

Abstract
INTRODUCTION
The alpha-synuclein (SNCA) gene has been implicated in the etiology of Parkinson’s disease (PD) and Dementia with Lewy Bodies (DLB).
METHODS
A computational analysis of SNCA-3’UTR to identify potential microRNA binding-sites, and quantitative real-time-PCR to determine their expression in isogenic iPSC-derived dopaminergic and cholinergic neurons as a model of PD and DLB, respectively. Additionally, deep sequencing analysis of SNCA-3’UTR of autopsy confirmed cases of PD, DLB, and normal followed by genetic association analysis of the identified variants.
RESULTS
We identified four miRNA binding-sites, and observed a neuronal-type specific expression profile for each miRNA in the different isogenic iPSC-derived neurons, i.e. dopaminergic and cholinergic. Furthermore, we found that the short-structural variant rs777296100-polyT was moderately associated with DLB but not with PD.
DISCUSSION
We suggest that the regulation of SNCA expression through miRNAs is neuronal-type specific, and possibly plays a part in the phenotypic heterogeneity of synucleinopathies. Furthermore, genetic variability in the SNCA gene may contribute to synucleinopathies in a pathology-specific manner.
Keywords: SNCA, DLB, PD, miRNA, iPSC-derived neurons, SNCA-3’UTR
1. BACKGROUND
Genome Wide Association studies (GWAS) of Parkinson’s disease (PD) reported that the 3' linkage disequilibrium (LD) block contains significantly associated SNPs, suggesting that PD-causal variants are located in or near the 3' region of the SNCA gene[1]. However, the molecular mechanism/s through which the 3' region of SNCA gene modulates the risk of developing sporadic PD remains to be determined. To date, accumulating evidence has been reported in both in vitro systems and in vivo models, suggesting that SNCA expression levels are critical for the development of the disease[2]. With the goal of understanding the genetic variation that underlies the observed association with the 3’ LD block of the SNCA gene we have been studying possible mechanisms that control gene expression in the context of the 3’ end of the gene. Previously we showed that SNPs tagging the SNCA 3' LD block have significant effects on the relative levels of SNCA112-mRNA. This splice variant lacks exon 5 (of six exons) and encodes a protein isoform with truncated C-terminus that is predicted to enhance aggregation [3, 4] and therefore may increase susceptibility to LB formation. Noteworthy is that SNCA has been reported to have a common short 3’UTR and a rare long (full length) 3’UTR isoform (ENST00000394986 and ENST00000420646, respectively; Ensembl transcript identification numbers). It has been suggested that common SNPs in the extended SNCA-3’UTR promote the accumulation and translation of the long 3’UTR transcripts and result in increased α-syn protein levels which are redirected away from synaptic terminals and towards the mitochondria, reminiscent of disease pathology. Collectively, cis-genetic regulation of post-transcriptional mechanisms in addition to transcriptional effects may explain the strong association signals between SNCA 3’LD region and PD[5].
Modulation of SNCA-mRNA levels by endogenous microRNAs (miRNAs) has been proposed as one of the post-transcriptional mechanisms of SNCA regulation. Two miRNAs – miR-7 and miR-153 – are abundantly expressed in the brain and in rodent cultured neurons, and were shown to affect SNCA expression levels[6–8]. It was demonstrated in cell-based models that both miR-7 and miR-153 interacted directly with their predicted binding sites and down-regulated SNCA-mRNA and protein levels showing an additive effect[7]. The role of miR-34b and miR-34c in the regulation of SNCA transcript levels was also investigated. In human brains, miR-34b and miR-34c are downregulated in PD[9, 10]. The effect of these miRNAs has been investigated in SH-SY5Y cells. Both miR-34b and miR-34c decrease SNCA-mRNA levels and α-syn protein by targeting the 3’UTR of SNCA-mRNA[11]. Furthermore, SNP rs10024743 in SNCA 3’UTR lies within a target site for miR-34b and was found to lower the miR-34b-mediated repression of the α-syn protein. This study suggested that down-regulation of miR-34b and miR-34c in the brain, as well as a SNP in the 3’UTR of SNCA gene can increase α-syn expression, possibly contributing to PD pathogenesis[11]. In contrast, a study that used a luciferase vector bearing the full length 3’UTR reported that miR-34b-3p led to increased expression, while miR-34b-3p specific inhibitor showed decreased luciferase levels, and speculated that the binding of miR34b-3p mediated translational induction of SNCA transcript with the long 3’UTR[5]. These effects were abolished by SNPs in the longer 3’UTR positioned within a miR-34b-3p predicted binding site. Nevertheless, the relevance of these diverse findings to the etiology of PD and other synucleinopathy disorders warrant further investigations.
The most common synucleinopathies share a common pathological hallmark, Lewy bodies (LBs) and Lewy-related neurites; however, each disease presents distinct characteristics. The cell types and brain regions containing the LBs differ, particularly in early disease stages, so that while LBs in dopaminergic (mDA) neurons are the primary early disease characteristic of PD [12, 13] [14], early stages of dementia with Lewy Bodies (DLB) has LBs primarily in the amygdala and cerebral cortex, as well as basal forebrain cholinergic neurons (BFCN)[15, 16][17, 18]. Interestingly, GWAS results reported that variants defining the genetic association of the SNCA gene with PD[1] are distinct from those SNCA variants associated with DLB[19].
Here we studied the contribution of the SNCA 3’UTR to synucleinopathies and to the heterogeneity of these disorders focusing on the comparison of PD to DLB, using two cohesive and complementary approaches. We determined the expression profiles of miRNAs that are predicted to interact with the SNCA 3’UTR in isogenic iPSCs-derived mDA and BFCN neurons that are primarily involved in PD and DLB, respectively. In addition, we performed a deep sequencing analyses of the SNCA 3’UTR using DNA samples from autopsy confirmed cases of PD, DLB, and matched normal controls, aiming to identify genetic variability in the 3’UTR that is associated, commonly or distinctively, with the neuropathological diagnoses.
2. METHODS
2.1. Computational analysis: prediction of potential miRNAs
The analysis of potential miRNA and their corresponding binding sites was performed using the sequence of the full length SNCA 3’UTR (Human SNCA ENST00000394989.2; 2,529bp; chr4: 90,645,250-90,647,778; GRCh37/hg19) using the TagetScan software (Release 7.0)[20].
2.2 Cell Culture and Neuronal Differentiation
Induced pluripotent stem cells (iPSCs) from an apparently healthy individual (GM23280) were purchased from Coriell cell repositories (http://ccr.coriell.org/). iPSCs from a patient with the triplication of the SNCA gene (Tri-SNCA, ND34391) were obtained from the NINDS Human Cell and Data Repository (https://nindsgenetics.org). GM23280 and ND34391 are characterized by a normal karyotype. iPSCs were cultured under feeder-independent conditions in mTeSRTM1 medium (StemCell Technologies) onto hESC-qualified Matrigel coated plates. Cells were passaged using Gentle Cell Dissociation Reagent (StemCell Technologies) according to the manufacturer’s manual.
The mDA derive from the Ventral Midbrain (MD), while the BFCN derive from the Medial Ganglionic Eminence (MGE) [21]. We therefore, used specific protocols to differentiate iPSCs to mDA and BFCN [22, 23].
The induction of mDA was performed following a modified protocol from Lin et al.[23]. Briefly, iPSCs were differentiated using an embryoid body-based protocol. iPSCs were dissociated with Accutase (StemCell Technologies) and seeded into Aggrewell 800 plates (10,000 cells per microwell; Stem Cell Technologies) in Neural Induction Medium (NIM - Stem Cell Technologies) supplemented with Y27632 (10 μM) to form Embryoid Bodies (EBs). EBs were replated after 5 days onto matrigel-coated plates in NIM. One day after EBs replating, NIM was supplemented with 200ng/mL SHH (Peprotech) leading to the formation of neural rosettes. On day 12, neural rosettes were selected with Neural Rosette Selection reagent (used per the manufacturer’s instructions, StemCell Technologies) and replated in matrigel-coated plates in N2B27 medium supplemented with 3 μM CHIR99021, 2 μM SB431542, 5 μg/ml BSA, 20 ng/ml bFGF, and 20 ng/ml EGF, leading to the formation of Neural Precursor Cells (NPCs). Differentiation of NPCs into mDA was initiated 1 day after passaging the NPCs on poly-L-ornithine/laminin-coated plates. NPC maintenance medium was substituted by final differentiation medium consisting of N2B27 medium supplemented with 100 ng/ml FGF8 (Peprotech), 2 μM Purmorphamine, 300 ng/ml Dibutyryl cAMP (db-cAMP), and 200 μM L-ascorbic acid (L-AA) for 14 days. From days 14, cells were fed with maturation medium consisting of 20 ng/ml GDNF, 20 ng/ml BDNF, 10 μM DAPT, 0.5 mM db-cAMP, and 200 μM L-AA. Medium was changed every other day.
The differentiation into BFCN was performed following a modified protocol of Crompton et al.[22]. Briefly, EBs were allowed to form into Aggrewell 800 plates in NIM. On day 5, EBs were replated and a daily medium changing was performed. From day 8, neural rosettes were grown into NEM (7 parts KO-DMEM to 3 parts F12, 2 mM Glutamax, 1% penicillin and streptomycin, supplemented with 2% B27 (all Life Technologies), plus 20 ng/ml FGF, 20 ng/ml EGF, 5 μg/ml heparin, 20 μM SB431542 and 10 μM Y27632[24]. On day 12, neural rosettes were selected with Neural Rosette Selection Reagent and replated in NEM onto Matrigel-coated plates. NEM contained 20 μM SB431542 and 10 μM Y27632, for the initial 10-day expansion period. On day 23, Y27632 was withdrawn and final differentiation was performed onto PLO/laminin coated plates in the presence of BrainPhys Medium (Stemcell Technologies) supplemented with N2, B27, BDNF, GDNF, L-ascorbic acid, and db-cAMP until day 45–50. Medium was changed every other day.
2.3 RNA extraction and cDNA synthesis
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) followed by purification with an RNeasy kit (Qiagen) used per the manufacturer’s protocol. RNA concentration was determined spectrophotometrically at 260 nm, while the quality of the purification was determined by 260 nm/280 nm ratio that showed values between 1.9 and 2.1, indicating high RNA quality.
cDNA was synthesized using MultiScribe RT enzyme (Applied Biosystems) using the following conditions: 10 min at 25°C and 120 min at 37°C.
2.4 miRNA extraction and cDNA synthesis
The extraction of miRNA at different stages of the differentiation was performed using the mirVana miRNA isolation Kit (Applied Biosystems) following the manufacturer’s instructions. miRNA concentration was determined as previously described. cDNA was synthesized using specific miRNA primers from the TaqMan microRNA Assay and reagents from the TaqMan MicroRNA Reverse Transcription kit (Invitrogen).
2.5 Real time PCR
Real-time PCR was used to quantify the levels of the neuronal markers (mRNAs) and the miRNAs. Briefly, triplicates of each sample were assayed by relative quantitative real-time PCR using TaqMan expression assays and the ABI QuantStudio 7 to determine the level of the message RNA (mRNA) of the neuronal markers, or miRNAs in the different cell-lines relative to mRNAs encoding housekeeping genes or control snoRNA, respectively. ABI MGB probe and primer set assays (Applied Biosystems) that were used to amplify the target neuronal markers and the house-keeping reference controls are listed in (Supplementary Table S1); and the target miRNAs and the reference snoRNA control are listed in (Supplementary Table S2). Each cDNA (10 ng) was amplified in duplicate in at least two independent runs (overall ≥ 4 repeats), using TaqMan Universal PCR master mix reagent (Applied Biosystems) and the following conditions: 2 min at 50 °C, 10 min at 95 °C, 40 cycles: 15 sec at 95 °C, and 1 min at 60°C. As a negative control for the specificity of the amplification, we used RNA control samples that were not converted to cDNA (no-RT) and no-cDNA/RNA samples (no-template) in each plate. No amplification product was detected in control reactions. Data were analyzed with a threshold set in the linear range of amplification. The cycle number at which any particular sample crossed that threshold (Ct) was then used to determine fold difference, whereas the geometric mean of the two control genes served as a reference for normalization. Fold difference was calculated as 2−ΔΔCt [25]; ΔCt=[Ct(target)-Ct (geometric mean of reference)]. ΔΔCt =[ΔCt(sample)]−[ΔCt(calibrator)]. The calibrator was a particular RNA sample, obtained from the control iPSC, used repeatedly in each plate for normalization within and across runs. The variation of the ΔCt values among the calibrator replicates was smaller than 10%.
2.6. Immunocytochemistry
For imaging, cells were plated onto Cells Imaging Coverglasses (Eppendorf, 0030742060). Cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% Triton-X100 prior to immunofluorescence staining. Cells were then blocked in 5% goat serum for 1 hour before incubating with primary antibodies overnight at 4°C (Supplementary Table S3). Following washes with PBS cell were incubated with secondary antibodies (Alexa fluor, Life Technologies) for 1 hour at room temperature. Nuclei were stained with NucBlue® Fixed Cell ReadyProbes® Reagent (ThermoFisher), following manufacturers’ instructions. Images were captured on the Leica SP5 confocal microscope.
2.7. Flow cytometry analysis
Flow cytometry analysis was performed at the NPC and the final neuronal stages. Cells were harvested by enzymatic dissociation using Accutase for 5 min at 37°C until cells were visibly detached. Cells were pelleted by centrifugation at 800 rpm for 3 minutes, resuspended in ice-cold PBS. Cells were fixed in ice-cold 1%PFA in PBS and permeabilized in 0.1% TritonX-100 in PBS. Cells were blocked in 10% Rabbit serum (Abcam) and 10% Mouse serum (Abcam) for 1h. Cells were then resuspended in blocking buffer containing the appropriate concentration of conjugated antibody according to the manufacturer’s specifications (see Supplementary Table S3 for the list of the antibodies used in this study). Cells were sorted with Becton Dickinson Canto II, and Becton Dickinson FacsCalibur, followed by analysis using FlowJo 10.2 (FlowJo, LLC)
2.8. Study samples
The study cohort (N=248) consisted of individuals with three autopsy-confirmed neuropathological diagnoses: (1) PD (N=70); (2) DLB (N=77); and (3) clinically and neuropathologically normal subjects (N=101) as controls for the genetic association analyses. The brain tissues were obtained through the Kathleen Price Bryan Brain Bank (KPBBB) at Duke University, the Banner Sun Health Research Institute, and Layton Aging & Alzheimer's Disease Center at Oregon Health and Science University. All samples were derived from Caucasian subjects. Demographic data and neuropathologic diagnoses for these samples are shown in Table 1. Neuropathologic phenotypes were determined in postmortem examination following standard well-established methods following the method and clinical practice recommendations of McKeith and colleagues [26, 27]. The density of the LB pathology (in a standard set of brain regions) received scores of mild, moderate, severe and very severe.
Table 1.
Demographic description of the study cohorts
| PD | DLB | Normal-control | |
|---|---|---|---|
| Total no. | 70 | 77 | 101 |
| White % | 100 | 100 | 100 |
| Male % | 65.7 | 63.6 | 61.4 |
| Age at Death (mean±S.E.M) | 79.8±0.8 | 79.2±1.0 | 73.3±0.8 |
2.9. Sanger sequencing of the 3’UTR amplified by PCR
Genomic DNA was extracted from brain tissues by the standard Qiagen protocol (Qiagen, Valencia, CA). DNA samples from 248 subjects were plated on 96-well plates for PCR and sequencing at Polymorphic DNA Technologies (Alameda, CA, USA). Briefly, SNCA exon 6 (2,563 bp) that contains the entire 3’UTR region (2,529bp, chr4: 90,645,250–90,647,778; GRCh37/hg19) was amplified in 10 amplicons by PCR using the Takara Taq Polymerase (Takara Mirus Bio, Inc., Madison, WI, USA) and 10 sets of forward and reverse primers listed in Supplementary Table S4. The Sequence determination was performed by the standard Sanger sequencing method using 10 sets of nested primers (forward and reverse primers corresponding to each amplicon, Supplementary Table S4), and the Big Dye, version 3.1 sequencing kit (Applied Biosystems, Foster City, CA, USA), following the manufacture’s protocol. The sequencing reaction products were run on an ABI 3730XL DNA sequencer with a 50-cm capillary array using standard run mode. The sequence data was analyzed using the proprietary software Agent (Celera) to align the 20 sequencing reads (Polymorphic DNA Technologies, Inc.).
2.10. Statistical association analyses
Statistical analyses were performed to assess the association between each genetic variant and PD or DLB pathologies. For each SNP or short tendon repeat (SSR), frequencies for each allele were calculated and stratified by diagnosis (DLB, PD or neuropathologically normal control (NC)) and the results were shown as a mosaic plot. Multivariate logistic regression analysis was used to test the association between each genetic variant and: (1) the diagnosis of PD and NC; (2) the diagnosis of DLB and NC controlling for age and sex. Nominal p values of 0.05 were considered significant and correction for multiple testing employed the Bonferroni method. All analyses were carried out using JMP statistical software, Version 12.0.1 (SAS Institute, Cary, NC).
Expression measurements of the different miRNAs and of the SNCA-mRNA in iPSC-derived cells represents three biological and technical replicates. Statistical significance of differences in expression between the iPSC-derived cells was analyzed by pairwise comparisons using the Tukey-Kramer HSD test (JMP Pro13, SAS).
3 RESULTS
3.1 TargetScan analysis revealed four highly conserved miRNA sites
We analyzed the full length SNCA-3’UTR (2,529bp; chr4: 90,645,250-90,647,778; GRCh37/hg19) to identify microRNA (miRNAs) conserved sites using TargetScan 7.0 (Fig. 1A). Four conserved sites for miRNA families broadly conserved among vertebrates were identified: miR-7-5p, miR-153-3p, miR-223-3p, miR-140-3p.1. All of the broadly conserved miRNA sites lie within a 526bp region (chr4:90,647,133-90,647,659) at the 5’ end of the SNCA 3’UTR. Three of the conserved miRNA sites, miR-7-5p, miR-153-3p and miR-140-3p.1, resides within the most prevalent short SNCA 3’UTR (575bp; chr4:90,647,204-90,647,778; GRCh37/hg19). For each putative miRNA, the predicted target sequence and position in the 3’UTR, the site type and the software calculated score values are summarized in Fig. 1B. We then used the miRcode database to browse the SNCA 3’UTR and found that miRCode predicted miRNA target sites for highly conserved families corresponds with three of the TargetScan 7.0 miRNAs that includes, miR-7-5p, miR-153-3p (high scores, indicate high sites conservation), and miR-223-3p (medium score)[28].
Figure 1.

(A) Schematic presentation of SNCA-3’UTR. UCSC genome browser view of the SNCA-3’UTR region (2,529bp), coordinates: chr4: 90,645,250–90,647,778 (GRCh37/hg19). Depicted tracks include: predicted microRNA target sites from TargetScan (7.0)[20]; 3’UTR from TargetScan (7.0)[20]; RefSeq genes; conservation; dbSNP. (B) miRNAs and their conserved sites within SNCA-3’UTR in vertebrates. #7mer-m8: An exact match to positions 2–8 of the mature miRNA (the seed + position 8); *7mer-A1: An exact match to positions 2–7 of the mature miRNA (the seed) followed by an 'A' ; The context++ score (CS) for a specific site is the sum of the contribution of 14 features as described[20].
We also examined the potential binding site of miR-34b-3p that was previously suggested to have a role in the regulation of SNCA expression by independent groups[5, 9–11]. TargetScan analysis showed three miR-34b-3p predicted sites, however all sites showed poor conservation and were conserved only in human, Chimp and Rhesus. Two of the miR-34b-3p sites are within the short SNCA-3’UTR region and the third site resides in the long 3’UTR. The summary of miR-34b-3p poorly conserved predicted binding sites is presented in Supplementary Table S5.
3.2 The expression levels of miRNAs and SNCA expression levels in isogenic iPSC-derived cholinergic and dopaminergic neurons
To evaluate the neuronal-type specific levels of the SNCA highly conserved miRNAs, we used the iPSC line GM23280 obtained from the Coriell collection of apparently healthy iPSC catalogue. GM23280 were differentiated into mDA and BFCN to model DLB- and PD-primarily affected neurons, respectively. The successful differentiation, in each stage and for each neuronal type, was validated by RealTime RT PCR using specific markers (Fig. 2A and Supplementary Table S1). In addition, we characterized each stage of the differentiation by immunofluorescent using specific markers and demonstrated expression of the specific corresponding marker protein in each stage (Fig. 2B–F). Furthermore, we determine the percentage of the specific cell-type by FACS analysis (Fig. 3A, B and C. Fig. 3A shows that 83.1% or 84.4% of the NPCs were double positive to Nestin and Forkhead box protein A2 (FOXA2) or to Nestin and NK2 homeobox 1(Nkx2.1), indicating successful differentiation to MD progenitors or MGE progenitors, respectively. We analyzed the final mDA for the mature neuronal marker Tubulin beta-3 chain (TUBB3) and the specific dopaminergic marker Tyrosine Hydrolase (TH) and showed that 28.3% of the cells co-expressed these markers (Fig. 3B). The analysis of the BFCN showed that 36.4% of the cells co-expressed the TUBB3 and the cholinergic specific marker, Vesicular Acetylcholine Transporter (VaChT) (Fig. 3C). We confirmed the absent of contamination with other neuronal types, particularly GABAergic neurons. The mature mDA and BFCN were immunostained with GABAergic neuronal marker, Glutamic Acid Decarboxylase (GAD67) and no positive cells were detected (Supplementary Fig. S1). All cell populations were also negative to Glial Fibrillary Acidic Protein (GFAP) validating the purity of both differentiated neuronal populations (Fig. 3B and 3C).
Figure 2. Cellular characterization of iPSC-derived cells by quantitative RT-PCR and immunofluorescence.

(A) iPSCs were differentiated into dopaminergic (mDA) and cholinergic (BFCN) neurons and each stage and neuronal type was confirmed using quantitative real-time RT-PCR for cellular specific markers (Supplementary Table S1). Levels of mRNAs were measured by real-time RT-PCR and calculated relatively to the geometric mean of GAPDH-mRNA and PPIA-mRNA reference controls using the 2−ΔΔCT method. Each column represents the mean of two biological and technical replicates. The error bars represent the Standard Error of the Mean (S.E.M.). (B–F) Immunofluorescence (IF) analyses of the cell types across differentiation. (B) The pluripotency markers POU5F1 (red) and NANOG (green) in iPSCs. (C) The NPCs marker, Nestin (red), and the marker expressed by floor plate progenitors, FOXA2 (red) in MD progenitors. (D) The dopaminergic marker, TH (green) and the mature neurons marker, TUBB3 (red) in mDA. Co-localization of the markers indicates the differentiation of mature dopaminergic neurons. (E) The NPCs marker, Nestin (red), and the marker indicating the pattern of basal forebrain cholinergic differentiation, Nkx2.1 (green) in MGE progenitors. (F) The cholinergic marker, ChAT (green) and the mature neuron marker TUBB3 (red) in BFCN. Co-localization of the markers indicates the differentiation of mature BFCN. DNA counterstaining in blue (B–F).
Figure 3. Cellular characterization of iPSC-derived cells by Fluorescence-activated cell sorting FACS).

((A–C) (FACS) profile of neural intracellular markers expressed during dopaminergic and cholinergic differentiation. Flow cytometric analysis for Nestin, FOXA2, Nkx2.1, TUBB3, GFAP, TH and VaChT are shown. (A) Combinatorial FACS analysis of Nestin and FOXA2 for MD progenitors (83.1% double positive), and Nestin and Nkx2.1 for MGE progenitors (84.4% double positive). (B) Combinatorial FACS analysis of TUBB3 and TH (28.3% double positive), and GFAP and TH for mDA(46.9% single positive). (C) Combinatorial FACS analysis of TUBB3 and VaChT (36.4% double positive), and GFAP and VaChT for BFCN (39.9% single positive).
The expression of miR-7-5p, miR-140-3p.1, miR-153-3p, and miR-223-3p was evaluated in iPSCs, NPCs and final neurons throughout the dopaminergic and cholinergic differentiation.
miR-7-5p levels were significantly higher in the mature mDA compared to the MD progenitors (p=0.045, Fig. 4A). Moreover, the expression levels of miR-7-5p in the mDA were significantly higher compared to the cholinergic differentiation, particularly MGE progenitors (p=0.003, Supplementary Table S6), and BFCN (p=0.004, Fig. 4A). This result suggests a possible role of the miR-7-5p in the mature mDA.
Figure 4. Expression profiles of the SNCA-3’UTR conserved miRNAs in iPSC derived cells.
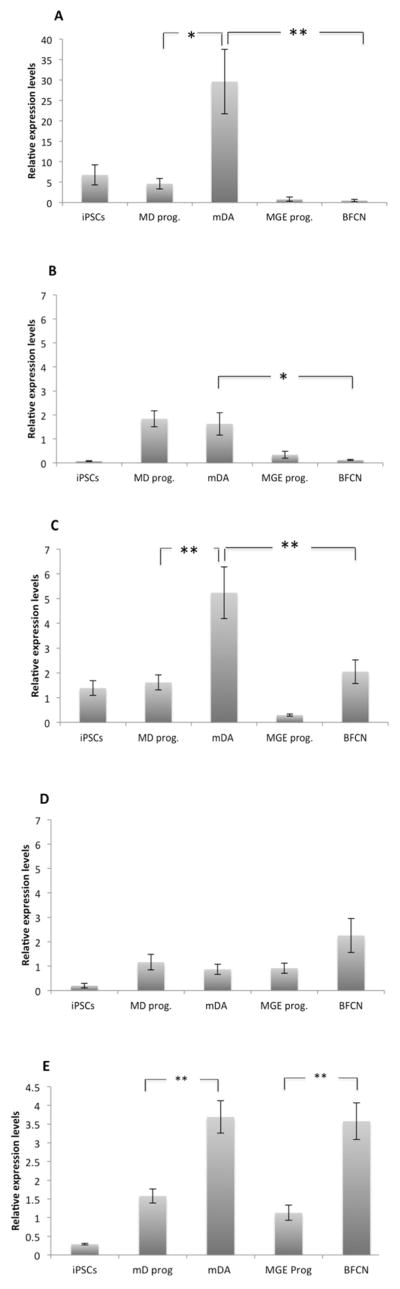
iPSCs were differentiated into dopaminergic (mDA) and cholinergic (BFCN) neurons. The expression of miR-7-5p (A), miR-153-3p (B), miR223-3p (C) and miR-140-3p.1 (D) was evaluated throughout the differentiation process. Levels of each miRNAs were measured by real-time RT-PCR and calculated relatively to the geometric mean of RNU6B-miRNA and RNU24-miRNA reference controls using the 2-ΔΔCT method. Each column represents the mean of three biological and technical replicates. The scale in the Fig.4A is different from the other scales. (E) The expression of SNCA-mRNA was evaluated during the differentiation process. Levels of SNCA-mRNA were measured by real-time RT-PCR and calculated relatively to the geometric mean of GAPDH-mRNA and PPIA-mRNA reference controls using the 2-ΔΔCT method. Each column represents the mean of three biological and technical replicates. The error bars represent the Standard Error of the Mean (S.E.M.). Significant differences are marked as following (*p<0.05, **p<0.01, Tukey-Kramer HSD test). In the figure, we denoted only the significant differences identified for the key comparisons: MD prog vs mDA; MGE prog. Vs BFCN, and mDA vs BFCN. P-values for all pairwise comparisons are summarized in Supplementary Table S6.
A similar pattern was observed for miR-153-3p and miR-223-3p. The levels of miR-153-3p were significantly elevated in the mature mDA in comparison to the BFCN (p=0.02, Fig. 4B). Similar trend was observed between the progenitor cell lines, MD and MGE progenitors (p=0.009). Also, the MD progenitors showed a significant differential pattern compared to the final BFCN (p=0.006) (Supplementary Table S6).
The levels of miR-223-3p were increased significantly in the mDA throughout the dopaminergic differentiation and also compared to the cholinergic cells (Fig. 4C). Specifically, our data showed that the mature mDA miR-223-3p levels were higher compared to the MD prog (p=0.0003, Fig. 4C), MGE prog (p<0.001, Supplementary Table S6) and BFCN (p=0.0044, Fig. 4C).
No significant differences were observed for the expression levels of miR-140-3p.1 by multiple comparison analysis (Fig. 4D). However, the levels of miR-140-3p.1 were higher in BFCN. Using t-test analysis, we found that the levels of miR-140-3p.1 were higher in the BFCN compared to mDA (p=0.016, Supplementary Table S6). We also observed a significant increased t miR-140-3p.1 expression throughout the cholinergic differentiation (p=0.0196, Supplementary Table S6).
In addition, we observed that within the same cell type and at the same differentiation stage the different miRNAs showed variability in their relative expression levels. For example, in the mature mDA, miR-7-5p showed the highest expression levels relative to the other miRs followed by miR-223-3p, and in the final BFCN, both miR-140-3p.1 and miR-223-3p were highly expressed compared to miR-7-5p and miR-153-3p (Fig. 4A–D). We repeated the miRNAs expression analysis using a cell-line form a PD patient with the SNCA triplication (SNCA-tri, Supplementary Fig. S2). The levels of the studied miRNAs were evaluated in the SNCA-tri iPSC-derived mature neurons, mDA and BFCN. Comparisons of miRNAs expression levels between mDA and BFCN derived from the healthy and the PD iPSC lines showed similar trends for miR-7-5p (mDA>BFCN), miR-153-3p (mDA>BFCN), and miR-140-3p.1 (mDA<BFCN) (Fig. S2A, B, and D, respectively). miR-223-3p didn’t showed significant difference in the SNCA-tri derived matured mDA compared to BFCN, however, an overall overexpression of miR-223-3p was observed in both mDA and BFCN derived from the SNCA-tri iPSC line compared to the healthy neuronal lines (Fig. S2C). Conversely, a ~10 fold decreased in the expression levels of miR-7-5p was observed in the SNCA-tri derived neurons compared to the mature neurons derived from the healthy iPSC line (Fig. S2A).
Next, we evaluated the levels of the SNCA-mRNA in the iPS-derived cells (Fig. 4E). The SNCA-mRNA levels significantly increased throughout the differentiation process for both cholinergic (p=0.0001) and dopaminergic (p=0.001) neurons (Fig. 4E). No significant differences were observed between mature mDA and BFCN (Supplementary Table S6). We also assessed the levels of α–syn protein in the iPSC-derived cells throughout the differentiation, and observed similar trends to those found for the SNCA-mRNA levels (Supplementary Fig. S3). The α–syn protein levels significantly increased throughout the differentiation process for both the cholinergic (p=0.0012) and the dopaminergic (p=0.0041) neurons (Supplementary Fig. S3F). Consistently with the mRNA analysis, we didn’t observe significant differences in α–syn expression between mature mDA and BFCN (Supplementary Fig. S3).
3.3 Sequence variations in SNCA 3’UTR
Next we examined the sequence of the entire 3’UTR (long form) in a cohort of individuals with three autopsy-confirmed neuropathological diagnoses, PD, DLB and normal controls. The SNCA 3’UTR was sequenced using the traditional Sanger sequencing method and several known and novel genetic variants were identified. A total of 7 genetic variants were identified that includes 5 single nucleotide polymorphisms (SNPs) and 2 short structural variants (SSVs) (Fig. 1A and Table 2). The known SNPs that were polymorphic in our cohort are: rs1045722, rs3857053, rs356165 and rs145304567. In addition, we identified rs17016074 in a heterozygote normal subject. The polymorphic SSVs in our entire cohort includes rs777296100 (SSR, poly-T), and a novel rare deletion ‘-ATTT’ was detected in 2 heterozygotes individuals at position chr4:90647199 of a PD and a DLB samples.
Table 2.
Association of variants in SNCA-3’UTR with DLB and PD
| Rs Number | Variant Type | Chr. location | Alleles minor/ major | Normal N=101 MAF |
DLB N=77 MAF |
PD N=70 MAF |
DLB OR (95% CI) |
PD OR (95% CI) |
DLB p value |
PD p value |
|---|---|---|---|---|---|---|---|---|---|---|
| rs1045722 | SNP | 4:90,645,671 | T/A | 0.095 | 0.084 | 0.085 | 1.62 (0.70–4.12) | 1.11 (0.52– 2.45) | 0.264 | 0.786 |
| rs3857053 | SNP | 4:90,645,674 | A/G | 0.095 | 0.084 | 0.085 | 1.59 (0.68–4.02) | 1.08 (0.50– 2.41) | 0.292 | 0.835 |
| rs356165 | SNP | 4:90,646,886 | C/T | 0.409 | 0.350 | 0.343 | 1.41 (0.88–2.27) | 1.25 (0.78– 1.99) | 0.149 | 0.353 |
| rs145304567 | SNP | 4:90,647,702 | A/C | 0.020 | 0.032 | 0.000 | 1.69 (0.38–7.51) | - | 0.475 | 0.052 |
| rs10716074 | SNP | 4:90,647,278 | T/C | 0.005 | 0.000 | 0.000 | - | - | 0.502 | 0.564 |
| rs777296100 | SSR | 4:90,646,470 | <20/ 21; 22+ | 0.090 | 0.060 | 0.079 | 1.18 (1.00–1.41) | 1.00 (0.87– 1.17) | 0.048 | 0.947 |
| DEL ATTT | deletion | 4:90,647,199 | -/ATTT | 0.000 | 0.013 | 0.014 | - | - | 0.143 | 0.166 |
SNP, single nucleotide polymorphism; SSR, simple sequence repeat; Chr. location based on GRCh37/hg19; N, total number of subjects; MAF, minor allele frequency; Bold P value, indicates significant association
3.4 The rs777296100-SSR associated with DLB but not with PD
The identified SNPs were not significantly associated with the neuropathologic diagnoses of disease compared to normal controls (see Table 2), however, the rarest variant (rs145304567-A, 2–3% in DLB and normal) was not detected in the PD group (n=70). A larger PD sample size will be needed to validate the absence of the A allele in PD.
The rs777296100 (dbSNP 147) poly-T site was highly variable presenting a broad spectrum of allele lengths, ‘T’s=16–24, and 28. The comparison between DLB and normal suggested a moderate association (p=0.047, Fig. 5A and Table 2), however this association would not be significant after correction for multiple testing (7 variants). The shorter allele lengths (17–20) were associated with normal classification, 21 ‘T’s was the change point and the longer allele lengths (T=22+) have a trend toward more DLB (Fig. 5A, B). No association was identified in an association analysis between PD and normal (p=0.947, Fig. 5C and Table 2). This poly-T resides within the long 3’UTR but is not included in the region of the short 3’UTR. Interestingly, the distance between rs777296100 and the closest position of predicted binding site of the most 3’ miR-223-3p is ~650bp. The proximity of the SSR may have an effect on RNA structure which may influence the accessibly of the miRNAs. Nonetheless, none of the identified genetic variants lies within the predicted target sequence of the studied miRNAs.
Figure 5. Mosaic plots of the proportion of individuals with diagnoses of PD or DLB compared to cognitively normal stratified by the number of rs777296100 poly-T bases.

Each column in the plot corresponds to specific poly-T lengths; the rows correspond to diagnoses and have an associated color code defined in the key at the far-right side of each plot (cognitively normal is red). The height of the color-coded block in each poly-T category shows the proportion of individuals with each specific neuropathological diagnosis that have associated rs777296100 (dbSNP 147) genotypes. The scale of the y-axis (far left) shows the proportions for each diagnosis, with the entire subset of individuals in a risk category corresponding to 1. The width of the columns are proportional to the group sample size. (A) Dementia with Lewy bodies (DLB) versus cognitively normal, (B) DLB versus cognitively normal with the poly-T lengths assigned to categories, (C) Parkinson’s disease (PD) versus cognitively normal. NOTE: rs777296100 (dbSNP 147) is denoted as rs33988309 dbSNP build 142.
4. DISCUSSION
In this paper, we studied the potential distinct contribution of SNCA-3’UTR, on both cis- and trans- levels, to DLB and PD. We established isogenic iPSCs derived cholinergic and dopaminergic neurons, two neuronal types that are differentially involved in DLB and PD, respectively. This model system is suitable in studying common and distinctive cell-specific regulatory mechanisms underlying synucleinopathies.
The modulation of SNCA-mRNA levels by endogenous miRNAs has been proposed as a post-transcriptional mechanism of SNCA expression[7–11]. We searched for miRNA families that are highly conserved among vertebrates, identified four sites – miR-7-5p, miR-153-3p, miR-223-3p, and miR-140-3p.1– and studied their involvement in pathology-relevant cells. The previously described roles of these four miRNAs are summarized in (Supplementary Table S7). In the mature BFCN, the levels of the miR-140-3p.1 and miR-223-3p were dominant. The role of miR223 in the brain has been investigated. miR-223 is expressed in the brain, regulates the differentiation of immature neurons[29], and exerts a neuroprotective role[30]. It was also reported that miR-223 is upregulated in PD and MSA compared to the controls [31]. However, miR-140-3p.1 has been understudied within the brain context. In contrast, in mature mDA miR-7-5p showed the highest expression by wide-margin, followed by miR-223-3p, and miR-153-3p. It has been reported that miR7 and miR153 regulate SNCA-mRNA expression levels[6, 7], and they are highly expressed in mouse brain and in brain cell types[6]. In our model system, although we observed extremely high levels of miR-7-5p in mDA, the levels of SNCA-mRNA were comparable to the isogenic BFCN in which the expression of miR-7-5p was much lower. Furthermore, the levels miR-7-5p are increased throughout dopaminergic differentiation but correlated with increased SNCA-mRNA levels, also in conflict with the role of miRNAs in down regulation of gene expression. Our results demonstrated that the elevated expression levels of miR-7-5p in mDA had no differential effect on SNCA-mRNA expression levels and therefore the presumed miR-7-5p downregulation effect is not clear, however, we cannot exclude the possibility that in mDA neurons, under physiological conditions, a combination of other factors promote SNCA upregulation, while increased levels of miR-7-5p maintains the basal SNCA-mRNA expression levels. Consistent with our data, it has been reported that the levels of miR-7 and miR-153 in mouse brain correspond directly to the expression of SNCA-mRNA, with high levels of SNCA-mRNA correlated with increased levels of miRNAs[7].
Genetic variants in miRNAs or miRNA-binding sites could affect miRNA function and contribute to disease risk[32, 33]. Polymorphisms within the SNCA 3’UTR have been reported, and specifically associated with PD [34–38]. Previous studies of the short 3’UTR of the SNCA gene didn’t find common variants within the binding sites of the highly conserved miR-7 and miR-153 in a cohort of sporadic PD cases[39]. A rare mutation within the miR-153 binding site in a patient with idiopathic PD has been reported[40]. A recent bioinformatics work predicted three PD associated SNPs (rs356165, rs3857053, and rs1045722) in the SNCA 3’UTR that overlap with poorly conserved miRNA-binding sites: miR-6082, miR-4693-5p, and miR-451a, respectively[32]. We extended on these studies and performed deep sequencing analysis of the full length SNCA 3’UTR that allow us to accurately determine both SNPs and SSVs in a cohort of individuals with PD, DLB and normal controls. However, in our smaller sample sets we didn’t detect significant association of these common SNPs with PD and DLB (Supplementary Table S5). We also didn’t detect any genetic variants in the binding sites of the studied miRNAs.
We found a moderate association of rs777296100 (dbSNP 147), a highly variable poly-T site with DLB. This poly-T doesn’t overlap with the highly conserved miRNAs we studied; however, the length of the poly-T site may change the RNA structure of the 3’UTR and thereby impact on the miRNA binding. It has been suggested that variants flanking miRNA-binding sites could alter the target structure and affect the accessibility of the miRNA-binding site[41]. Nevertheless, we cannot exclude the involvement of other regulatory mechanisms in the role of the 3’UTR. Such mechanisms could be the selection of different polyadenylation sites that determine the length of the 3’UTR with an effect on the mRNA stability and localization [5, 42, 43]. Collectively the results of our and other studies imply that common variations may not affect the binding sites of the predicted highly conserved miRNAs. However, it is possible that rare variants may influence the binding affinity of the highly-conserved miRNAs and by that influence SNCA-mRNA expression levels and disease-risk. Alternatively, common or rare variants may also affect the binding of the poorly conserved miRNAs[32]. Insights into these lines of mechanisms warrant further investigations in a larger cohort of subjects for the discovery of rare variants, and using biological system, such as the isogenic iPSC-derived system developed in this work to determine expression profile of the poorly conserved miRNAs.
The sample sizes of the study cohorts are relatively small as a consequence of the scarcity of well-characterized DLB neuropathological samples, that introduced a limitation for our study. Bonferroni correction for 7 genetic variants would set a significance threshold of 0.01 and the genetic association results we reported for DLB with rs777296100 fall short of this level and therefore have to be considered suggestive.
We suggest that cell-specific mechanisms regulate SNCA gene expression and involved the contribution of cis and trans acting factors[2].Here as a pathology distinct cis factor we discovered a genetic variant in SNCA 3’UTR that specifically affect DLB risk and as a trans factor we found differential expression of miRNAs in pathology-relevant cells. Neuronal-type specific mechanisms regulating SNCA expression may contribute, at least in part, to the heterogeneity of synucleionpathies.
Supplementary Material
RESEARCH IN CONTEXT.
Systematic review: The authors reviewed the Literature using Pubmed, meeting abstracts and presentations. While genetic variants in the SNCA gene are associated with PD and DLB, the common and diverse molecular mechanisms by which they exert their role are not fully understood. Interpretation: Our findings generated the hypothesis that SNCA-mRNA levels are differentially regulated by miRNAs in a neuronal-type specific manner. This may provide an example for a differential molecular mechanism underlying DLB vs PD. Future directions: the manuscript proposed a framework for in depth investigations in the following directions: (a) Exploring the effect of the identified DLB-associated polyT on SNCA-mRNA levels in neurons from DLB compared to PD patients; (b) Understanding how genetic variants in the SNCA 3’UTR, distant-from of the miRNA binding sites, may affect the miRNA-binding; (c) Identifying rare causal genetic variations in the SNCA 3’UTR using a larger clinical cohorts.
Highlights.
Four highly conserved miRNAs target sites were identified in SNCA 3’UTR
These miRNAs showed differential expression in cholinergic vs. dopaminergic neurons.
Differential miRNA regulation of SNCA may contribute to heterogeneity in DLB vs PD.
A polyT variant within SNCA 3’UTR is distinctively associated with DLB but not with PD.
Acknowledgments
FUNDING: This work was funded in part by the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) [R01 NS085011 to O.C.].
We thank Mr. Thomas Ribar the director of the iPSC-core facility at Duke University for his assistance with the iPSCs work, Dr. Maria Blasi for her guidance in the FACS analysis, and Dr. Yasheng Gao the microscopy specialist of the Light Microscopy Core Facility at Duke University for technical assistance.
We thank the Layton Aging & Alzheimer’s Disease Center at Oregon Health and Science University, and the Brain and Body Donation Program (BBDP) at the Banner Sun Health Research Institute (BSHRI) for providing us with the brain tissues. The Layton Aging & Alzheimer’s Disease Center is supported by NIH/NIA P30 AG08017 to the Oregon Alzheimer’s Disease Center. The Banner Sun Health Research Institute Brain and Body Donation Program has been supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson's Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research.
ABBREVIATIONS
- α-syn
alpha-synuclein protein
- BFCN
Basal Forebrain Cholinergic Neurons
- DLB
Dementia with Lewy Bodies
- EBs
Embryoid Bodies
- GWAS
Genome Wide Association Studies
- iPSCs
induced Pluripotent Stem Cells
- LB
Lewy Bodies
- LD
Linkage Disequilibrium
- PD
Parkinson’s Disease
- MD
Ventral Midbrain
- mDA
Dopaminergic Neurons
- MGE
Medial Ganglionic Eminence
- miRNAs
microRNAs
- miR-7-5p
microRNA-7-5p
- miR-140-3p.1
microRNA-140-3p.1
- miR-153-3p
microRNA-153-3p
- miR-223-3p
microRNA-223-3p
- MSA
Multiple System Atrophy
- NEM
Neural Expansion Medium
- NIM
Neural Induction Medium
- NPC
Neural Precursor Cells
- SNCA
α-synuclein
- SNPs
Single Nucleotide Polymorphisms
- SSVs
Short Structural Variants
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tagliafierro L, Chiba-Falek O. Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics. 2016 doi: 10.1007/s10048-016-0478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beyer K. Alpha-synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta neuropathologica. 2006;112:237–51. doi: 10.1007/s00401-006-0104-6. [DOI] [PubMed] [Google Scholar]
- 4.Lee HJ, Choi C, Lee SJ. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J Biol Chem. 2002;277:671–8. doi: 10.1074/jbc.M107045200. [DOI] [PubMed] [Google Scholar]
- 5.Rhinn H, Qiang L, Yamashita T, Rhee D, Zolin A, Vanti W, et al. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson's disease pathology. Nat Commun. 2012;3:1084. doi: 10.1038/ncomms2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Junn E, Lee KW, Jeong BS, Chan TW, Im JY, Mouradian MM. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc Natl Acad Sci U S A. 2009;106:13052–7. doi: 10.1073/pnas.0906277106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doxakis E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J Biol Chem. 2010;285:12726–34. doi: 10.1074/jbc.M109.086827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fragkouli A, Doxakis E. miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Frontiers in cellular neuroscience. 2014;8:182. doi: 10.3389/fncel.2014.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minones-Moyano E, Porta S, Escaramis G, Rabionet R, Iraola S, Kagerbauer B, et al. MicroRNA profiling of Parkinson's disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum Mol Genet. 2011;20:3067–78. doi: 10.1093/hmg/ddr210. [DOI] [PubMed] [Google Scholar]
- 10.Villar-Menendez I, Porta S, Buira SP, Pereira-Veiga T, Diaz-Sanchez S, Albasanz JL, et al. Increased striatal adenosine A2A receptor levels is an early event in Parkinson's disease-related pathology and it is potentially regulated by miR-34b. Neurobiology of disease. 2014;69:206–14. doi: 10.1016/j.nbd.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 11.Kabaria S, Choi DC, Chaudhuri AD, Mouradian MM, Junn E. Inhibition of miR-34b and miR-34c enhances alpha-synuclein expression in Parkinson's disease. FEBS Lett. 2015;589:319–25. doi: 10.1016/j.febslet.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cookson MR. The biochemistry of Parkinson's disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 13.Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 14.Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 15.Grothe MJ, Schuster C, Bauer F, Heinsen H, Prudlo J, Teipel SJ. Atrophy of the cholinergic basal forebrain in dementia with Lewy bodies and Alzheimer's disease dementia. J Neurol. 2014;261:1939–48. doi: 10.1007/s00415-014-7439-z. [DOI] [PubMed] [Google Scholar]
- 16.Hall H, Reyes S, Landeck N, Bye C, Leanza G, Double K, et al. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson's disease. Brain. 2014;137:2493–508. doi: 10.1093/brain/awu193. [DOI] [PubMed] [Google Scholar]
- 17.Marui W, Iseki E, Nakai T, Miura S, Kato M, Ueda K, et al. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195:153–9. doi: 10.1016/s0022-510x(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 18.Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, et al. Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta neuropathologica. 2009;117:613–34. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bras J, Guerreiro R, Darwent L, Parkkinen L, Ansorge O, Escott-Price V, et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet. 2014;23:6139–46. doi: 10.1093/hmg/ddu334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015:4. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu H, Zhang SC. Specification of neuronal and glial subtypes from human pluripotent stem cells. Cell Mol Life Sci. 2011;68:3995–4008. doi: 10.1007/s00018-011-0770-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crompton LA, Byrne ML, Taylor H, Kerrigan TL, Bru-Mercier G, Badger JL, et al. Stepwise, non-adherent differentiation of human pluripotent stem cells to generate basal forebrain cholinergic neurons via hedgehog signaling. Stem Cell Res. 2013;11:1206–21. doi: 10.1016/j.scr.2013.08.002. [DOI] [PubMed] [Google Scholar]
- 23.Lin L, Goke J, Cukuroglu E, Dranias MR, VanDongen AM, Stanton LW. Molecular Features Underlying Neurodegeneration Identified through In Vitro Modeling of Genetically Diverse Parkinson's Disease Patients. Cell Rep. 2016;15:2411–26. doi: 10.1016/j.celrep.2016.05.022. [DOI] [PubMed] [Google Scholar]
- 24.Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, et al. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141–52. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (San Diego, Calif. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 26.McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53:902–5. doi: 10.1212/wnl.53.5.902. [DOI] [PubMed] [Google Scholar]
- 27.McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 28.Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 2012;28:2062–3. doi: 10.1093/bioinformatics/bts344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harraz MM, Xu JC, Guiberson N, Dawson TM, Dawson VL. MiR-223 regulates the differentiation of immature neurons. Mol Cell Ther. 2014:2. doi: 10.1186/2052-8426-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang C, Ji B, Cheng B, Chen J, Bai B. Neuroprotection of microRNA in neurological disorders (Review) Biomed Rep. 2014;2:611–9. doi: 10.3892/br.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallelunga A, Ragusa M, Di Mauro S, Iannitti T, Pilleri M, Biundo R, et al. Identification of circulating microRNAs for the differential diagnosis of Parkinson's disease and Multiple System Atrophy. Frontiers in cellular neuroscience. 2014;8:156. doi: 10.3389/fncel.2014.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghanbari M, Darweesh SK, de Looper HW, van Luijn MM, Hofman A, Ikram MA, et al. Genetic Variants in MicroRNAs and Their Binding Sites Are Associated with the Risk of Parkinson Disease. Hum Mutat. 2016;37:292–300. doi: 10.1002/humu.22943. [DOI] [PubMed] [Google Scholar]
- 33.Ghanbari M, Ikram MA, de Looper HW, Hofman A, Erkeland SJ, Franco OH, et al. Genome-wide identification of microRNA-related variants associated with risk of Alzheimer's disease. Sci Rep. 2016;6:28387. doi: 10.1038/srep28387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 35.Mizuta I, Satake W, Nakabayashi Y, Ito C, Suzuki S, Momose Y, et al. Multiple candidate gene analysis identifies alpha-synuclein as a susceptibility gene for sporadic Parkinson's disease. Hum Mol Genet. 2006;15:1151–8. doi: 10.1093/hmg/ddl030. [DOI] [PubMed] [Google Scholar]
- 36.Mueller JC, Fuchs J, Hofer A, Zimprich A, Lichtner P, Illig T, et al. Multiple regions of alpha-synuclein are associated with Parkinson's disease. Ann Neurol. 2005;57:535–41. doi: 10.1002/ana.20438. [DOI] [PubMed] [Google Scholar]
- 37.Ross OA, Gosal D, Stone JT, Lincoln SJ, Heckman MG, Irvine GB, et al. Familial genes in sporadic disease: common variants of alpha-synuclein gene associate with Parkinson's disease. Mech Ageing Dev. 2007;128:378–82. doi: 10.1016/j.mad.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sotiriou S, Gibney G, Baxevanis AD, Nussbaum RL. A single nucleotide polymorphism in the 3'UTR of the SNCA gene encoding alpha-synuclein is a new potential susceptibility locus for Parkinson disease. Neurosci Lett. 2009;461:196–201. doi: 10.1016/j.neulet.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitt I, Wullner U, van Rooyen JP, Khazneh H, Becker J, Volk A, et al. Variants in the 3'UTR of SNCA do not affect miRNA-433 binding and alpha-synuclein expression. Eur J Hum Genet. 2012;20:1265–9. doi: 10.1038/ejhg.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim HJ, Park G, Jeon BS, Park WY, Kim YE. A mir-153 binding site variation in SNCA in a patient with Parkinson's disease. Mov Disord. 2013;28:1755–6. doi: 10.1002/mds.25505. [DOI] [PubMed] [Google Scholar]
- 41.Liu C, Rennie WA, Carmack CS, Kanoria S, Cheng J, Lu J, et al. Effects of genetic variations on microRNA: target interactions. Nucleic Acids Res. 2014;42:9543–52. doi: 10.1093/nar/gku675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yeh HS, Yong J. Alternative Polyadenylation of mRNAs: 3'-Untranslated Region Matters in Gene Expression. Mol Cells. 2016;39:281–5. doi: 10.14348/molcells.2016.0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.