arrhythmic effects of noncardiac medications: lessons from macrolide antibiotics. J Am Coll Cardiol 66: 2185–2188. [DOI] [PubMed] [Google Scholar]
- Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM et al. (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov 14: 475–486. [DOI] [PubMed] [Google Scholar]
- Woosley RL, Romero K (2013). Assessing cardiovascular drug safety for clinical decision‐making. Nat Rev Cardiol 10: 330–337. [DOI] [PubMed] [Google Scholar]
- Xi B, Wang T, Li N, Ouyang W, Zhang W, Wu J et al. (2011). Functional cardiotoxicity profiling and screening using the xCELLigence RTCA cardio system. J Lab Autom 16: 415–421. [DOI] [PubMed] [Google Scholar]
- Yu Z, IJzerman AP, Heitman LH (2015). Kv 11.1 (hERG)‐induced cardiotoxicity: a molecular insight from a binding kinetics study of prototypical Kv 11.1 (hERG) inhibitors. Br J Pharmacol 172: 940–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Liu J, van Veldhoven JPD, Ijzerman AP, Schalij MJ, Pijnappels DA et al. (2016). Allosteric modulation of Kv11.1 (hERG) channels protects against drug‐induced ventricular arrhythmias. Circ Arrhythm Electrophysiol 9: e003439. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zou B, Yu H, Moretti A, Wang X, Yan W et al. (2012). Modulation of hERG potassium channel gating normalizes action potential duration prolonged by dysfunctional KCNQ1 potassium channel. Proc Natl Acad Sci U S A 109: 11866–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, D'Aniello C, Verkerk AO, Wrobel E, Frank S, Ward‐van Oostwaard D et al. (2014). Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange‐Nielsen syndrome: disease mechanisms and pharmacological rescue. Proc Natl Acad Sci U S A 111: E5383–E5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Forrester JV, McCaig CD (1999). A small, physiological electric field orients cell division. Proc Natl Acad Sci U S A 96: 4942–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R, Millrod MA, Zambidis ET, Tung L (2016). Variability of action potentials within and among cardiac cell clusters derived from human embryonic stem cells. Sci Rep 6: 18544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann WH, Schneiderbanger K (2002). Tissue engineering of a differentiated cardiac muscle construct. Circulation 90: 223–230. [DOI] [PubMed] [Google Scholar]