

Abstract
The lower efficacy of opioids in neuropathic pain may be due to the increased activity of pronociceptive systems such as substance P. We present evidence to support this hypothesis in this work from the spinal cord in a neuropathic pain model in mice. Biochemical analysis confirmed the elevated mRNA and protein level of pronociceptive substance P, the major endogenous ligand of the neurokinin-1 (NK1) receptor, in the lumbar spinal cord of chronic constriction injury (CCI)-mice. To improve opioid efficacy in neuropathic pain, novel compounds containing opioid agonist and neurokinin 1 (NK1) receptor antagonist pharmacophores were designed. Structure–activity studies were performed on opioid agonist/NK1 receptor antagonist hybrid peptides by modification of the C-terminal amide substituents. All compounds were evaluated for their affinity and in vitro activity at the mu opioid (MOP) and delta opioid (DOP) receptors, and for their affinity and antagonist activity at the NK1 receptor. On the basis of their in vitro profiles, the analgesic properties of two new bifunctional hybrids were evaluated in naive and CCI-mice, representing models for acute and neuropathic pain, respectively. The compounds were administered to the spinal cord by lumbar puncture. In naive mice, the single pharmacophore opioid parent compounds provided better analgesic results, as compared to the hybrids (max 70% MPE), raising the acute pain threshold close to 100% MPE. On the other hand, the opioid parents gave poor analgesic effects under neuropathic pain conditions, while the best hybrid delivered robust (close to 100% MPE) and long lasting alleviation of both tactile and thermal hypersensitivity. The results presented emphasize the potential of opioid/NK1 hybrids in view of analgesia under nerve injury conditions.
Keywords: Hybrid peptides, neuropathic pain, acute pain, NK1 antagonists, opioid agonism
Graphical Abstract
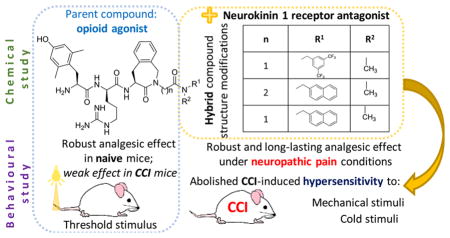
INTRODUCTION
Neuropathic pain is a chronic pain state which results from a lesion or pathology of the central or peripheral nervous system; it affects up to 7–8% of the general population.3 Even though a variety of agents are used to reduce pain in neuropathy, less than half of patients report effective pain relief.4 However, one particular strategy, the design of drugs with bi- or even multifunctional activity, offers a promising strategy to treat neuropathic pain. This strategy addresses the complex nature of this pain type, which is associated with pathological over-activation of endogenous pronociceptive systems as a maladaptive response to the somatosensory system injury.5,6 Bi- or multifunctional agents are able to interact independently with two or more targets (such as different types of receptors) to achieve the desired therapeutic effect.7–10 In particular, hybrid compounds designed as potential therapeutic agents in neuropathic pain are able to activate the opioid system—to achieve an antinociceptive effect—and hamper the activation of pronociceptive systems, to correct an imbalance between anti- and pronociception after repetitive opioid administration. In consequence, such hybrids are expected to provide an efficacious analgesic effect with lowered risk of adverse events.
In previous studies on rats, we obtained promising results for hybrids acting as opioid agonists and neurokinin-1 (NK1) receptor antagonists.2 In the present study, we performed biochemical analyses in mice under neuropathic pain conditions, which provide a basis for the observations described above: the inefficacy of morphine to alleviate neuropathic pain is not due to a reduced expression of the mu opioid peptide (MOP) receptor, but rather related to counteraction due to an increased release of the pronociceptive substance P.11 This conclusion motivates further studies to design new opioid agonists/NK1 receptor antagonists to treat neuropathic pain, especially with regard to the influence of the relative potencies of the included pharmacophores on in vivo efficacy. Therefore, in the current study, eight new hybrids were prepared (compounds 6–13, RCCHM1–8, Table 1), and on the basis of their in vitro pharmacological profile, the analgesic action of two of them in mice was compared to that of a reference opioid 1 (AN81) and hybrid 3 (SBCHM1), both of which were tested previously on rats.2 This comparison allowed a selection of the best hybrid structures for testing of effective analgesia in mouse models of acute and neuropathic pain. Moreover, it also provides insight into correlations between chemical structure and analgesic efficacy in different behavioral tests.
Table 1.
Structure of the Reference Compounds 1–5 and of the New Hybrids 6–13
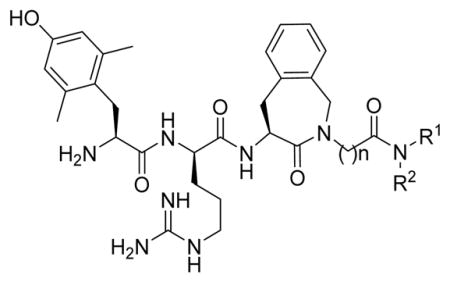
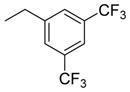
| |||
|---|---|---|---|
|
| |||
| Compound number (code) | n | R1 | R2 |
| 1 (AN81)1 | 1 | H | H |
| 2 (KGOP1)2 | 2 | H | H |
| 3 (SBCHM1)1 | 1 |
|
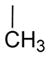
|
| 4 (KGCHM2)2 | 2 |
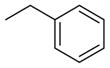
|
|
| 5 (KGCHM5)2 | 1 |
|
|
|
| |||
| 6 (RCCHM1) | 2 |
|
|
| 7 (RCCHM2) | 2 |
|
|
| 8 (RCCHM8) | 1 |
|
|
| 9 (RCCHM4) | 2 |
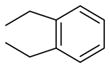
|
|
| 10 (RCCHM5) | 1 |
|
|
| 11 (RCCHM7) | 1 |
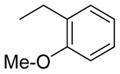
|
|
| 12 (RCCHM3) | 2 |
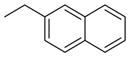
|
|
| 13 (RCCHM6) | 1 |
|
|
RESULTS AND DISCUSSION
Development of Neuropathic Pain and Biochemical Changes in the Spinal Cord, at a Selected Time Point after Chronic Constriction Injury (CCI) to the Sciatic Nerve in Mice
Development of Neuropathic Pain
At 24 h after performing the chronic constriction injury (CCI), mice developed hypersensitivity to tactile (von Frey test) and thermal (cold plate test) stimuli (Figure 1, panels A and B, respectively). This was manifested as a significant decrease of the response threshold in comparison with naive mice. Seven days after performing the nerve damage, CCI-mice reacted to tactile stimuli (paw withdrawal) in the von Frey test at the mean force of 1.05 ± 0.3 g (cutoff: 6 g). Tactile stimuli up to 6 g are neutral for healthy mice, and hence naive animals do not react to it. Paw withdrawal of CCI-mice in the cold plate test was observed on average after 5.6 ± 0.2 s (cutoff: 30 s). Naive mice react after 26.9 ± 1.7 s on average. The greatest manifestation of hypersensitivity symptoms was observed between the 7th and 14th day after nerve injury (Figure 1).
Figure 1.

Development of hypersensitivity to tactile stimuli, as measured by the von Frey test (panel A; cutoff: 6 g) and thermal stimuli, as measured by the cold plate test (panel B; cutoff: 30 s) at different time-points in naive (black dotted line) and CCI-subjected (red line) mice.
Changes in mRNA and Protein Levels of MOP, DOP, and NK1 Receptors, and in Substance P Level in the Lumbar Spinal Cord of Mice at Day 7 after CCI
To investigate biochemical changes that parallel behavioral hypersensitivity and clarify the causes of the inefficacy of opioids to treat neuropathic pain, a quantification of the mu-opioid (MOP), delta-opioid (DOP) neurokinin 1 (NK1) receptors and substance P mRNA abundance and their protein level was performed using qRT-PCR and Western Blot methods in the lumbar part of the spinal cord of mice, on the seventh day after CCI.
The qRT-PCR results on day 7 following CCI revealed no changes in MOP and DOP receptor mRNA level in the spinal cord. Whereas the protein expression level was also not altered in the case of the MOP receptor, DOP receptor protein (p < 0.05 vs control) levels increased significantly on the injury site. A slight increase on the contralateral side did not reach significance (Figure 2D). A decrease in the NK1 receptor mRNA level in the lumbar spinal cord, on the ipsilateral (p < 0.001 vs control) and contralateral (p < 0.05 vs control) side was observed. However, the protein level of NK1 receptor was significantly diminished only on the ipsilateral side (p < 0.05 vs control) (Figure 2F). The level of substance P mRNA was increased on the ipsilateral (p < 0.01 vs control), but not the contralateral side, which was accompanied by a corresponding change in protein level (p < 0.05 vs control) (Figure 2G,H).
Figure 2.

The mRNA (left panel) and protein (right panel) level of MOP, DOP and NK1 receptors and substance P in lumbar spinal cord of CCI-exposed mice, on the 7th day after surgical procedure. The data are presented as the mean ± SEM. Intergroup differences were analyzed using ANOVA followed by Bonferroni’s multiple comparison tests. *p < 0.05; **p < 0.01; ***p < 0.001 indicate significant differences compared with naive mice.
The injury-induced development of neuropathic pain is clearly accompanied by changes in mRNA and protein levels of both the opioid and neurokinin systems, as measured on day 7 after injury. It should be noted that there is also a report demonstrating an increase of SP-encoding gene TAC1 mRNA level in rats at day 3 after injury in the dorsal root ganglia (DRG), without changes in the spinal cord.12 The changes demonstrated in our experiments on mice support the pronociceptive activity of substance P. The increase in levels of both mRNA and especially functionally active substance P on the injury side is balanced by a lower expression of the NK1 receptor, seen as a lower level of the mRNA on both sides and of protein on the lesion side. This indicates an increased activity of the pronociceptive system in the development of injury-induced neuropathic pain. In contrast, there is a significant increase of the DOP receptor protein on the ipsilateral side after damage. Moreover, neither mRNA nor the protein of MOP receptor changes at the spinal cord level at day 7 after sciatic nerve injury. Obara et al. did not observe any changes in mRNA of MOP in the spinal cord of CCI-rats at days 3 and 14 after injury.13 However, several lines of evidence have shown a reduction of MOP receptor at the spinal cord level after nerve injury in rats and mice.14,15 The observed discrepancies may result from the different methods of spinal cord dissection used to collect the lumbar part for biochemical analysis. In the mentioned work of Obara,13 no significant changes in mRNA of DOP receptor (3 and 14 days after CCI) were observed, which is in line with data presented here for day 7. Nonetheless, this report is the first to show that protein levels of DOP receptor are elevated in the ipsilateral side of the spinal cord of CCI-mice 7 days after injury. In 2007, Kabli and Cahill observed an upregulation of the DOP protein level in DRG 14 days after sciatic nerve injury.16 It was suggested that this increase was correlated with DOP trafficking.
Our results support the hypothesis of altered NK1 system activation under neuropathic pain conditions, which together with no changes in MOP receptor activity, increases the importance of the pronociceptive system. Altogether, these findings provide the rationale for the development of bifunctional opioid compounds containing an NK1 system-targeting component as drug candidates for neuropathic pain treatment.
Compound Design and Synthesis
As a starting point for the current study, bifunctional hybrids 3 (SBCHM1, H-Dmt-D-Arg-Aba-Gly-NMe-3′,5′-(CF3)2-Bn) and 4 (KGCHM2, H-Dmt-D-Arg-Aba-β-Ala-NMeBn), which are a combination of an opioid agonist pharmacophore and NK1 receptor antagonist pharmacophore to attenuate pronociceptive action, were selected.1,2 Both compounds have a N-methylated amide group in the C-terminal NK1 pharmacophore, which is essential for affinity and antagonism at the NK1 receptor.1 Whereas secondary amide groups adopt exclusively a trans conformation, tertiary amides exist as a mixture of cis and trans isomers with a reduced activation energy for rotation.17 Since a large percentage of cis isomers (18–22%) was found in an earlier study of NK1-ligands, this configuration might be important for activity.18,19 Hence, the introduction of N-alkyl groups larger than methyl may result in higher populations of the cis amide bond. We previously showed that the introduction of an N-isobutyl group resulted in a loss of affinity at the NK1 receptor.2 Therefore, the smaller cyclopropyl group was evaluated in an attempt to avoid unfavorable steric interactions with the NK1R (compounds 6 to 8, RCCHM1/2/8, Table 1). Additionally, a conformational constraint was introduced by using the isoindoline amide (compounds 9 and 10; RCCHM4/5). This leads to identical cis and trans isomers and reduces the number of rotatable bonds, which could improve membrane permeability.20 Aside from the bis(trifluoromethyl)-benzyl substituent, the ortho-methoxybenzyl group often occurs in small molecule NK1 receptor antagonists,21 and hence, this substituent was investigated as well. Finally, because NK1 receptor antagonists have been shown to mostly possess two key aromatic systems, which are in a stacked “L” or “T” configuration,21 we investigated whether a change from the C-terminal benzyl amide to naphtalen-2-yl-methyl amide could improve stacking of the benzene ring within the amino-benzazepinone (Aba) scaffold with the larger naphthalene ring (cf. compounds 12 and 13).
The syntheses of the opioid tetrapeptide Dmt-D-Arg-Aba-Gly-NH2 1 (AN81) and the reference hybrid 3 (SBCHM1) were reported previously.1 For the preparation of hybrids 6–13, protected tetrapeptide analogues Boc-Dmt-D-Arg(Pbf)-Aba-Gly-OH and Boc-Dmt-D-Arg(Pbf)-Aba-β-Ala-OH were assembled as reported by a standard solid phase method.2 Next, the secondary amines were coupled to the two Boc-protected tetrapeptide analogues using DIC/HOBt. These secondary amines were prepared either by reductive amination of cyclopropylamine with benzaldehyde, using sodium cyanoborohydride, or by alkylation of cyclopropylamine with 2′,5′-bis(trifluoromethyl)benzyl chloride, or of methylamine with 2-(bromomethyl)naphtalene. The isoindoline amine was commercially available.
Binding and in Vitro Activity at the MOP, DOP, and NK1 Receptors
All compounds were evaluated for their affinity and in vitro activity at the MOP and DOP receptors, and for their affinity and antagonist activity (except for the parent opioid 1 and 2 compounds) at the NK1 receptor. The results are summarized in Table 2.
Table 2.
Binding and in Vitro Activity at the Opioid and NK1 Receptorsa
| compd no. | structure | NK1Rb | NK1R | GPId | MVDd | MOPe | DOPe |
|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
||
| Ki (nM) | pA2c | IC50 (nM) | IC50 (nM) | Ki (nM) | Ki (nM) | ||
| 1f | H-Dmt-D-Arg-Aba-Gly-NH2 | nd | nd | 0.32 | 0.42 | 0.15 | 0.60 |
| 2g | H-Dmt-D-Arg-Aba-β-Ala-NH2 | nd | nd | 0.80 | 0.24 | 1.34 | 17.0 |
| 3f | H-Dmt-D-Arg-Aba-Gly-NMe-3′,5′-(CF3)2-Bn | 0.5 | 7.80 | 8.51 | 43.3 | 0.42 | 10.4 |
| 4g | H-Dmt-D-Arg-Aba-β-Ala-NMe-Bn | 13.0 | 6.44 | 1.86 | 2.16 | 0.08 | 0.28 |
| 5g | H-Dmt-D-Arg-Aba-Gly-NMe-Bn | 1530 | nd | 5.97 | 8.64 | 0.09 | 2.35 |
| 6 | H-Dmt-D-Arg-Aba-β-Ala-NcPr-3′,5′-(CF3)2-Bn | 735 | 5.36 | 18 | 16 | 47 | 190 |
| 7 | H-Dmt-D-Arg-Aba-β-Ala-NcPr-Bn | 520 | 5.20 | 19 | 6.6 | 2.7 | 66 |
| 8 | H-Dmt-D-Arg-Aba-Gly-NcPr-Bn | 95 | 5.50 | 10 | 11.2 | 0.30 | 68 |
| 9 | H-Dmt-D-Arg-Aba-β-Ala-isoindoline | >10 000 | 0.25 | 5 | 2.1 | 0.20 | 16 |
| 10 | H-Dmt-D-Arg-Aba-Gly-isoindoline | 3495 | 3.16 | 7.3 | 8.0 | 0.06 | 81 |
| 11 | H-Dmt-D-Arg-Aba-Gly-NMe-2′-OMe-Bn | 501 | 4.84 | 5.0 | 2.4 | 0.90 | 26 |
| 12 | H-Dmt-D-Arg-Aba-β-Ala-NMe-2′-methylenenaphthyl | 612 | 7.01 | 9.0 | 4.0 | 0.40 | 44 |
| 13 | H-Dmt-D-Arg-Aba-Gly-NMe-2′-methylenenaphthyl | 709 | 5.44 | 3.0 | 10.0 | 2.8 | 228 |
Results of the compounds selected for in vivo testing are shown in bold. nd: not determined.
Binding affinities of compounds for hNK1 receptors were determined by displacement of [3H]substance P from binding sites in hNK1 receptors expressed in CHO cells.
The pA2 values were calculated using Schild’s equation.22
The GPI functional assay is representative of MOP receptor activation, whereas the MVD is a DOP receptor-representative assay.
Binding affinities of compounds for MOP and DOP receptors were determined by displacement of [3H]DAMGO ([D-Ala2,NMePhe4,Gly-ol5]enkephalin) and [3H]DSLET ([D-Ser2,Leu5]enkephalin-Thr6), respectively, from rat brain membrane binding sites.
Data from Ballet et al.1
Data from Guillemyn et al.2
Our previous studies showed that when Gly4 (n = 1) in 3 was substituted by a β-alanine residue (n = 2) and the bis-trifluoromethyl substituents were removed to give 4, binding affinities to MOP and DOP receptors improved. Also, the activity in the GPI and MVD assays (representative of MOP and DOP receptor agonism, respectively) improved in 4, but affinity for the NK1 receptor and antagonism at the NK1 receptor was reduced (Ki shifted from 0.5 to 13 nM and pA2 shifted from 7.80 to 6.44).2 Moreover, we had demonstrated that the replacement of the N-methyl (R2) substituent by a bulky isobutyl substituent in either 3 or 4 resulted in a dramatic loss in NK1 receptor affinity.2 Here, we observe that replacement of the N-methyl in the β-Ala-containing 4 by a N-cyclopropyl group, which is smaller than isobutyl, unfortunately also resulted in reduced binding and activity at the opioid receptors, especially at DOP in 6 and 7. Concomitantly, binding affinity and antagonism at the NK1 receptor substantially dropped. The same substitution in the Gly-containing analogue 52 gave 8 with somewhat reduced MOP receptor affinity (Ki = 0.3 versus 0.09 nM), but especially reduced DOP receptor affinity (Ki = 68 versus 2.35 nM) and increased, but still rather low NK1 affinity (Ki 95 versus 1530 nM) with a very modest pA2 value of 5.50. Additionally, the N-methyl-benzyl substituent in 4 was replaced by an isoindoline group. This modification, leading to 9, results in identical cis/trans isomers and limits the amount of rotatable bonds (nRot reduced by two units),20 which could potentially improve binding and absorption. Surprisingly, it resulted in a detrimental drop in NK1 antagonism (pA2: 6.44 to 0.25), while retaining good activity and affinity at the opioid receptors. The glycine equivalent, 10, similarly has strongly reduced NK1 receptor affinity and antagonism, but importantly it yielded a ligand with a binding affinity for MOP receptor of 60 pM, while DOP receptor affinity was lower than that of 9 (Ki of 81 vs 16 nM). Addition of a methoxy-group in ortho-position of the N-benzyl moiety in 11 did not result in good affinity and antagonism at the NK1 receptor, but again good activity and affinity was observed at the opioid receptors. As mentioned above, the insertion of larger aromatic groups might lead to increased aromatic stacking to provide the relative “L” or ‘T” orientations of these groups, which was reported to be required for potent antagonism at the NK1 receptor.21 The NK1 receptor antagonism of 12, containing a naphthyl instead of a phenyl substituent, was in line with this hypothesis. Comparison of the N-methylbenzyl 4 with the naphthyl derivative 12 indeed showed an increased pA2 (from 6.44 to 7.01), despite a lower NK1 receptor affinity, while retaining low nanomolar activity at the opioid receptors. However, the Gly4 analogue 13 displayed a 2 orders of magnitude lower NK1 receptor antagonism (pA2 of 7.01 vs 5.44). It can be concluded that the nature of the N-substituent in these hybrids is crucial for the NK1 receptor, whereas the opioid receptors are much more tolerant to changes.
Based on these results, the naphthyl substituted analogues 12 and 13 were selected for further in vivo evaluation. Their properties were compared to the reference opioid parent 1 and hybrid 3.
Behavioral Study
Effect of the Selected Hybrids and Their Parent Compounds on Motor Performance after i.t. Administration in Naive Mice
A dose of 1 μg/5 μL per animal (i.e., ca. 1 nmol of hybrids 3, 12, and 13, and 1.3 nmol for opioids 1 and 2) was chosen to assess its influence on motor performance of naive mice in the RotaRod test (Figure 3). Vehicle, hybrids 3, 12, and 13, and their reference opioid compounds 1 and 2 were administered intrathecally; after 30 min, motor performance was assessed by the RotaRod test. None of the hybrids influenced motor performance of naive mice. In contrast, reference opioid compounds 1 and 2 significantly hampered motor performance at the dose of ca. 1.3 nmol.
Figure 3.
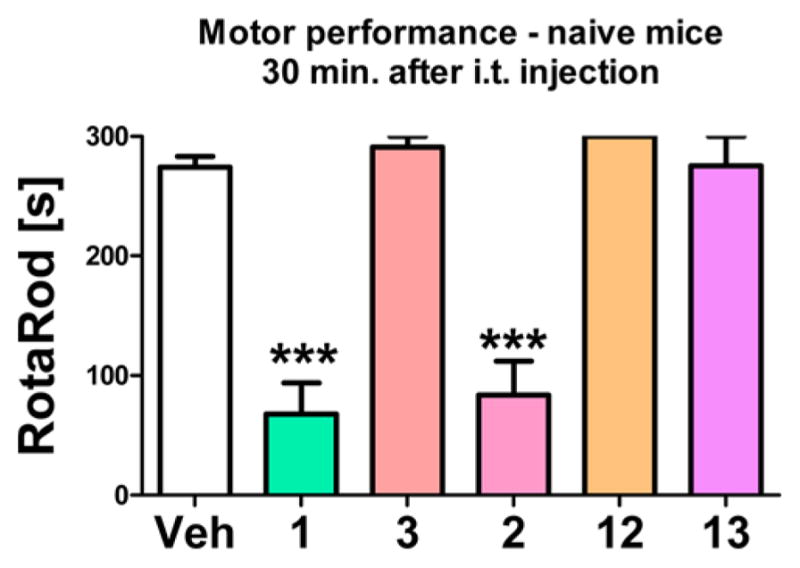
Effect of intrathecal (i.t.) administration of ca. 1–1.3 nmol of reference opioid compound 1 and its hybrid 3 and reference opioid compound 2 and its hybrids 12 and 13 on motor performance, as compared to Vehicle (Veh)-treated group, measured 30 min after administration by the RotaRod test in naive mice.
The Antinociceptive Effect of Selected Hybrids and Their Parent Opioid Compounds after i.t. Administration in Naïve Mice
The effect of hybrid 3 and its reference opioid 1, and hybrids 12 and 13, and their reference opioid 2 on the pain threshold to a thermal stimulus was investigated in naive mice after administration to the spinal cord. Opioid agonist 1 was able to raise the latency of the response to thermal stimuli (as measured by the tail-flick test) at a dose as low as 0.0013 nmol. At the highest dose tested (1.3 nmol), it provided a robust analgesic effect (close to 100% MPE) lasting up to 180 min after administration (Figure 4A left panel). In contrast, the effect of hybrid 3 proved to be weaker than its parent compound (Figure 4B left panel).
Figure 4.

Effect of intrathecal (i.t.) administration of the reference opioid 1 and its hybrid 3 (A+F, B+G), and reference opioid 2 and its hybrids 12 and 13 (C+H, D+I, E+J) on pain threshold as measured by the tail-flick test in naive mice (left panel), and hypersensitivity as measured by the von Frey and cold plate tests in CCI-exposed mice (right panel). The hypersensitivity tests were performed on mice (6–8 animals per group) between the 7th and 14th day after chronic constriction injury (CCI). Intergroup differences were analyzed by ANOVA with Bonferroni’s multiple comparison post hoc test. The results are presented as percentages of the maximal possible effect (%MPE). The horizontal line in the figure separates parents and hybrid compounds including Gly4 vs β-Ala4 residues. The doses are presented in nmol/5 μL/animal. *p < 0.05, **p < 0.01, ***p < 0.001 vs vehicle-treated group (Veh).
Opioid agonist 2 also gave longer-lasting analgesia at doses 0.1–1 nmol, though the effect provided by the highest 1 nmol dose tested was no greater than 80% MPE (Figure 4C left panel). The two hybrids 12 and 13 gave long-lasting analgesic effects in acute pain, yet weaker responses were noticed (60–70% MPE) in spite of raising the doses up to ca. 10 nmol (Figure 4D,E left panel). This is in line with the weaker agonism at the opioid receptors of the hybrids, compared to 1. Among all hybrids, 13 was the one that provided the longest lasting effect (p < 0.001 vs vehicle at 180 min after administration), even though the effect did not rise above 50% MPE. (Figure 4E left panel).
The Antinociceptive Effect of Selected Hybrids and Their Parent Compounds after i.t. Administration in CCI-Subjected Mice
All investigated compounds influenced the response latency in the neuropathic pain model. As expected, the opioid structures 1 and 2 demonstrated a limited efficacy in both tests (Figure 4F,H right panel). Reference opioid 1 raised the response threshold in both tests, yet a weak analgesic effect was observable: the response was not higher than 30% MPE in spite of significant dose increments in the cold plate test, and not higher than 40% MPE in the von Frey test (Figure 4F right panel). In the case of compound 2, which is the reference opioid for hybrids 12 and 13, no influence on the response threshold in the von Frey test was observed at all. In addition, its analgesic action in the cold plate test was seriously limited, no more than 20% MPE (Figure 4H right panel).
Hybrid 3 was the weakest analgesic of the examined hybrids, but it was still able to raise the response latency to ca. 60% MPE (Figure 4G right panel). Hybrid 13 proved to be the most efficient analgesic in both tests. Application of the lowest tested dose (i.e., 0.1 nmol) raised the response threshold to ca. 70% MPE in the von Frey test. This dose did not significantly influence the response in the cold plate test, but raising the dose to 10 nmol resulted in a robust analgesic effect (close to 100% MPE) for both tactile and thermal stimuli (Figure 4J right panel). The effect of hybrid 12 was weaker in both tests (Figure 4I right panel).
Nonetheless, hybrid 3 was the only compound (p < 0.05) showing activity up to 180 min in the von Frey test when administered at the moderate dose of approximately 1 nmol (Figure 4G right panel). Hybrids 12 and 13, at the equivalent moderate dose, provided a shorter lasting effect, not observable 180 min after administration (Figure 4I,J right panel). Of note, 13 was the only compound that provided an effect at 180 min after administration in the cold plate test (Figure 4J, right panel), but only at the highest dose of 10 nmol.
We then examined the effects of opioid parents and hybrid compounds in an acute pain model in naive mice. In contrast to the neuropathic pain results, the opioid parent structure 1 (AN81) induced a superb analgesic effect in the acute pain model. It was efficacious at doses as low as 0.001 nmol/5 μL/animal (p < 0.05 vs vehicle) and it provided almost 100% MPE at the dose of 1 μg. The effect of this dose was still very robust (>80% MPE) at 180 min after i.t. administration. The opioid parent structure 2 demonstrated similar properties. However, both of the reference opioids demonstrated significantly hampered action in the neuropathic pain model: 1 exhibited a modest effect in the von Frey test (ca. 40% MPE at 30 min), where 2 was not efficacious at all, and showed a poor effect in the cold plate test (<30% MPE). This effect was no longer present 30 min after administration. This indicates that both 1 and 2 lose their efficacy under neuropathic pain conditions, which stands in line with the results of previous experiments on rats,2 as well as with the well-documented phenomenon of limited analgesic activity of opioids under neuropathic pain conditions.23–26 All hybrid compounds provided a lowered analgesic effect (<70% MPE) in the tail-flick test up to 180 min after administration, as compared to the parent opioids. Compound 3 was the strongest analgesic among all hybrids at the highest dose 9.5 nmol/5 μL/animal (p < 0.001 at 30 min after administration). On the other hand, hybrids 12 and 13 were able to provide an analgesic effect at the relatively low dose 0.1 nmol up to 180 min after administration. In contrast, 3 provided the weakest effect in the neuropathic pain model, as compared to 12 and 13. At the highest dose 9.5 nmol, hybrid 3 was more potent in the cold plate test than in the von Frey test 30 min after administration. In the von Frey test, 3 was the only compound that induced an analgesic effect at the dose 1 μg (ca. 1 nmol) up to 180 min after administration.
Summing up, the addition of the NK1 receptor antagonist pharmacophore to the opioid sequence counteracted the loss of efficacy in a neuropathic pain model, as seen by the differences in opioid parent structure activity in naive and neuropathic mice (Figure 4A, C). It is very interesting that hybrid 3 also loses its effectiveness, although its effect at a short time after administration is close to that in naive mice. In contrast to hybrid 3, two hybrids of the novel series (i.e., compounds 12 and 13) exhibit stronger effects in neuropathic pain than in naive mice. Paradoxically, these two hybrids have a much weaker binding to the NK1 receptor compared to 3, with similar binding to the opioid receptors. It can be assumed that spinal NK1 receptors are less significant for that effect than receptors located in other structures of the pain pathway, but this would require further investigation.
Taking into consideration all behavioral results, 13 seems to exhibit the most beneficial pharmacological profile as an analgesic drug: it provides a moderate (~60% MPE) effect in attenuating acute pain, as measured at three time points after administration. In addition, its analgesic effect in CCI-exposed mice is highly efficacious. It produced a robust response in both the von Frey and cold plate tests, with the effect of the highest dose (10 nmol/5 μL/animal) close to 100% MPE in both tests.
As observed before, the best analgesic compounds have high MOP/DOP receptor agonist potency and efficacy, but to be an effective analgesic for neuropathic pain it must be combined with an NK1 receptor antagonist. In contrast to the MOP/DOP potency and efficacy, NK1 potency and efficacy does not necessarily need to be very high.2,27 The same applies to motor performance, wherein the hybrids do not show impaired motor performance as clearly manifested for the parent compounds. It seems that for both these opioid effects the NK1 component is important, but not the strength of its (in vitro) antagonism. This is important information when searching for new analgesics to be used for neuropathic pain treatments.
CONCLUSIONS
In the present study we demonstrated the analgesic effect of bifunctional hybrids, both in naive mice and in a neuropathic pain mouse model. Our results support the concept of altered NK1 system activation under neuropathic pain conditions which, in parallel with no changes in MOP receptor mRNA level, increases the importance of the NK1 pronociceptive system. These findings justify the strategy to use opioid compounds, containing an NK1 system-targeting component.
We have studied the influence of the N-terminal amide substituents in opioid agonist–NK1 receptor antagonist hybrids. Whereas these substituents do not dramatically influence the affinity and agonist activity at the MOP and DOP receptors, their influence on NK1 receptor affinity and antagonism is pronounced. The isoindoline-substituted analogues 9 and 10 are very potent MOP/DOP receptor agonists, but their activity at the NK1 receptor is abolished. The NMe-2-methylenenaphthyl analogues 12 and 13 have the best balance of opioid agonism/NK1 receptor antagonism as determined in the in vitro study.
As a consequence, we assessed the analgesic effects of three distinct opioid agonist–NK1 receptor antagonist hybrid compounds 12, 13, and 3, after intrathecal (i.t.) administration in mice in an acute and neuropathic pain model. Each of the tested compounds demonstrates unique analgesic properties, as determined in the different tests. Two hybrids of the novel series (i.e., compounds 12 and 13) exhibit stronger effects in the neuropathic pain models, as compared to an antinociceptive assessment in naive mice. Paradoxically, these two hybrids have a much weaker NK1 receptor binding affinity as compared to the initial lead structure 3, with similar binding affinities at the opioid receptors. It can be suggested that spinal NK1 receptors are less significant for that effect than receptors located in other structures of the pain pathway, but this would require further investigation. As previously demonstrated, the combination of a MOP/DOP agonist with a NK1 antagonist in a bifunctional compound can delay or suppress analgesic tolerance,28,29 and transport an otherwise nonpermeable NK1 antagonist into the brain.2 In addition, the present results demonstrate that weak NK1 antagonism is sufficient for potent analgesia in neuropathic pain models. This avoids the use of high doses of a NK1 antagonist alone, which has been shown to induce neurotoxicity in rats.27
In brief, the addition of the NK1 receptor antagonist pharmacophore to the opioid sequence counteracted the loss of efficacy in a neuropathic pain model, as seen in differences in opioid parent structure activity in naive and neuropathic mice. Altogether, these findings justify a continued search for dual opioid agonists-NK1 receptor antagonists, which in our hands emerged in the discovery of compound 13, a compound which is efficacious in both acute and neuropathic pain models in mice.
METHODS
General
Thin-layer chromatography (TLC) was performed on glass plates precoated with silica gel 60F254 (Merck, Darmstadt, Germany) using the mentioned solvent systems. Purification of organic molecules was done with flash chromatography (Davisil LC60A, 40–63 μm). Mass spectrometry (MS) was performed on a Micromass Q-Tof Micro spectrometer with electrospray ionization (ESI). Data collection and spectrum analysis was done with Masslynx software. Analytical RP-HPLC was performed using a Waters 717plus autosampler, a Waters 1525 binary HPLC pump and a Waters 2487 dual absorbance wavelength detector (Milford, MA), on a Grace (Deerfield, IL) Vydac RP C18 column (25 cm × 4.6 mm × 5 μm) using UV detection at 215 nm. The mobile phase was a mixture of water and acetonitrile both containing 0.1% TFA. The used gradient runs from 3 to 100% acetonitrile in 20 min at a flow rate of 1 mL/min. Preparative RP-HPLC purification was done on a Gilson (Middleton, WI) HPLC system with Gilson 322 pumps, controlled by the software package Unipoint, and a reversed phase C18 column (DiscoveryBIO SUPELCO Wide Pore C18 column, 25 cm × 2.21 cm, 5 μm) using a gradient that increased by 1%/min of acetonitrile in water (both containing 0.1% TFA) until the product eluted. After purification, the purity of all compounds was evaluated as being more than 95% by analytical RP-HPLC. All fractions were lyophilized using a Flexy-Dry lyophilizer (FTS Systems, Warminster, PA).
Peptide Synthesis
The protected tetrapeptides Boc-Dmt-D-Arg(Pbf)-Aba-Gly-OH and Boc-Dmt-D-Arg(Pbf)-Aba-β-Ala-OH were synthesized by Nα-Fmoc solid phase methodology on 2-chlorotrityl resin, including the construction of the Aba-constraint on solid phase by use of phthaloyl protected ortho-formyl phenylalanine, as described in detail previously.2 After cleavage of the fully protected peptide acid, it was coupled to the corresponding benzyl amines in solution using DIC/HOBt. Final deprotection was performed with the mixture TFA/TES/H2O (95/2.5/2.5) for 3 h. The peptides were purified by preparative HPLC and characterized by high resolution mass spectrometry (see the Supporting Information). Purity was >95% as determined by analytical HPLC.
Functional NK1 Receptor Assay30
Cell Line and Culture Conditions, Aequorin Charging Protocol
The Chinese hamster ovary K1 (CHO-K1) cell line, stably expressing human NK1 receptor, was transfected with an apoaequorin expression vector (pER2) using Fugene6 (Roche Applied Science). Transfected cells in the mid-log phase were loaded with coelenterazine and diluted 10-fold as described in detail elsewhere.2
Aequorin Luminescence Assay
A dilution series of peptide agonist (substance P, Sigma) ranging from 10−11 to 10−4 M was distributed in a white 96-well plate, after which the synthetic compounds were added to these wells to obtain the desired concentrations (ranging from 10−8 to 10−4 M). A negative control sample (BSA medium only) was included in each row of the 96-well plate. Measurement started at the moment of injection of 50 μL of the coelenterazine-loaded cell suspension, containing 2.5 × 104 cells. Light emission was measured every second for 30s, after which 50 μL of 10 nM ATP solution (positive control) was injected.2
Data Analysis
Luminescence data (peak integration) were calculated as described previously2 using MikroWin 2000 software (Berthold). Statistical and curve-fitting analyses were performed using Prism 4.0 (GraphPad) software. Data were expressed in percentage (% RLU) of the maximal luminescence that was detected with 10−4 M SP (without antagonist). The competitive nature of antagonism was evaluated using the Schild plot method.22 All antagonists analyzed in this study provided linear regression plots and were considered competitive. The pA2 values were calculated using the Schild’s equation.22
hNK1-CHO Membrane Preparation for Binding Assay
The hNK1-CHO cells were cloned locally and grown in a humidified CO2 (5%) atmosphere at 37 °C in a Forma Scientific incubator (Thermo Forma, Series II Water Jacketed) in a Dulbecco’s DMEM-F12 50:50 medium (Corning Cellgro) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Life Technologies) and 1× PenStrep solution (Gibco, Life Technologies). Confluent cell monolayers were harvested with 1× Dulbecco’s PBS containing 5 mM EDTA. Cells were centrifuged in a Sorvall ST 16R centrifuge (ThermoScientific) at 1500 rpm for 3 min. The supernatants were aspirated and the remaining pellets were placed at −80 °C. On the day of the experiments, cell pellets were thawed and homogenized with a Teflon-glass tissue homogenizer in ice-cold 50 mMTris-HCl (pH 7.4) buffer containing 5 mM MgCl2. The membrane fractions were collected by centrifugation in a Sorvall RC 5C Plus (ThermoScientific) at 13 000 rpm for 25 min at 4 °C and the resulting pellets were resuspended in 50 mM Tris-HCl (pH 7.4), 5 mM MgCl2 buffer. Protein contents of the pellet suspensions were determined by the BCA method (Bio-Rad, Bio-Rad Laboratories).
[3H]Substance-P Competition Binding Assay
The test compounds were dissolved in DMSO and stored in 10 mM aliquots at −20 °C. Serial dilutions of the compounds were prepared in the binding buffer (50 mMTris-HCl, pH 7.4, 5 mM MgCl2). Serial dilutions of the competitor ligands were coincubated with 45 μg of membrane protein and 0.5–0.8 nM [3H]Substance-P (36.5 Ci/mmol, PerkinElmer) in a final volume of 200 μL at RT for 1 h. Total binding was determined in the absence of competitors while nonspecific binding was defined in the presence of 10 μM Substance-P (final concentration). Substance-P (Tocris, Tocris Bioscience, UK) was also used as a control to validate the binding assay conditions. The reactions were terminated by rapid filtration onto UniFilter GF/B 96-well plates (PerkinElmer), previously presoaked in cold 1% polyethylenimine solution (Sigma-Aldrich). Plates were washed three times with cold water and then dried at RT for overnight. Filter bound radioactivity was measured with a MicroBeta 2450 microplate reader (PerkinElmer) using a microscintillation cocktail (PerkinElmer).
Data Analysis
Experiments were carried out in duplicate in 96-well plates, and are the average of at least three independent experiments. The IC50 and Ki values for each test compound were calculated by nonlinear regression, and standard error of means (SEM) is represented. Data analyses were performed via GraphPad Prism4 software (Graph-Pad, San Diego, CA). The Ki was calculated using the previously determined Kd of the [3H]Substance-P in this cell line by saturation binding.
Functional GPI and MVD Assays
The guinea pig ileum (GPI)31 and mouse vas deferens (MVD)32 bioassays were carried out as described in detail elsewhere.33,34 A dose–response curve was determined with [Leu5]enkephalin as standard for each ileum and vas preparation and IC50 values of the compounds being tested were normalized according to a published procedure.35
Opioid Receptor Binding Assays
Opioid receptor binding studies were performed as described in detail elsewhere.33 Binding affinities for MOP and DOP receptors were determined by displacing, respectively, [3H]DAMGO (Multiple Peptide Systems, San Diego, CA) and [3H]DSLET (Multiple Peptide Systems) from rat brain membrane binding sites, and κ opioid receptor binding affinities were measured by displacement of [3H]U69,593 (Amersham) from guinea pig brain membrane binding sites. Incubations were performed for 2 h at 0 °C with [3H]DAMGO and [3H]DSLET at respective concentrations of 0.72 and 0.78 nM. IC50 values were determined from log-dose displacement curves, and Ki values were calculated from the IC50 values by means of the equation of Cheng and Prusoff,36 using values of 1.3 and 2.6 nM for the dissociation constants of [3H]DAMGO and [3H]-DSLET, respectively.
Animals
Male Albino Swiss CD-1 IGS mice (30–35 g) obtained from Charles River Breeding Laboratories, Germany, were housed 10 mice per cage (lined with sawdust) under a standard 12/12 h light/dark cycle (lights on at 6:00 am) with food and water available ad libitum. All experiments were performed according to the recommendations of IASP, the NIH Guide for Care and Use of Laboratory Animals, and were approved (1214/2015) by the local Bioethics Committee (Cracow, Poland).
Neuropathic Pain Model
Chronic constriction injury (CCI) model was performed according to Bennet and Xie37 and used in our laboratory.38,39 The surgical procedure was performed under isoflurane anesthesia. An incision was made below the mouse’s right hipbone, and the sciatic nerve was exposed. Three ligatures with 4/0 silk thread were made around the nerve distal to the sciatic notch with 1 mm spacing, until a brief twitch in the respective hind limb was observed. Mice were tested for the presence of characteristics for neuropathy. All CCI mice developed allodynia and hyperalgesia. The behavioral experiments were conducted on the seventh to 14th day following the CCI surgical procedure.
Biochemical Analysis
qRT-PCR Analysis of Gene Expression
Spinal cord tissue for biochemical analysis was collected on the seventh day after the CCI. Lumbar (L4–L6) fragments of the spinal cord were removed immediately after the mice were decapitated, and both the ipsilateral and contralateral parts were dissected. The tissue samples were placed in individual tubes containing the tissue storage reagent RNAlater (Qiangen Inc.) and stored at −70 °C for RNA isolation. Total RNA was extracted using TRIzol reagent (Invitrogen), as previously described. RNA concentrations and quality were measured using a NanoDropND-1000 spectrometer (NanoDrop Technologies). Reverse transcription was performed on 1 μg of total RNA using Omniscript reverse transcriptase (Quiagen Inc.) at 37 °C for 60 min. Reverse transcription (RT) reactions were carried out in the presence of an RNase inhibitor (RNasin, promega) and an oligo (dT16) primer (Quiagen Inc.). The cDNA was diluted 1:10 with water, and for each reaction, ca. 50 ng of cDNA synthesized from the total RNA of an individual animal was used for the quantitative real-time PCR (qPCR) reaction. qPCR was performed using Assay-On-Demand TaqMan probes according to the manufacturer’s protocol (Applied Biosystems), and the reactions were run on an iCycler device (BioRad, Hercules). The following TaqMan primers and probes were used: Mm00446-968_m1 (Hprt, hypoxanthine guanine phosphoribosyl transferase), Mm00436892_m1 (Tac1r, NK1 receptor), Mm01166996_m1 (Tac1, substance P), Mm01188089_m1 (Oprm1, MOP receptor), and Mm01180757_m1 (Oprd1, DOP receptor). The expression of Hprt was measured in the lumbar spinal cord of mice as measured on days 7 and 14 following CCI procedure and quantified to control for variations in cDNA amounts. Hprt did not significantly change for CCI-exposed mice and therefore served as an adequate housekeeping gene (data not shown). The cycle threshold values were calculated automatically by iCycler IQ 3.0 software using default parameters. The RNA abundance was calculated as 2−(threshold cycle).
Western Blot
The ipsilateral and contralateral dorsal parts of the spinal cord (L4–L6) from naive and CCI-exposed mice (7th day after injury) were collected in RIPA (Radio-Immunoprecipitation Assay) buffer with protease and phosphatase inhibitor cocktails (Sigma-Aldrich). Tissue lysates were cleared by centrifugation (14 000g for 30 min at 4 °C) and protein concentration in the supernatant was determined using the BCA Protein Assay Kit (Sigma-Aldrich). Samples containing 25 μg of protein were heated in a loading buffer (4× Laemmli Buffer, Bio-Rad) for 5 min at 98 °C and then were resolved on 4–15% Criterion TGX precast polyacrylamide gels (Bio-Rad, Poland). The proteins were transferred to Immune-Blot PVDF membranes (Bio-Rad, Poland) with semidry transfer (30 min, 25 V) and then the membranes were blocked for 1h at RT using 5% nonfat, dry milk (Bio-Rad) in Tris-buffered saline. Next, the membranes were washed in TBST (Tris-buffered saline with 0.1% Tween-20) and incubated overnight at 4 °C with the following primary antibodies: rabbit polyclonal NK1 receptor (Novus Biologicals) 1:500; MOP receptor (Abcam) 1:500, DOP receptor (LifeSpan) 1:500 and substance P (Santa Cruz) 1:250 or mouse polyclonal GAPDH (Millipore) 1:5000. The membranes were then incubated for 1 h in horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies at a dilution of 1:5000. All antibodies were diluted in the SignalBoost Immunoreaction Enhancer Kit (Merck Millipore Darmstadt). The membranes were washed four times for 5 min each with TBST. The Clarity Western ECL Substrate (Bio-Rad, Poland) was used to detect immunocomplexes, which were visualized using a Fujifilm LAS-4000 FluorImager system. The Fujifilm Multi Gauge software was used to quantify the relative levels of immunoreactivity. The immunoblots shown are representative of four to six individual samples.
Drug Administration
Three designed compounds containing both NK1 receptor antagonist and MOP and DOP receptor-agonist pharmacophores, 3 (SBCHM1), 12 (RCCHM3), and 13 (RCCHM6), and the reference compounds containing only pure opioid pharmacophore, 1 (AN81) and 3 (KGOP1), were used in the study. Drugs were dissolved in water for injections (Polpharma, Poland) and administered intrathecally (i.t.) in 5 μL dose volume through lumbar puncture between L5 and L6 vertebrae to nonanesthetized mice, according to the Hylden and Wilcox model40 described with modifications by Fairbanks.41 The i.t. injections were performed with disposable 30 gauge 1/2 in. needles (Becton Dickinson and Company, Rutherford, NJ) matched to a 25 μL syringe (Hamilton, Reno, NV). Behavioral tests were performed 30, 90, and 180 min after drugs administration.
Behavioral Tests
Von Frey Test
Mechanical sensitivity to non-noxious stimuli was measured by applying a set of calibrated nylon monofilaments (0.6–6 g; Stoelting), in serial increments on a tested hind paw plantar surface, until a behavioral response was observed. Responses considered as pain behavior included paw withdrawal, shaking, and licking.
Cold Plate Test
Sensitivity to noxious thermal stimuli was assessed with usage of Cold/Hot Plate Analgesia Meter, Columbus Instruments. The temperature of the plate was kept at 2 °C, and the cutoff latency was 30 s. The mice were placed on the cold plate, and the time until the hind paw was lifted was recorded.
Tail-Flick Test
The threshold of nociceptive pain to a thermal stimulus was assessed by tail-flick latency determined by a tail-flick analgesic meter (Analgesia Meter; Ugo Basile, Italy). In this test, the beam of light was focused on the dorsal tail surface 1 cm from the tip of the tail. The baseline was determined at 2.5–4.5 s. The cutoff time was 9 s.
RotaRod Test
The motor performance of naive mice was assessed by the RotaRod test (Mouse Rota-Rod, Ugo Basile, Italy). Mice were placed on a horizontal rod which was rotating at accelerating speed, starting with 2 rpm and reaching 40 rpm within 300 s. The main experiment was conducted after three training sessions, lasting 300 s each. The time was recorded until a mouse fell off the rod. RotaRod test was conducted 30 min after drug administration.
Supplementary Material
Acknowledgments
Funding
S.B. and D.T. thank the Research Foundation–Flanders for the financial support. The work of J.S., K.P.B., J.M., and B.P. was supported by the National Science Centre, Poland, via MAESTRO 2012/06/A/NZ4/00028 grant and statutory funds Institute of Pharmacology Polish Academy of Sciences; J.S. is a holder of a KNOW scholarship sponsored by the Ministry of Science and Higher Education, Poland. The work of PWS was supported by grants from the U.S. National Institute on Drug Abuse (NIH, DA004443) and the Canadian Institutes of Health Research (MOP-89716). Research performed by J.V.D. and J.V.B. was supported by the Interuniversity Attraction Poles (IAP) program (Belgian Science Policy Grant (P7/40).
ABBREVIATIONS
- Aba
4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one
- CCI
chronic constriction injury
- DIC
N,N′-diisopropylcarbodiimide
- DOP
delta opioid peptide
- Dmt
2′,6′-dimethyltyrosine
- GPI
guinea pig ileum
- HOBt
1-hydroxybenzotriazole
- MOP
mu opioid peptide
- mRNA
mRNA
- MPE
maximum possible effect
- MVD
mouse vas deferens
- NK1
neurokinin-1
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- qRT-PCR
quantitative real-time polymerase chain reaction
- GAPDH
glyceralde-hyde 3-phosphate dehydrogenase
Footnotes
Author Contributions
J.S., K.P.-B., J.M., and B.P. were in charge of performing and designing the biochemical and animal experiments. R.C., K.G., A.M., D.T., and S.B. have designed and prepared the hybrid opioid-NK1R ligands. N.N.C., C.L., and P.W.S. carried out and supervised the functional tissue assays (GPI and MVD). A.K., J.S., J.V.D., and J.V.B. have performed the NK1R binding and activity assays, respectively. D.T., P.W.S., S.B., and B.P. were in charge of writing and editing the manuscript.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.7b00226.
Chemistry procedures and compound characterization (PDF)
References
- 1.Ballet S, Feytens D, Buysse K, Chung NN, Lemieux C, Tumati S, Keresztes A, Van Duppen J, Lai J, Varga E, et al. Design of novel neurokinin 1 receptor antagonists based on conformationally constrained aromatic amino acids and discovery of a potent chimeric opioid agonist-neurokinin 1 receptor antagonist. J Med Chem. 2011;54(7):2467–2476. doi: 10.1021/jm1016285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guillemyn K, Kleczkowska P, Lesniak A, Dyniewicz J, Van der Poorten O, Van den Eynde I, Keresztes A, Varga E, Lai J, Porreca F, et al. Synthesis and biological evaluation of compact, conformationally constrained bifunctional opioid agonist–neurokinin-1 antagonist peptidomimetics. Eur J Med Chem. 2015;92:64–77. doi: 10.1016/j.ejmech.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bouhassira D, Attal N. Translational neuropathic pain research: a clinical perspective. Neuroscience. 2016;338:27–35. doi: 10.1016/j.neuroscience.2016.03.029. [DOI] [PubMed] [Google Scholar]
- 4.Jackson KC. Pharmacotherapy for neuropathic pain. Pain practice. 2006;6(1):27–33. doi: 10.1111/j.1533-2500.2006.00055.x. [DOI] [PubMed] [Google Scholar]
- 5.Przewlocki R, Przewlocka B. Opioids in neuropathic pain. Curr Pharm Des. 2005;11(23):3013–3025. doi: 10.2174/1381612054865055. [DOI] [PubMed] [Google Scholar]
- 6.Vanderah TW. Pathophysiology of pain. Med Clin North Am. 2007;91(1):1–12. doi: 10.1016/j.mcna.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Kleczkowska P, Lipkowski A, Tourwe D, Ballet S. Hybrid opioid/non-opioid ligands in pain research. Curr Pharm Des. 2013;19(42):7435–7450. doi: 10.2174/138161281942140105165646. [DOI] [PubMed] [Google Scholar]
- 8.Schiller PW. Bi-or multifunctional opioid peptide drugs. Life Sci. 2010;86(15):598–603. doi: 10.1016/j.lfs.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dvoracsko S, Stefanucci A, Novellino E, Mollica A. The design of multitarget ligands for chronic and neuropathic pain. Future Med Chem. 2015;7(18):2469–2483. doi: 10.4155/fmc.15.156. [DOI] [PubMed] [Google Scholar]
- 10.Labianca R, Sarzi-Puttini P, Zuccaro SM, Cherubino P, Vellucci R, Fornasari D. Adverse effects associated with non-opioid and opioid treatment in patients with chronic pain. Clin Drug Invest. 2012;32:53–63. doi: 10.2165/11630080-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Recio S, Gascón P. Biological and pharmacological aspects of the NK1-receptor. BioMed Res Int. 2015;2015:1. doi: 10.1155/2015/495704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsumura T, Sakai A, Nagano M, Sawada M, Suzuki H, Umino M, Suzuki H. Increase in hemokinin-1 mRNA in the spinal cord during the early phase of a neuropathic pain state. Br J Pharmacol. 2008;155(5):767–774. doi: 10.1038/bjp.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Obara I, Parkitna JR, Korostynski M, Makuch W, Kaminska D, Przewlocka B, Przewlocki R. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain. 2009;141(3):283–291. doi: 10.1016/j.pain.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 14.Popiolek-Barczyk K, Makuch W, Rojewska E, Pilat D, Mika J. Inhibition of intracellular signaling pathways NF-κB and MEK1/2 attenuates neuropathic pain development and enhances morphine analgesia. Pharmacol Rep. 2014;66(5):845–851. doi: 10.1016/j.pharep.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Zhou X-L, Yu L-N, Wang Y, Tang L-H, Peng Y-N, Cao J-L, Yan M. Increased methylation of the MOR gene proximal promoter in primary sensory neurons plays a crucial role in the decreased analgesic effect of opioids in neuropathic pain. Mol Pain. 2014;10(1):51. doi: 10.1186/1744-8069-10-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kabli N, Cahill CM. Anti-allodynic effects of peripheral delta opioid receptors in neuropathic pain. Pain. 2007;127(1):84–93. doi: 10.1016/j.pain.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 17.Pierson NA, Chen L, Russell DH, Clemmer DE. Cis–trans isomerizations of proline residues are key to bradykinin conformations. J Am Chem Soc. 2013;135(8):3186–3192. doi: 10.1021/ja3114505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saulitis J, Mierke DF, Byk G, Gilon C, Kessler H. Conformation of cyclic analogs of substance P: NMR and molecular dynamics in dimethyl sulfoxide. J Am Chem Soc. 1992;114(12):4818–4827. [Google Scholar]
- 19.Levian-Teitelbaum D, Kolodny N, Chorev M, Selinger Z, Gilon C. 1H-nmr studies of receptor-selective substance P analogues reveal distinct predominant conformations in DMSO-d6. Biopolymers. 1989;28(1):51–64. doi: 10.1002/bip.360280108. [DOI] [PubMed] [Google Scholar]
- 20.Veber DF, Johnson SR, Cheng H-Y, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45(12):2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- 21.Takeuchi Y, Shands EB, Beusen DD, Marshall GR. Derivation of a three-dimensional pharmacophore model of substance P antagonists bound to the neurokinin-1 receptor. J Med Chem. 1998;41(19):3609–3623. doi: 10.1021/jm9700171. [DOI] [PubMed] [Google Scholar]
- 22.Schild H. pA, a new scale for the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2(3):189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricardo Buenaventura M, Rajive Adlaka M, Nalini Sehgal M. Opioid complications and side effects. Pain Physician. 2008;11:S105–S120. [PubMed] [Google Scholar]
- 24.Eisenberg E, Suzan E. Drug combinations in the treatment of neuropathic pain. Curr Pain Headache Rep. 2014;18(12):1–8. doi: 10.1007/s11916-014-0463-y. [DOI] [PubMed] [Google Scholar]
- 25.Marion Lee M, Sanford Silverman M, Hans Hansen M, Vikram Patel M. A comprehensive review of opioid-induced hyperalgesia. Pain Physician. 2011;14:145–161. [PubMed] [Google Scholar]
- 26.McNicol ED, Midbari A, Eisenberg E. Opioids for neuropathic pain. Cochrane Library. 2013 doi: 10.1002/14651858.CD006146.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Misterek K, Maszczynska I, Dorociak A, Gumulka S, Carr D, Szyfelbein S, Lipkowski A. Spinal co-administration of peptide substance P antagonist increases antinociceptive effect of the opioid peptide biphalin. Life Sci. 1994;54(14):939–944. doi: 10.1016/0024-3205(94)00494-3. [DOI] [PubMed] [Google Scholar]
- 28.Largent-Milnes T, Yamamoto T, Nair P, Moulton J, Hruby V, Lai J, Porreca F, Vanderah T. Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. British journal of pharmacology. 2010;161(5):986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foran SE, Carr DB, Lipkowski AW, Maszczynska I, Marchand JE, Misicka A, Beinborn M, Kopin AS, Kream RM. A substance P-opioid chimeric peptide as a unique nontolerance-forming analgesic. Proc Natl Acad Sci U S A. 2000;97(13):7621–7626. doi: 10.1073/pnas.130181897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Janecka A, Poels J, Fichna J, Studzian K, Vanden Broeck J. Comparison of antagonist activity of spantide family at human neurokinin receptors measured by aequorin luminescence-based functional calcium assay. Regul Pept. 2005;131(1):23–28. doi: 10.1016/j.regpep.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Paton WD. The action of morphine and related substances on contraction and on acetylcholine output of coaxially stimulated guinea-pig ileum. Br J Pharmacol Chemother. 1957;12(1):119–127. doi: 10.1111/j.1476-5381.1957.tb01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Henderson G, Hughes J, Kosterlitz H. A new example of a morphine-sensitive neuro-effector junction: adrenergic transmission in the mouse vas deferens. Br J Pharmacol. 1972;46(4):764–766. doi: 10.1111/j.1476-5381.1972.tb06901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiller PW, Lipton A, Horrobin DF, Bodanszky M. Unsulfated C-terminal 7-peptide of cholecystokinin: a new ligand of the opiate receptor. Biochem Biophys Res Commun. 1978;85(4):1332–1338. doi: 10.1016/0006-291x(78)91149-x. [DOI] [PubMed] [Google Scholar]
- 34.DiMaio J, Nguyen TM, Lemieux C, Schiller PW. Synthesis and pharmacological characterization in vitro of cyclic enkephalin analogs: effect of conformational constraints on opiate receptor selectivity. J Med Chem. 1982;25(12):1432–1438. doi: 10.1021/jm00354a008. [DOI] [PubMed] [Google Scholar]
- 35.Waterfield AA, Leslie FM, Lord JA, Ling N, Kosterlitz HW. Opioid activities of fragments of β-endorphin and of ites leucine65-analogue. Comparison of the binding properties of methionine-and leucine-enkephalin. Eur J Pharmacol. 1979;58(1):11–18. doi: 10.1016/0014-2999(79)90334-0. [DOI] [PubMed] [Google Scholar]
- 36.Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 37.Bennett GJ, Xie Y-K. A peripheral mononeurop-athy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33(1):87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 38.Mika J, Osikowicz M, Rojewska E, Korostynski M, Wawrzczak-Bargiela A, Przewlocki R, Przewlocka B. Differential activation of spinal microglial and astroglial cells in a mouse model of peripheral neuropathic pain. Eur J Pharmacol. 2009;623(1):65–72. doi: 10.1016/j.ejphar.2009.09.030. [DOI] [PubMed] [Google Scholar]
- 39.Mika J, Wawrzczak-Bargiela A, Osikowicz M, Makuch W, Przewlocka B. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain, Behav, Immun. 2009;23(1):75–84. doi: 10.1016/j.bbi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 40.Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67(2–3):313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 41.Fairbanks CA. Spinal delivery of analgesics in experimental models of pain and analgesia. Adv Drug Delivery Rev. 2003;55(8):1007–1041. doi: 10.1016/s0169-409x(03)00101-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.