

Abstract
Background
Distinguishing alveolar rhabdomyosarcoma (ARMS) from embryonal rhabdomyosarcoma (ERMS) has historically been of prognostic and therapeutic importance. However, classification has been complicated by shifting histologic criteria required for an ARMS diagnosis. Children’s Oncology Group (COG) studies after IRS-IV, which included the height of this diagnostic shift, showed both an increased number of ARMS and an increase in the proportion of fusion-negative ARMS. Following diagnostic standardization and histologic re-review of ARMS cases enrolled during this era, analysis of low-risk (D9602) and intermediate-risk (D9803) rhabdomyosarcoma (RMS) studies showed that fusion status rather than histology best predicts prognosis for patients with RMS. This analysis remains to be completed for patients with high-risk RMS.
Procedure
We re-reviewed cases on high-risk COG studies D9802 and ARST0431 with an enrollment diagnosis of ARMS. We compared the event-free survival (EFS) and overall survival by histology, PAX-FOXO1 fusion, and clinical risk factors (Oberlin score) for patients with metastatic RMS using the log-rank test.
Results
Histology re-review resulted in reclassification as ERMS for 12% of D9802 cases and 5% of ARST0431 cases. Fusion-negative RMS had a superior EFS to fusion-positive RMS; however, poorer outcome for metastatic RMS was most related to clinical risk factors including age, primary site, and number of metastatic sites.
Conclusions
In contrast to low- or intermediate-risk RMS, in metastatic RMS, clinical risk factors have the most impact on patient outcome. PAX-FOXO1 fusion is more common in patients with a high Oberlin score, but fusion status is not an independent biomarker of prognosis.
Keywords: ARST0431, D9802, fusion status, high risk, histology, rhabdomyosarcoma
1 INTRODUCTION
Between 1995 and 2005, there was a shift in the histologic criteria required for a diagnosis of alveolar rhabdomyosarcoma (ARMS). This shift followed publication of the International Classification of Rhabdomyosarcoma (ICR), which was the first prognostically relevant classification system for rhabdomyosarcoma (RMS).1 The ICR identified three prognostic groups: botryoid and spindle cell RMS had a superior prognosis, conventional embryonal rhabdomyosarcoma (ERMS) had an intermediate prognosis, and ARMS had a poor prognosis. However, the ICR also broadened the diagnostic criteria for ARMS to include the solid variant and tumors with only focal alveolar histology, when previously a diagnosis of ARMS required >50% classic alveolar histology. This change increased both the frequency of ARMS diagnoses and the proportion of FOXO1 fusion-negative ARMS.2–4
Studies have examined the effect of this histologic shift in patients with low-risk clinical characteristics enrolled on Children’s Oncology Group (COG) Study D9602, as well as in patients with intermediate-risk RMS enrolled on COG Study D9803.4–6 Fifty-two percent of cases originally enrolled on D9602 with a diagnosis of ARMS were reclassified as ERMS, whereas 33% of cases enrolled on D9803 were similarly reclassified. Upon re-review using current diagnostic criteria, the percentage of ARMSn on D9803 decreased from 37 to 18% of total ARMS diagnoses, a ratio more closely approaching earlier reports.4
These reviews validated the current criteria for ARMS, standardized the diagnosis of ARMS across COG studies, and allowed definitive analysis of the prognostic significance of histology and fusion status for patients with RMS. As predicted by retrospective gene expression studies showing that ARMSn and ERMS are molecularly indistinguishable, analysis of these prospective studies confirmed that the presence of a PAX-FOXO1 fusion drives unfavorable outcome for children with RMS.5–8 Based on these data, we hypothesized that (1) a significant number of high-risk, metastatic ARMS would be reclassified as other histologic subtypes and (2) fusion status would have similar prognostic significance for these high-risk patients.
2 MATERIALS AND METHODS
2.1 Case selection
COG study D9802 enrolled 127 patients with metastatic RMS between September 1999 and February 2004.9 Sixteen patients were ineligible and four had a central review diagnosis of undifferentiated sarcoma. Of the remaining 107 patients with RMS, 23 patients originally diagnosed as ARMS did not have material available for re-review. Given the high reclassification rate observed on contemporaneous low- and intermediate-risk studies, the prior central pathology diagnosis of ARMS was not considered reliable for the 23 cases without material for review and these cases were excluded from the analysis.4,5 Two additional cases had no central review diagnosis and no material available for re-review, resulting in 82 total cases from D9802 available for outcome analysis (Table 1).
TABLE 1.
D9802 and ARST0431 cases included in outcome analysis
| Enrolled | Excluded | Central review diagnosis | Re-review diagnosis | Total included | |
|---|---|---|---|---|---|
| D9802 | 127 | 45 | – | 82 | 82 |
| ARST0431 | 109 | 13 | 34 | 62 | 96 |
ARST0431 enrolled 109 patients with metastatic RMS (64 ARMS, 36 ERMS, and nine other) between July 2006 and June 2008.10 Sixty-two cases had material available for histologic re-review. Thirty-four cases that lacked material for re-review had a documented central pathology review diagnosis. Because the current criteria for ARMS were used during the conduct of ARST0431,4 these cases were included in the study group. In total, 96 cases from ARST0431 were included in the outcome analysis (Table 1).
2.2 Histology review
Four pathologists (ERR, MAA, LAT, and DMP) participated in the re-review. Cases were re-classified as ARMS (solid or classic patterns), ERMS (typical, dense or botryoid), spindle cell/sclerosing RMS, mixed RMS (defined as separate, discrete areas of ARMS and ERMS histology), or RMS not otherwise specified (NOS) if the material was too small, crushed, or necrotic for definitive histologic classification.4 In cases difficult to classify, diffuse myogenin reactivity favored a diagnosis of ARMS over ERMS. Myogenin immunohistochemistry was available for 60 D9802 cases and 58 ARST0431 cases. Nuclear myogenin expression was scored from 0 to 4+ based on the following percentages of tumor cells: 0 (absent expression), 1+ (<10%), 2+ (10–50%), 3+ (>50–90%), and 4+ (>90%).11 The histologic type was determined prior to FOXO1 fusion status testing.
2.3 Fusion testing
In most cases (81 of 85), fusion status was determined by FISH to detect rearrangements of the FOXO1 (13q14), PAX3 (2q35), and PAX7 (1p36) loci using unstained formalin-fixed, paraffin-embedded tissue sections, as previously described.12 In the other four cases, fusion status was previously determined by quantitative reverse transcription PCR to detect expression of a PAX3-FOXO1 or PAX7-FOXO1 fusion transcript, as previously described.13 Fusion testing was not performed in the remaining cases due to a lack of available material.
2.4 Patient characteristics and statistical analyses
Event-free survival (EFS) was defined as the time from study entry to the first occurrence of disease progression, disease relapse, or death. For those not experiencing one of these events, EFS was censored at last contact. Estimates of overall survival (OS) and EFS as time-to-event distributions were calculated using the Kaplan–Meier method, and distributions were compared using log-rank tests.
Clinical characteristics were reviewed to generate an overall risk score based on the factors presented by Oberlin et al.14 Using this scheme, four risk factors (age <1 or ≥10 years, presence of bone or bone marrow disease, unfavorable primary site and ≥3 metastatic sites) were each assigned 1 point. Patients with a score of 0 or 1 were considered low risk, while scores ≥2 were considered high risk. The EFS was compared between fusion statuses, after stratification by risk category.
3 RESULTS
3.1 Histologic Re-review
Histology re-review was performed for 86 RMS cases enrolled on D9802, although four were later deemed ineligible for the final outcome analysis (Supplementary Fig. S1A). For cases with an original central pathology diagnosis of ARMS (n = 73), the diagnosis was confirmed in 58 cases (79%). Nine cases were reclassified as ERMS (12%), one was reclassified as mixed RMS (1%), and five were reclassified as RMS NOS due to lack of sufficient material. The original diagnosis was confirmed for 10 cases initially diagnosed as ERMS and three cases initially diagnosed as RMS NOS. We did not identify any cases with spindle cell/sclerosing morphology.
Histology re-review was performed for 62 cases enrolled on ARST0431 with an original diagnosis of ARMS. A diagnosis of ARMS was confirmed for 57 (92%), while three cases (5%) were reclassified as ERMS. Two cases (3%) were reclassified as RMS NOS due to insufficient material. No cases with an original diagnosis of ERMS were reviewed for this cohort, and no cases with spindle cell/sclerosing morphology were identified.
3.2 Fusion status
Fusion status was determined for 63% of confirmed ARMS cases (29 enrolled on D9802 and 51 enrolled on ARST0431), but material was not available for fusion testing in the remaining cases (Table 2). Five cases reclassified as ERMS also had material available for fusion testing. One D9802 case reclassified as RMS NOS was fusion negative, while fusion status was unknown for the remaining four cases; one ARST0431 case reclassified as RMS NOS was fusion positive, while fusion status was unknown for the other case. RMS NOS cases were excluded from the final outcome analyses, however (Supplementary Fig. S1B). Fusion status was unknown for all other remaining cases, including 48 confirmed ARMS (31 from D9802 and 17 from ARST0431). For either study, ERMS cases lacking fusion data were considered to be fusion negative.
TABLE 2.
Histology and fusion status for D9802 and ARST0431 cases
| Histology, fusion | D9802 | ARST0431 | Total |
|---|---|---|---|
| ARMS, negative | 5 | 7 | 12 |
| ARMS, PAX3 | 18 | 42 | 60 |
| ARMS, PAX7 | 6 | 2 | 8 |
| ARMS, unknown | 31 | 17 | 48 |
| ERMS | 22 | 28 | 50 |
| Total | 82 | 96 | 178 |
Twelve (15%) of the confirmed ARMS cases with available fusion data were fusion negative (five from D9802 and seven from ARST0431), whereas 68 (85%) of confirmed ARMS cases with known fusion status were fusion positive (Table 2). A PAX3-FOXO1 fusion was identified in 60 confirmed ARMS (18 from D9802 and 42 from ARST0431). A PAX7-FOXO1 fusion was present in only eight confirmed ARMS cases (six from D9802 and two from ARST0431). Thus, 88% of fusion-positive ARMS had a PAX3-FOXO1 rearrangement versus 12% with a PAX7-FOXO1 rearrangement. Five reclassified ERMS cases with known fusion status were fusion negative.
3.3 Clinical features
Clinical characteristics for the 178 patients included in the outcome analysis showed a predominance of male patients (52%, n = 94), age ≥ 10 years (70%, n = 125), with bone or bone marrow disease (52%, n = 94). Primary tumor sites included extremity (26%, n = 46), retroperitoneum/perineum (24%, n = 42), parameningeal (11%, n = 19), trunk (10%, n = 18), genitourinary, nonbladder/prostate (10%, n = 18), bladder/prostate (5%, n = 10), intrathoracic (4%, n = 7), head and neck (3%, n = 5), and other (7%, n = 13) (Supplementary Table S1).
Using the Oberlin risk groups,14 51 patients had a score of 0 or 1 (nine fusion-positive RMS (RMSp), 33 fusion-negative RMS (RMSn), nine fusion status unknown), 48 patients had a score of 2 (23 RMSp, 12 RMSn, 13 fusion status unknown), 47 patients had a score of 3 (24 RMSp, 11 RMSn, 12 fusion status unknown), and 32 had a score of 4 (12 RMSp, 6 RMSn, 14 fusion status unknown) (Table 3). Patients with fusion-positive tumors were significantly more likely to have a higher risk score than those patients with fusion-negative tumors (P < 0.0001).
TABLE 3.
Distribution by Oberlin risk score for ARMS cases with known fusion status and presumed fusion negative ERMS (D9802 and ARST0431)
| Total
|
Fusion positive
|
Fusion negative
|
|||||
|---|---|---|---|---|---|---|---|
| Risk score | N (%) | 5-Year EFS (95% CI) |
N (%) | 5-Year EFS (95% CI) |
N (%) (ARMSn/ERMS) |
5-Year EFS (95% CI) |
Long-rank P value |
| 0,1 | 42 (32%) | 9(21%) | 33 (79%) | ||||
|
| |||||||
| 52.4(17–87.8) | 27.8 (0–60.5) | (4/29) | 58.5 (21.6–95.4) | 0.07 | |||
|
| |||||||
| 2 | 35 (27%) | 23 (66%) | 12 (34%) | ||||
|
| |||||||
| 22.8 (3.2–42.5) | 15.8 (0–32.3) | (2/10) | 41.7(10.5–72.9) | 0.42 | |||
|
| |||||||
| 3 | 35 (27%) | 24 (69%) | 11 (31%) | ||||
|
| |||||||
| 6.3 (0–18.2) | 4.7 (0–13.8) | (4/7) | 9.1(0–26.1) | 0.70 | |||
|
| |||||||
| 4 | 18 (14%) | 12 (67%) | 6 (33%) | ||||
|
| |||||||
| 0 | 0 | (2/4) | 0 | 0.72 | |||
EFS, event-free survival.
3.4 Outcome
Five-year EFS for the entire cohort (n = 178) was 22% (95% confidence interval [CI]: 16–29%). There was a difference in EFS by study: 5-year EFS for D9802 and ARST0431 was 15% (95% CI 8–23%) and 30% (95% CI 20–40%), respectively (P = 0.0014) (Fig. 1A). The difference in EFS was restricted to patients with ERMS: 5-year EFS was 23% for D9802 (95% CI 8–41%) compared to 59% for ARST0431 (95% CI 39–75%) (P = 0.012), which is accounted for by the exclusion of patients less than 10 years of age with metastatic ERMS on D9802 (Fig. 1B). There was no significant difference in 5-year EFS for patients with ARMS: D9802, 12% (95% CI 5–21%) versus ARST0431, 16% (95% CI 8–27%) (P = 0.057) (Fig. 1C).
FIGURE 1.
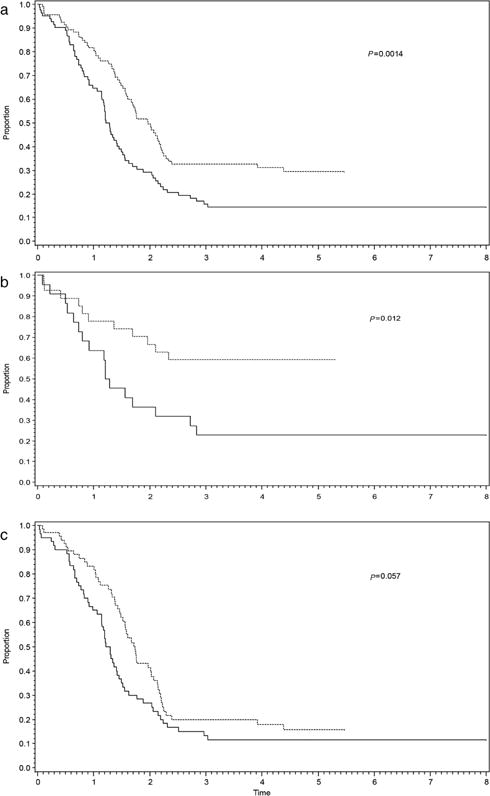
Event-free survival for D9802 (solid lines) and ARST0431 (dashed lines) including (a) all patients, (b) ERMS only, and (c) ARMS only
When analyzed by histology and fusion status, the 5-year EFS between all histologic subsets were as follows: ERMS = 43% (95% CI 29–56%), fusion negative ARMS = 29% (95% CI 7–56%), ARMS-unknown = 17% (95% CI 8–30%), ARMS-PAX7 = 17% (95% CI 1– 51%), and ARMS-PAX3 = 8% (95% CI 3–17%) (P = 0.020) (Fig. 2A). No statistically significant differences in OS were seen among these subsets (P = 0.14). Additionally, although there was a quantitative difference in EFS between fusion negative and fusion positive cases with exclusively alveolar histology (including both PAX3 and PAX7-FOXO1 rearrangements), this difference was not statistically significant (P = 0.22). After grouping cases into RMSp and RMSn categories, the 5-year EFS for RMSp (n = 68) was 9% (95% CI 3–18%) versus RMSn (n = 62) of 40% (95% CI 28–52%) (P = 0.0012) (Fig. 2B). OS for RMSp was approximately 20% versus 50% for RMSn (P = 0.029).
FIGURE 2.
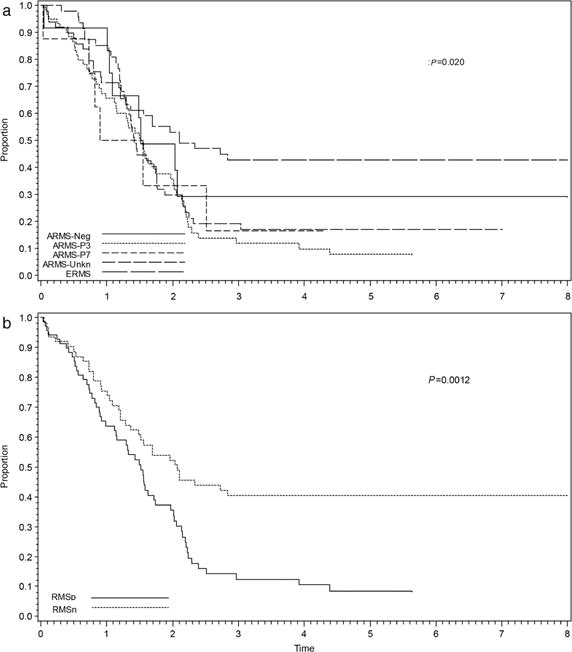
Event-free survival (EFS) by histology and fusion status for D9802 and ARST0431 cases combined including (a) all histologic subtypes and (b) fusion status alone
Using the 80 ARMS cases with known fusion status and 50 presumed fusion negative ERMS cases, EFS was examined based on fusion status and Oberlin risk score. Patients with fusion positive tumors were more likely to have a higher risk score (≥2) than patients with fusion negative tumors (P < 0.0001). The 5-year EFS by Oberlin risk score alone was: Oberlin 0 or 1, 52.4% (95% CI 17–87.8%); Oberlin 2, 22.8% (95% CI 3.2–42.5%); Oberlin 3, 6.3% (95% CI 0–18.2%); and Oberlin 4, 0% (P < 0.0001) (Fig. 3). Factoring in fusion status has no significant effects on the 5-year EFS when stratified by Oberlin score (Table 3). For patients with a low-risk score (Oberlin 0 or 1), there was a trend toward inferior outcome for RMSp as compared to RMSn; however, overall the Oberlin score remained predictive independent of fusion status for patients with metastatic RMS.
FIGURE 3.
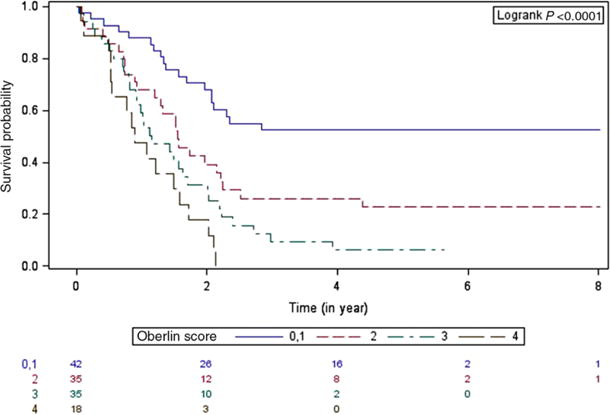
Product-limit survival estimates with number of subjects at risk. Event-free survival by Oberlin risk score
4 DISCUSSION
Previous studies have addressed histology and fusion status in low-and intermediate-risk RMS.4–6 This study provides the first analysis for high-risk RMS enrolled in two consecutive COG studies using current histologic criteria. As seen in D9602 and D9803, histology re-review resulted in a decrease in the number of total ARMS diagnoses, although to a lesser degree than seen on previous studies. However, in contrast to these earlier studies of low- and intermediate-risk RMS, FOXO1 fusion status is associated with higher clinical risk score, but is not an independent predictor of prognosis for patients with metastatic RMS. The histologic shift that accompanied the ICR increased the number of ARMS diagnoses on COG studies that enrolled patients from 1997 to 2005.4,5 Previous histologic review of cases originally diagnosed as ARMS and enrolled on low-risk (D9602) or intermediate-risk (D9803) RMS clinical trials during this period resulted in a reclassification as ERMS for 52% and 33% of low- and intermediate-risk cases, respectively. However, only 12% of original ARMS was reclassified as ERMS on the concurrent high-risk study D9802, a statistically significant difference confirming less frequent reclassification of ARMS cases in higher risk groups (P < 0.001). The rate of reclassification of ARMS in the subsequent high-risk study ARST0431 (following modification of the ICR criteria) also decreased, with only 5% of original ARMS diagnoses being reclassified as ERMS.
It is interesting that the histology of high-risk ARMS appears more classic in higher risk groups, as there is a lower rate of reclassification in higher risk disease. The reason for this is unclear. Here and in previous reviews, we have noted that ARMS cases with PAX7-FOXO1 often have more varied histologic patterns.15 PAX7 fusions also seem to be overrepresented on low-risk studies (27% on D9602) but still account for 15–20% of ARMS on intermediate- and high-risk studies (16% on D9803 and 20% on D9802).4,5 Similarly, among RMSp alone, PAX7 fusions are overrepresented in RMS with low-risk clinical features (46% on D9602 compared with 19% on D9803 and 25% on D9802). However, the overall percentage of ARMSn remains stable across all risk groups at 15–25% of confirmed ARMS.
Applying current diagnostic criteria to prospective clinical trials of high-risk RMS with mature outcome data allows for analysis of the relative prognostic significance of histology and fusion status for patients with metastatic RMS. Within these high-risk cohorts, PAX-FOXO1 fusion status was associated with a higher clinical risk score, and fusion positivity may contribute to the increased aggressiveness. However, once metastatic disease is established, clinical features seem to remain the most important factor in risk-stratifying patients with metastatic RMS. As determined by the Oberlin et al.’s criteria, a higher risk score predicted poor outcomes for patients with metastatic disease. After adjusting for Oberlin risk score, FOXO1 fusion status did not offer additional predictive value. Poor outcome for the highest risk patients with a risk score of 2, 3, or 4 (most of whom have RMSp) is associated with older age, unfavorable primary sites, the presence of bone and/or bone marrow involvement, and the number of sites of metastatic disease.
Potential weaknesses of the study include the availability of fusion data for only 65% of ARMS enrolled on the study, which could lead to erroneous conclusions when evaluating a “convenience cohort.”16 There were few fusion negative ARMS cases, and a larger analysis is needed. Additionally, fusion data for patients with metastatic ERMS were not available for many cases; however, fusion-positive ERMS is very rare.3,5 The superior outcome for patients with metastatic ERMS also provides support that fusion-positive ERMS is exceedingly rare for high-risk patients as well. Future studies will include analysis of FOXO1 fusion status for all histologic subtypes of RMS, allowing a more rigorous examination of this group.
This study resolves the variation in histologic classification for a generation of COG RMS studies, providing standardized diagnoses for all patients with ARMS enrolled during that era by current histologic criteria. Additionally, our data demonstrate that FOXO1 fusion status is associated with a higher Oberlin score for patients with metastatic RMS, but the these clinical risk factors remain the most predictive marker of poor outcome for patients with metastatic RMS. This study provides the foundation for future analysis of risk stratification with the addition of molecular data across all risk groups, including recent generations of COG studies.
Supplementary Material
Acknowledgments
St. Baldrick’s Foundation Consortium Research Grant; Grant number: 244660; Grant sponsor: Children’s Oncology Group; Grant numbers: U10CA180886, U10CA180899, U10CA098543, and U10CA098413; Grant sponsor: National Cancer Institute.
Abbreviations
- ARMS
alveolar rhabdomyosarcoma
- COG
Children’s Oncology Group
- EFS
event-free survival
- ERMS
embryonal rhabdomyosarcoma
- ICR
International Classification of Rhabdomyosarcoma
- OS
overall survival
- RMS
rhabdomyosarcoma
- RMSn
fusion-negative RMS
- RMSp
fusion-positive RMS
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
All studies were conducted in accordance with institutional review board approved protocols.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
References
- 1.Newton WA, Gehan EA, Webber BL, et al. Classification of rhabdomyosarcomas and related sarcomas. Pathologic aspects and proposal for a new classification–an Intergroup Rhabdomyosarcoma Study. Cancer. 1995;76:1073–1085. doi: 10.1002/1097-0142(19950915)76:6<1073::aid-cncr2820760624>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 2.Barr FG, Smith LM, Lynch JC, et al. Examination of gene fusion status in archival samples of alveolar rhabdomyosarcoma entered on the Inter-group Rhabdomyosarcoma Study-III trial: A report from the Children’s Oncology Group. J Mol Diagn. 2006;8:202–208. doi: 10.2353/jmoldx.2006.050124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sorensen PH, Lynch JC, Qualman SJ, et al. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhab-domyosarcoma: A report from the children’s oncology group. J Clin Oncol. 2002;20:2672–2679. doi: 10.1200/JCO.2002.03.137. [DOI] [PubMed] [Google Scholar]
- 4.Rudzinski ER, Teot LA, Anderson JR, et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: A report from the soft tissue sarcoma committee of the Children’s Oncology Group. Am J Clin Pathol. 2013;140:82–90. doi: 10.1309/AJCPA1WN7ARPCMKQ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnold MA, Anderson JR, Gastier-Foster JM, et al. Histology, fusion status, and outcome in alveolar rhabdomyosarcoma with low-risk clinical features: A report from the Children’s Oncology Group. Pediatr Blood Cancer. 2016;63:634–639. doi: 10.1002/pbc.25862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Skapek SX, Anderson J, Barr FG, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: A children’s oncology group report. Pediatr Blood Cancer. 2013;60:1411–1417. doi: 10.1002/pbc.24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davicioni E, Anderson MJ, Finckenstein FG, et al. Molecular classification of rhabdomyosarcoma–genotypic and phenotypic determinants of diagnosis: A report from the Children’s Oncology Group. Am J Pathol. 2009;174:550–564. doi: 10.2353/ajpath.2009.080631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Williamson D, Missiaglia E, de Reynies A, et al. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma. J Clin Oncol. 2010;28:2151–2158. doi: 10.1200/JCO.2009.26.3814. [DOI] [PubMed] [Google Scholar]
- 9.Pappo AS, Lyden E, Breitfeld P, et al. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: The Children’s Oncology Group. J Clin Oncol. 2007;25:362–369. doi: 10.1200/JCO.2006.07.1720. [DOI] [PubMed] [Google Scholar]
- 10.Weigel BJ, Lyden E, Anderson JR, et al. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: A report from the Children’s Oncology Group. J Clin Oncol. 2016;34:117–122. doi: 10.1200/JCO.2015.63.4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rudzinski ER, Anderson JR, Lyden ER, et al. Myogenin, AP2beta, NOS-1, and HMGA2 are surrogate markers of fusion status in rhabdomyosarcoma: A report from the soft tissue sarcoma committee of the Children’s Oncology Group. Am J Surg Pathol. 2014;38:654–659. doi: 10.1097/PAS.0000000000000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishio J, Althof PA, Bailey JM, et al. Use of a novel FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of alveolar rhabdomyosarcoma. Lab Invest. 2006;86:547–556. doi: 10.1038/labinvest.3700416. [DOI] [PubMed] [Google Scholar]
- 13.Barr FG, Qualman SJ, Macris MH, et al. Genetic heterogeneity in the alveolar rhabdomyosarcoma subset without typical gene fusions. Cancer Res. 2002;62:4704–4710. [PubMed] [Google Scholar]
- 14.Oberlin O, Rey A, Lyden E, et al. Prognostic factors in metastatic rhabdomyosarcomas: Results of a pooled analysis from United States and European cooperative groups. J Clin Oncol. 2008;26:2384–2389. doi: 10.1200/JCO.2007.14.7207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parham DM, Barr FG. Classification of rhabdomyosarcoma and its molecular basis. Adv Anat Pathol. 2013;20:387–397. doi: 10.1097/PAP.0b013e3182a92d0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg AR, Skapek SX, Hawkins DS. The inconvenience of convenience cohorts: rhabdomyosarcoma and the PAX-FOXO1 biomarker. Cancer Epidemiol Biomarkers Prev. 2012;21:1012–1018. doi: 10.1158/1055-9965.EPI-12-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.