

Abstract
Background
Posterior fossa (PF) ependymomas (EPNs) in infants less than 1 year of age (iEPN-PF) have a poorer clinical outcome than EPNs in older children. While radiation therapy is the standard of care for the latter, it is withheld in infants to avoid neurotoxicity to immature brain. It is unknown whether the adverse outcome in iEPN-PFs is due to treatment differences or aggressive biology. We examined this question using molecular profiling.
Methods
Six anaplastic iEPN-PFs were subjected to transcriptomic analysis and FISH for p16 loss and gains of 1q, and compared with anaplastic PF EPNs from older children. Results were validated by immunohistochemistry (IHC).
Results
All six iEPN-PFs were grouped within EPN PF subgroup A (PFA). E2F targets and G2M checkpoint were identified as the most enriched gene sets in iEPN-PF, which was validated in a larger independent cohort. Accordingly, MIB-1 IHC demonstrated a higher mitotic rate in iEPN-PFs than noninfant anaplastic EPN PFA. Genetic and protein analyses demonstrated that p16 loss and low p16 protein expression is a hallmark of iEPN-PF, and that none harbored 1q gains. Kaplan–Meier analysis confirmed the poorer clinical outcome of the iEPN-PF cohort.
Conclusions
Biological differences, characterized by loss of p16 expression without gains of 1q in iEPN-PFs, as well as deregulated E2F target gene transcription, are indicative of deregulated p16-CDK4/6-pRB-E2F pathway activity. This may underlie the poor clinical outcome seen in this group of iEPN-PFs, rather than the withholding of radiation therapy. Results suggest a potential actionable therapy for iEPN-PF, namely cyclin-dependent kinase 4/6 (CDK4/6) inhibitors.
Keywords: E2F, ependymoma, infant, mitosis, p16
1 INTRODUCTION
Ependymomas (EPNs) are the third most common brain tumor in the pediatric population, with two-thirds located in the posterior fossa (PF) and one-third in the supratentorial (ST) compartment.1 EPNs are histologically graded as either World Health Organization (WHO) grade II or III (anaplastic) tumors.2 However, the histological distinction between grades II and III can be difficult.3,4
Recent large-scale genomic, transcriptomic, and methylation array profiling studies have elucidated significant differences in EPNs from different anatomic sites with respect to the molecular features and prognosis, despite histopathological similarities.5–9 A recent multiinstitutional and multivariate analysis by Pajtler et al. of 500 EPNs resulted in a proposed molecular classification of EPNs into nine distinct molecular subgroups.9 Two subgroups were identified within the PF: PF subgroup A (PFA) and PF subgroup B (PFB).7,8
Aside from the molecular subgrouping, copy number gains of the long arm of chromosome 1 (1q) and extent of surgical resection were the only remaining independent prognostic factors.9 PFA tumors harbored the highest frequencies of gain of 1q, which significantly influenced the overall survival (OS) within this subgroup. Interestingly, despite an increased frequency of gains of 1q in ST RELA-fusion EPNs, 1q gain did not adversely impact OS as significantly as it did in the PFA.9 However, homozygous loss of the cyclin-dependent kinase inhibitor 2A (CDKN2A) locus (on chromosome 9p21) has consistently been shown to be of poor prognostic significance and has been reported to be more frequent within ST RELA-fusion EPNs.1,5,6,9
EPNs occurring within the infant period (age <1 year) are associated with a particularly poor survival rate of only 42–55% at 5 years after diagnosis, despite the fact that infants typically handle surgery well.10 While postoperative radiation is standard-of-care therapy for EPNs in older children, in infants radiation is withheld until 12 months of age to avoid toxicity to the developing brain. The poor survival in infant EPN has been attributed to withholding radiotherapy. However, few previous studies have focused efforts on studying any potential biological differences in infant EPNs due to their relative infrequency. Indeed, in the recent study by Pajtler et al., only 12 of 168 EPNs (7.1%) for which age was reported occurred in infants less than 1 year of age.9 The subset of infant EPNs confined to the posterior fossa (iEPN-PF) was even smaller (n = 6).9
The current study compares iEPN-PFs with PF EPNs in older children of similar PF group and histological grade. Our cohort of six iEPN-PFs was examined for transcriptomic characteristics and the known prognostic genetic markers,1q gain and p16 loss. These features were compared to EPN in older children and correlated with clinical outcome. Elucidation of biological characteristics unique to iEPN-PF may be a critical step to the development of effective therapies for this particularly vulnerable infant subgroup, which has not been specifically addressed in other studies.9
2 MATERIALS AND METHODS
2.1 Patient cohort
All studies were conducted in compliance with local and federal research protection guidelines and institutional review board regulations (Colorado Multiple Institutional Review Board [COMIRB] #95-500 and 05-149). Retrospective review of the neurooncology database at Children’s Hospital Colorado was conducted to select EPNs at our institution for whom frozen material of suitable quality for microarray analysis was available. Eligible patients had been initially seen, received neurosurgical resection of tumor, or been seen in clinical follow-up between 1997 and 2015, with closure date of study in 2016. This search yielded six infant patients with EPN (defined as <12 months of age). Cases were diagnosed and graded according to WHO tumor classification guidelines.2 Clinical progression was determined by the neurooncologist on the study, which was also responsible for patient follow-up and care (NKF), and included evaluation of neuroimaging features.
2.2 Routine histology and immunohistochemistry
Tissues were fixed in 10% buffered formalin and cut at 5 μm. Immunohistochemistry (IHC) was performed on formalin-fixed, paraffin-embedded tumor tissue sections. IHCs that had originally been utilized for diagnostic purposes at the time of initial assessment varied, but in most cases included glial fibrillary acidic protein (GFAP; Dako Corporation, Carpinteria, CA, polyclonal, 1:2,500, no antigen retrieval), MIB-1 (Dako Corporation; monoclonal, 1:400 dilution, antigen retrieval), and p16 (Ventana, monoclonal, predilute, antigen retrieval). MIB-1 counts were performed manually based on 5,000 cells. Scoring for p16 was conducted using previously published methods (strong >50% of cells positive, moderate 10–50% of cells, weak <10%, or negative).11
2.3 Transcriptomic analysis
EPN transcriptomic study cohort data were obtained from a prior study of EPN molecular subgroups conducted by our laboratory.12 These gene expression microarray data are available through the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database13 (GEO Series accession number GSE66354; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE66354). All patient samples were obtained with consent (COMIRB 95–500) and processed identically using the Human Genome U133plus2 Array (Affymetrix) platform as described previously.12
Clustering analysis was done using transcriptomic data from 50 primary pediatric EPN patients for whom samples were available at our institution; this data set included the six iEPN-PFs. Within the expanded pediatric EPN data set, nine tumors had been ST and 41 had been infratentorial. These 50 samples were assigned to consensus molecular subgroups using nonnegative matrix factorization (NMF) of transcriptomic profiles available through the Broad Institute Gene Pattern platform. Ontology tools Database for Annotation, Visualization, and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov) and Gene Set Enrichment Analysis (GSEA) were used to assess enrichment of gene sets. Gene sets found to be enriched in our cohort of iEPN-PF were validated in an independently published EPN data set from the German Cancer Research Center, Heidelberg, Germany (GSE64415). This validation cohort contained 12 iEPNs and 156 noninfant, older pediatric EPNs for which age was reported.
2.4 FISH
FISH was conducted to evaluate the copy number status of p16 (CDKN2A, 9p21) and 1q25. Briefly, dual-color FISH probe sets, manufactured by Vysis (Abbott Laboratories Inc., Des Plaines, IL), were used for loss of heterozygosity (LOH) studies of chromosome 1q25 (Spectrum Green) and 9p21 (Spectrum Orange) with nine centromere as internal control. DNA copy number alterations (CNAs) were further examined in a large independent cohort of molecularly classified EPN that was recently published by Pajtler et al.9 CNAs, identified by examination of the combined intensity values of Illumina 450K methylation probes, were matched with patient age and molecular subtype for comparative analyses.
2.5 Survival analysis
Kaplan–Meier plots and log-rank (Mantel–Cox) tests were used to estimate survival probability. OS was calculated from the date of initial diagnosis to death from any cause. Patients who had not had an event (death or progression) at the time of analysis were censored.
2.6 Statistical analysis
Statistical analyses were performed using Prism (GraphPad) and Excel (Microsoft) software. For all tests, significance was defined as P-value < 0.05.
3 RESULTS
3.1 iEPN-PF: Patient characteristics and pathologic features
Six EPN were identified in patients <1 year of age (four male, two female) (Table 1). All iEPN-PF cases showed similar histologic features including relatively monomorphic cells with round to oval nuclei, speckled chromatin, perivascular pseudorosettes with GFAP-positive fibrillary processes oriented radially around vessels, and often showed hyalinization of tumor vessels. These tumors showed no histological dissimilarity to anaplastic PF EPN in noninfants in our comparison cohort. All six iEPN-PFs met histologic criteria for anaplastic WHO grade III, with mitotic counts ≥5 mitoses per 10 high power (40×) microscopic fields (Figs. 1A–1D).
TABLE 1.
Anaplastic EPN PFA patient demographics, dates of primary tissue diagnosis, clinical outcome, IHC, and FISH data
| Case | Infant | Sex | Age at diagnosis | Clinical details | p16 | +1q | MIB-1(%) | |
|---|---|---|---|---|---|---|---|---|
| IHC | FISH | |||||||
| 1 | Yes | M | 10 months | DOD after 23 months, GTR, XRT and lapatinib, one recurrence | Weak | Normal | Normal | 37 |
| 2 | Yes | M | 8 months | DOD after 48 months, GTR, XRT, multiple recurrences | Weak | Normal | Normal | 59 |
| 3 | Yes | F | 7 months | DOD after 33 months, GTR, XRT, multiple recurrences, chemo | Negative | LOH | Normal | 54 |
| 4 | Yes | F | 6 months | DOD after 98 months, GTR, XRT, two recurrences | Negative | Normal | Normal | 51 |
| 5 | Yes | M | 11 months | Living after 11 months, GTR, XRT, chemo | Negative | LOH | Normal | 40 |
| 6 | Yes | M | 4 months | DOD after 4 months, STR, oral palliative etoposide | Weak | – | – | 75 |
| 7 | No | F | 31 months | Living after 13 years, STR, XRT, chemo | Weak | – | – | 36 |
| 8 | No | M | 37 months | Living after 9 years, GTR, XRT, chemo | Weak | Normal | Normal | 32 |
| 9 | No | M | 45 months | Living after 4 years, GTR, XRT, chemo, multiple recurrences | Moderate | Normal | Gain | 30 |
| 10 | No | F | 22 months | Living after 5 years, GTR, XRT, chemo, single recurrence | Moderate | Normal | Normal | 13 |
| 11 | No | M | 32 months | Living after 15 years, GTR, XRT, chemo, multiple recurrences | Moderate | Normal | Normal | 47 |
| 12 | No | M | 72 months | DOD after 17 months, STR, XRT, chemo, drop metastases | Moderate | – | Gain | 45 |
| 13 | No | M | 204 months | Living after 5 years, STR, XRT, chemo, drop metastasis | Moderate | – | Gain | 35 |
Chemo, protocol chemotherapy regimen; DOD, died of disease; GTR, gross total resection; LOH, loss of heterozygosity; STR, subtotal resection; XRT, radiation therapy; “–”, used when data are not available.
FIGURE 1.
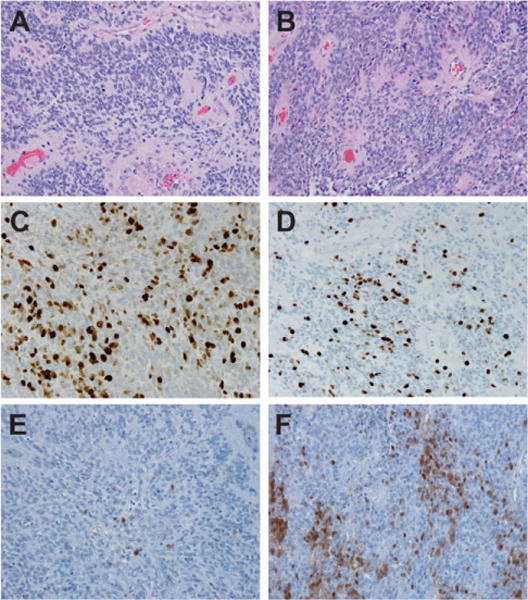
Histological features of infant EPN. (A) Anaplastic ependymoma from an infant case demonstrating perivascular pseudorosettes, increased mitotic figures, and monomorphic round to oval nuclei with speckled chromatin (H&E, 200×). (B) Anaplastic ependymoma from a noninfant case demonstrating similar features seen in part (A) (H&E, 200×). (C) MIB-1 immunohistochemistry demonstrating elevated cell cycle activity in an infant case (MIB-1, 200×). (D) MIB-1 immunohistochemistry demonstrating elevated cell cycle activity in a noninfant case (MIB-1, 200×). (E) Immunohistochemistry for p16 in an infant case with weak expression (p16, 200×). (F) Immunohistochemistry for p16 in a noninfant case with moderate to strong expression (p16, 200×)
3.2 All PF iEPN-PFs cluster with EPN PFA
Unbiased clustering analysis (NMF) was performed on transcriptomic profiles from primary tumor samples of iEPN-PFs (n = 6) and compared with 44 noninfant pediatric EPNs in our larger EPN tumor bank (all deposited in GEO: series accession number GSE66354). NMF analysis identified three molecular subgroups—ST, PFA, and PFB as described previously.12 All six iEPN-PFs were clustered within the PFA (Supplementary Fig. S1).
3.3 iEPN-PF harbor unique transcriptomic signatures compared to subgroup and grade-matched EPN in older patients
Based on our observation that all iEPN-PF were in PFA, we first narrowed our comparison group further to only PFA cases. Subsequent analyses of iEPN-PF transcriptome profiles were restricted to comparison to the 15 noninfant PFA EPN (median age = 3) we had identified within our larger EPN tumor bank of 44 cases. Following this first round of restrictions, we further focused only on the subset of 15 that were similarly graded as anaplastic, WHO grade III noninfant PFA in order to generate a completely concordant comparison group. This was done because ontological analysis using DAVID had revealed genes that had been significantly overexpressed greater than twofold in iEPN-PF versus noninfant PFA EPN (P < 0.05). Thus, the analysis had identified mitosis and cell cycle regulators as predominant roles of genes over-expressed in iEPN-PFs versus noninfant PFA EPN. Given that the histological classification of anaplasia in EPN is paralleled by a higher mitotic index (≥5 mitoses per 10 high power [40×] microscopic fields), we addressed this potentially confounding factor by further restricting transcriptomic analyses to histological grade-matched EPN PFA. This further narrowed our comparisons to seven noninfant anaplastic, WHO grade III, PFA cases.
Transcriptomic analysis compared all six anaplastic iEPN-PF to noninfant anaplastic EPN PFA (n = 7). These groups were subjected to a more-thorough GSEA ontological analysis to identify differential enrichment of well-defined biological states and processes using Molecular Signatures Database Hallmark gene sets (n = 50). Even when adjusted to compare only anaplastic EPN PFA, the ontologies associated with iEPN-PF were, similar to the preliminary analysis, largely related to mitosis and cell cycle (Table 2). Notably, most enriched gene sets were Hallmark_E2F_targets (normalized enrichment score [NES] = 2.14) and Hallmark_G2M_checkpoint (NES = 2.20) (both FDR q < 0.0001) (Figs. 2A and 2B).
TABLE 2.
Enriched molecular signatures database (MSigDB): Hallmark gene sets in denver and heidelberg cohort iEPN-PFs versus older EPN as measured by GSEAa
| Gene set | NES | NOM p-val | FDRq-val | FWER p-val |
|---|---|---|---|---|
| Denver cohort: up in iEPN-PF (n = 6) versus older anaplastic EPN PFA (n = 7) | ||||
| HALLMARK_G2M_CHECKPOINT | 2.20 | <0.0001 | <0.0001 | <0.0001 |
| HALLMARK_E2F_TARGETS | 2.14 | <0.0001 | <0.0001 | <0.0001 |
| HALLMARK_SPERMATOGENESIS | 1.74 | <0.0001 | 0.003 | 0.01 |
| HALLMARK_MITOTIC_SPINDLE | 1.68 | <0.0001 | 0.007 | 0.03 |
| HALLMARK_DNA_REPAIR | 1.56 | 0.001 | 0.025 | 0.116 |
| Heidelberg cohort: up in iEPN (n = 12) versus older EPN (n = 156) | ||||
| HALLMARK_E2F_TARGETS | 2.27 | <0.0001 | <0.0001 | <0.0001 |
| HALLMARK_HYPOXIA | 2.13 | <0.0001 | <0.0001 | <0.0001 |
| HALLMARK_G2M_CHECKPOINT | 2.08 | <0.0001 | <0.0001 | <0.0001 |
| HALLMARK_PANCREAS_BETA_CELLS | 1.81 | 0.0028 | 0.0051 | 0.008 |
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 1.68 | <0.0001 | 0.011 | 0.021 |
The top five enriched gene sets as ranked by NES are shown.
NES, normalized enrichment score; NOM, nominal; FDR, false discovery rate; FWER, family-wise error rate.
FIGURE 2.
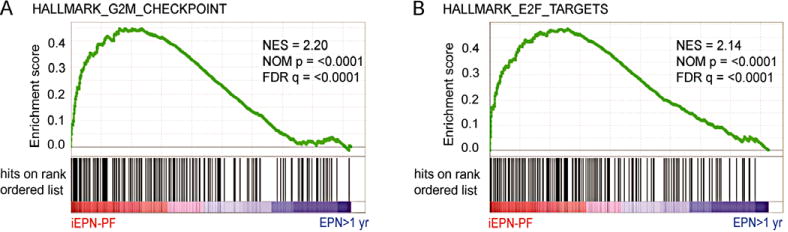
Increased cell cycle related gene expression distinguishes infant EPN from older anaplastic EPN PFA. GSEA plots demonstrating enrichment of (A) Hallmark_E2F_Targets and (B) Hallmark_G2M_Checkpoint gene sets in iEPN-PF. NES, normalized enrichment score; FDR, false discovery rate
Gene sets enriched in our cohort of iEPN-PF were validated in an independent data set (GSE64415). As this data set contained infant EPN from both PF and ST sites, we compared iEPNs (n = 12; six PF, six ST) to noninfant EPN (n = 156; median 12 years, 109 PF and spine, 47 ST) from all anatomic locations.9 GSEA analysis of the German cohort validated our finding that Hallmark_E2F_target (NES = 2.27) and Hallmark_G2M_checkpoint (NES = 2.08) gene sets were the most enriched gene sets in infant EPN (FDR q < 0.0001) (Table 2).
The upregulation of mitotic activity in iEPN-PF suggested by the enrichment of numerous mitosis-associated gene sets was confirmed by immunohistochemical MIB-1 labeling indices, showing significantly higher (P = 0.01) rates in iEPN-PF (mean = 53%) than the noninfant PFA EPNs (mean = 34%) (Table 1).
3.4 Patient demographics and clinical outcome
A review of patients presenting at our institution between 1997 and 2015 identified 50 EPN patients of which six were less than 1 year of age (12%). Details of iEPN-PF patient gender, age at diagnosis, and clinical details along with noninfant anaplastic EPN PFA control cohort are provided in Table 1. The male:female ratio of the iEPN-PF cohort was 2:1. The median age at diagnosis was 7.7 months (range: 4–11 months).
Kaplan–Meier analysis confirmed the poor clinical outcome of the iEPN-PF (n = 6) compared to noninfant anaplastic EPN PFA (n = 7), with a hazard ratio of 6.2 (log rank P = 0.029) (Fig. 3A). When EPNs PFB (n = 20) were included in this analysis, the survival curves for noninfant EPN PFA of any histological grade and EPN PFB were overlapping, suggesting that the particularly adverse survival in iEPN-PF has a significant influence on the well-documented adverse risk associated with EPN PFA as a whole (Fig. 3B).
FIGURE 3.
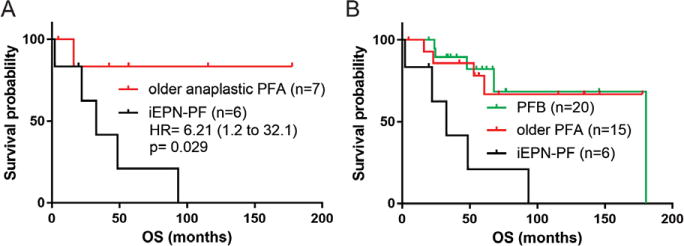
Infant EPN convey significantly worse survival. (A) Kaplan–Meier plot demonstrating a significant difference in estimated survival probability in iEPN-PF (GpA infant) versus the noninfant anaplastic EPN PFA (older PFA) cases. (B) When EPN PFB cases are superimposed, they demonstrate the same survival as noninfant EPN PFA cases and both had statistically significant better survival than the iEPN-PF cases alone
Survival difference could not be attributed to the extent of surgical resection: gross total resection was achieved in five of six iEPN-PFs and in four of seven of the noninfant anaplastic EPN PFA. All cases of anaplastic EPN PFA (infant and noninfant), with the exception of a single iEPN-PF patient, had received adjuvant radiation therapy when they passed the age of 1 year (Table 1). The exceptional case (case 6) had received only palliative oral etoposide chemotherapy. As stated above, in all iEPN-PFs, cranial radiation had been delayed/withheld in all infants until the age of 1 year.
3.5 iEPN-PF harbor p16 losses but not 1q gain
Gains of 1q, associated with poor outcome in EPNs of any age in most series, were found in 10 of 20 tested (50%) by FISH (assessable EPN PFA and PFB combined). In contrast, none of the five iEPN-PF tests showed 1q gain, a surprising finding given their poor outcome (Fisher’s exact test, P = 0.061). Examination of a large independent cohort of EPN for which CNA data were available corroborated this observation, showing that 1q was not affected in any (0/6) infant anaplastic EPN (all PFA), but 1q gain was seen in 42 of 151 (28%) of older anaplastic EPN (PFA and PFB combined), significantly by Fisher’s exact test (P = 0.0006).
However, FISH showed that two of five (40%) assessable iEPN-PF cases did harbor LOH in p16 (Table 1). For comparison, the frequency of p16 loss in noninfant cases identified no p16 losses in five of five EPN PFA regardless of histological grade, and only one of five cases of EPN PFB that could be assessed. Contingency analysis of the frequency of p16 loss in iEPN-PF versus noninfants (EPN PFA and PFB combined) did not demonstrate significance (Fisher’s exact test, P = 0.24). Despite the small case numbers, the association of this pathogenic genetic alteration with iEPN-PF warranted further investigation.
To address this, we further examined CNA data from the independent cohort of EPN, as described above, to identify loss of p16 in DNA. In this cohort, p16 genetic losses were only identified in ST EPN harboring RELA-fusion (14/88), which were generally associated with large-scale alterations of chromosome 9, and no genetic losses of p16 were identified in any EPN PFA or PFB examined, including infant EPN. An earlier study of p16 status in EPN (n = 101) by FISH analysis identified p16 loss in approximately 25% of infratentorial and ST EPN that was not significantly associated with site of disease.14 Given this discrepancy, we suspect that, unlike larger changes such as 1q gain, potentially smaller genetic alterations of p16 cannot be easily assessed by CNA analysis of Illumina 450K methylome data.
IHC for p16 protein expression was therefore performed to further identify possible differences in iEPN-PF versus noninfant anaplastic PFA EPN (Figs. 1E and 1F). As detailed in Table 1, the IHC expression of p16 was reduced in iEPN-PF when compared to the noninfant anaplastic EPN PFAs, with complete loss of IHC expression observed in three of six iEPN-PFs, and two of three with complete loss also harbored heterozygous loss of p16 by FISH analyses (cases 3 and 5). Weak expression of p16 was observed in the remaining three iEPN-PF. Collectively, these data suggest impaired activity of this tumor-suppressive protein in all iEPN-PF. In contrast, noninfant pediatric anaplastic EPN PFA cases did not reveal any p16 loss by FISH, or complete loss of protein by IHC, again with strong concordance between the two techniques. Moderate p16 protein expression was observed in five of seven noninfant EPNs, with the remaining two showing weak expression. Comparison of p16 protein expression in iEPN-PF versus noninfants anaplastic EPN PFA was performed by contingency analysis. This demonstrated a significant association of low p16 (defined as negative or weak expression) in iEPN-PF (Fisher’s exact test, P = 0.021).
Complete loss of p16 protein expression in the iEPN-PF that harbored p16 LOH suggests that in these cases, a second silencing event at the p16 locus has occurred, resulting in complete loss of function of the gene. Such silencing events include genetic point mutations and epigenetic alterations. A precedent for epigenetic silencing of p16 in iEPN-PF is the finding that p16 is epigenetically silenced in atypical teratoid/rhabdoid tumors (AT/RT), a pediatric brain tumor that, similar to iEPN-PF, occurs predominantly in children less than 3 years of age. p16 was shown to be silenced in AT/RT via a process that involves chromatin methylation and remodeling.15,16 Epigenetic silencing may also underlie loss of p16 protein expression in a portion of cells in noninfant EPN that showed moderate to strong expression (Fig. 1F), despite a lack of apparent genetic alterations in these samples.
4 DISCUSSION
Despite advances in neurosurgery and postoperative adjuvant therapy, prognosis remains relatively poor for pediatric EPNs, with 5-year survival rates ranging from 39 to 64%, and 5-year progression-free survival (PFS) from 23 to 45%.17 The clinical behavior of EPNs is highly variable, with late relapses occurring up to 15 years after initial treatment and local recurrences in 50% of patients even with gross total resection.18 Up to 40% of patients have incurable tumors.19,20
iEPN-PFs have a particularly poor prognosis.17,19–21 The aim of this study was to further explore the biology of iEPN-PFs, all of which in our study grouped within PFA were WHO grade III tumors. This PFA sub-grouping was anticipated by Pajtler et al. who showed that PFA EPNs were almost exclusively found in young children and resulted in a very poor prognosis compared to other EPN molecular subgroups.9 Indeed, PFA EPNs paralleled the ST RELA-fusion subgroup in terms of similar poor OS and event-free survival/PFS rates.9 Our study demonstrates that iEPN-PFs potentially make a major contribution to the adverse OS and PFS within the PFA, as the OS data approach near-equivalence for the PFA and PFB when the iEPN-PFs are separated out.
Overexpression of cell cycle and mitosis-related genes and gene sets for PF tumors and the PFA EPN has been previously demonstrated.7,9,22 However, the current study further shows that these gene sets appear primarily overexpressed within the PFA iEPN-PF cohort when compared to the noninfant pediatric PFA EPN cohort. Increased MIB-1 (Ki-67) is known to be associated with worse outcomes for EPN and is particularly associated with PF tumors.23–25 We have demonstrated overexpression of mitosis and cell cycle related gene sets within the PFA iEPN-PF group versus the noninfant PFA EPN group, as well as a statistically significant higher average MIB-1 rate than the noninfant pediatric group, despite identical WHO grade III (P = 0.01).
Further genomic, transcriptomic, and immunohistochemical analyses of iEPN-PF identified a potential mechanism underlying the elevated mitotic activity in these tumors. Loss of p16 was shown to be more frequent in iEPN-PFs, whereas the high-risk genetic feature of 1q gain was absent. Loss of p16 is known to be associated with a poor outcome in patients with ST RELA-fusion tumors and is infrequently observed within the other subgroups of EPNs.5,9,24 Copy number gains of 1q were identified to be the most common copy number variation within primary and recurrent EPNs and, in some studies, have been shown to serve as an independent marker for tumor progression.1,26 Additionally, Pajtler et al. noted gains of 1q to indicate a significantly worse outcome within the PFA as a whole.9 However, no iEPN-PF tumors in our iEPN-PF cohort had gains of 1q, despite having worse clinical outcomes as compared to PFA noninfant EPNs. In contrast, in our study, nine of 19 assessable noninfant EPN PF tumors harbored gains of 1q. This finding was corroborated in a larger validation cohort that again showed that no anaplastic iEPN-PF harbored 1q gains. This was an unexpected finding in light of the cited literature. A possible explanation lies in the functional impairment of tumor suppressor p16. These findings pave the way for larger multiinstitutional cohort studies to further examine this question.
The tumor suppressor protein p16 is an important cell cycle regulator that inhibits the activity of CDK4 and CDK6. Loss of p16, a frequent event in a wide variety of tumors, leads to deregulated CDK4/6 activity, resulting in phosphorylation of retinoblastoma (RB) protein. Phosphorylation of RB allows for dissociation of the transcription factor E2F from the RB/E2F complex and subsequent transcription of E2F target genes that are responsible for progression through the G0 phase of the cell cycle.27 Collectively, our findings suggest that deregulation of the p16-CDK4/6-pRB-E2F pathway might underlie the aggressive biology of iEPN-PF, demonstrated by the genomic loss of p16 and transcriptomic enrichment of mitosis-related gene sets, and in particular, genes encoding cell cycle related targets of E2F transcription factors. This negative impact may surpass any gain of 1q as an adverse prognostic factor, at least in iEPN-PFs.
The finding of p16 loss additionally represents a potential therapeutic vulnerability in iEPN-PF, as tumors harboring p16 loss are potentially sensitive to CDK4/6 inhibitors.28,29 Deregulation of the p16-CDK4/6-pRB-E2F pathway has been described in other brain tumors, notably adult glioblastoma where therapeutic targeting of this pathway with CDK4/6 inhibitor palbociclib is being explored preclinically and clinically.30,31 Of relevance to potential treatment of iEPN-PF, palbociclib was well tolerated and showed stabilization of tumor growth in pediatric intracranial growing teratoma syndrome.32 To our knowledge, however, no trials are ongoing for the use of palbociclib in EPNs of any type.
Supplementary Material
Acknowledgments
The authors thank Dr. Dexiang Gao for statistical expertise and Mrs. Diane Hutchinson for expert manuscript preparation. The Genomics and Microarray Core receives direct funding support from the National Cancer Institute through the University of Colorado Cancer Center Support Grant (P30CA046934).
Funding Information
Grant sponsor: Tanner Seebaum Foundation; Morgan Adams Foundation; Hyundai Hope on Wheels; National Cancer Institute; Grant number: P30CA046934.
Abbreviations
- AT/RT
atypical teratoid/rhabdoid tumor
- CDK4/6
cyclin-dependent kinase 4/6
- CDKN2A
cyclin-dependent kinase inhibitor 2A
- CNA
copy number alteration
- COMIRB
Colorado Multiple Institutional Review Board
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- EPN
ependymoma
- FDA
Food and Drug Administration
- FDR
false discovery rate
- GEO
Gene Expression Omnibus
- GFAP
glial fibrillary acidic protein
- GSEA
Gene Set Enrichment Analysis
- iEPN-PF
infant posterior fossa ependymoma
- IHC
immunohistochemistry
- LOH
loss of heterozygosity
- NES
normalized enrichment score
- NMF
nonnegative matrix factorization
- OS
overall survival
- PF
posterior fossa
- PFA
PF subgroup A
- PFB
PF subgroup B
- PFS
progression-free survival
- RB
retinoblastoma
- ST
supratentorial
- WHO
World Health Organization
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
Presented in platform presentation format at the 100th annual fall meeting of the Society for Pediatric Pathology, Toronto, Ontario, Canada on October 2, 2015. Winner of the Harry B. Neustein award for innovative technological advances in the study of pediatric pathology.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
References
- 1.Kilday JP, Rahman R, Dyer S, et al. Pediatric ependymoma: biological perspectives. Mol Cancer Res. 2009;7(6):765–786. doi: 10.1158/1541-7786.MCR-08-0584. [DOI] [PubMed] [Google Scholar]
- 2.Louis DO, Hiroko W, Otmar WC. WHO Classification of Tumours of the Central Nervous System. 4th. Lyon France: Internation Agency for Research on Cancer; 2007. [Google Scholar]
- 3.Ellison DW, Kocak M, Figarella-Branger D, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed. 2011;10:7. doi: 10.1186/1477-5751-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tihan T, Zhou T, Holmes E, Burger PC, Ozuysal S, Rushing EJ. The prognostic value of histological grading of posterior fossa ependymomas in children: a Children’s Oncology Group study and a review of prognostic factors. Mod Pathol. 2008;21(2):165–177. doi: 10.1038/modpathol.3800999. [DOI] [PubMed] [Google Scholar]
- 5.Korshunov A, Witt H, Hielscher T, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28(19):3182–3190. doi: 10.1200/JCO.2009.27.3359. [DOI] [PubMed] [Google Scholar]
- 6.Mendrzyk F, Korshunov A, Benner A, et al. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin Cancer Res. 2006;12(7 Pt 1):2070–2079. doi: 10.1158/1078-0432.CCR-05-2363. [DOI] [PubMed] [Google Scholar]
- 7.Wani K, Armstrong TS, Vera-Bolanos E, et al. A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol. 2012;123(5):727–738. doi: 10.1007/s00401-012-0941-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20(2):143–157. doi: 10.1016/j.ccr.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. doi: 10.1016/j.ccell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gatta G, Botta L, Rossi S, et al. Childhood cancer survival in Europe 1999–2007: results of EUROCARE-5–a population-based study. Lancet Oncol. 2014;15(1):35–47. doi: 10.1016/S1470-2045(13)70548-5. [DOI] [PubMed] [Google Scholar]
- 11.Raabe EH, Lim KS, Kim JM, et al. BRAF activation induces transformation and then senescence in human neural stem cells: a pilocytic astrocytoma model. Clin Cancer Res. 2011;17(11):3590–3599. doi: 10.1158/1078-0432.CCR-10-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Griesinger AM, Josephson RJ, Donson AM, et al. Interleukin-6/STAT3 pathway signaling drives an inflammatory phenotype in Group A ependymoma. Cancer Immunol Res. 2015;3:1165–1174. doi: 10.1158/2326-6066.CIR-15-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajaram V, Leuthardt EC, Singh PK, et al. 9p21 and 13q14 dosages in ependymomas. A clinicopathologic study of 101 cases. Mod Pathol. 2004;17(1):9–14. doi: 10.1038/modpathol.3800029. [DOI] [PubMed] [Google Scholar]
- 15.Wilson BG, Wang X, Shen X, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- 17.Zacharoulis S, Levy A, Chi SN, et al. Outcome for young children newly diagnosed with ependymoma, treated with intensive induction chemotherapy followed by myeloablative chemotherapy and autologous stem cell rescue. Pediatr Blood Cancer. 2007;49(1):34–40. doi: 10.1002/pbc.20935. [DOI] [PubMed] [Google Scholar]
- 18.Sowar K, Straessle J, Donson AM, Handler M, Foreman NK. Predicting which children are at risk for ependymoma relapse. J Neurooncol. 2006;78(1):41–46. doi: 10.1007/s11060-005-9072-2. [DOI] [PubMed] [Google Scholar]
- 19.Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res. 2014;20(22):5630–5640. doi: 10.1158/1078-0432.CCR-14-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouffet E, Foreman N. Chemotherapy for intracranial ependymomas. Childs Nerv Syst. 1999;15(10):563–570. doi: 10.1007/s003810050544. [DOI] [PubMed] [Google Scholar]
- 21.Paulino AC, Wen BC, Buatti JM, et al. Intracranial ependymomas: an analysis of prognostic factors and patterns of failure. Am J Clin Oncol. 2002;25(2):117–122. doi: 10.1097/00000421-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Perez-Ramirez M, Hernandez-Jimenez AJ, Guerrero-Guerrero A, et al. Genomics and epigenetics: a study of ependymomas in pediatric patients. Clin Neurol Neurosurg. 2016;144:53–58. doi: 10.1016/j.clineuro.2016.02.041. [DOI] [PubMed] [Google Scholar]
- 23.McLendon RE, Lipp E, Satterfield D, et al. Prognostic marker analysis in pediatric intracranial ependymomas. J Neurooncol. 2015;122(2):255–261. doi: 10.1007/s11060-014-1711-z. [DOI] [PubMed] [Google Scholar]
- 24.Li AM, Dunham C, Tabori U, et al. EZH2 expression is a prognostic factor in childhood intracranial ependymoma: a Canadian Pediatric Brain Tumor Consortium study. Cancer. 2015;121(9):1499–1507. doi: 10.1002/cncr.29198. [DOI] [PubMed] [Google Scholar]
- 25.Bennetto L, Foreman N, Harding B, et al. Ki-67 immunolabelling index is a prognostic indicator in childhood posterior fossa ependymomas. Neuropathol Appl Neurobiol. 1998;24(6):434–440. doi: 10.1046/j.1365-2990.1998.00143.x. [DOI] [PubMed] [Google Scholar]
- 26.Kilday JP, Mitra B, Domerg C, et al. Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the Children’s Cancer Leukaemia Group (CCLG), Societe Francaise d’Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP) Clin Cancer Res. 2012;18(7):2001–2011. doi: 10.1158/1078-0432.CCR-11-2489. [DOI] [PubMed] [Google Scholar]
- 27.Khleif SN, DeGregori J, Yee CL, et al. Inhibition of cyclin D-CDK4/CDK6 activity is associated with an E2F-mediated induction of cyclin kinase inhibitor activity. Proc Natl Acad Sci USA. 1996;93(9):4350–4354. doi: 10.1073/pnas.93.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konecny GE, Winterhoff B, Kolarova T, et al. Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res. 2011;17(6):1591–1602. doi: 10.1158/1078-0432.CCR-10-2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166(3):740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cen L, Carlson BL, Schroeder MA, et al. p16-Cdk4-Rb axis controls sensitivity to a cyclin-dependent kinase inhibitor PD0332991 in glioblastoma xenograft cells. Neuro Oncol. 2012;14(7):870–881. doi: 10.1093/neuonc/nos114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schroder LB, McDonald KL. CDK4/6 inhibitor PD0332991 in glioblastoma treatment: does it have a future? Front Oncol. 2015;5:259. doi: 10.3389/fonc.2015.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schultz KA, Petronio J, Bendel A, Patterson R, Vaughn DJ. PD0332991 (palbociclib) for treatment of pediatric intracranial growing teratoma syndrome. Pediatr Blood Cancer. 2015;62(6):1072–1074. doi: 10.1002/pbc.25338. [DOI] [PubMed] [Google Scholar]
- 33.Wu Z, Irizarry RA, Gentleman R, Martinez-Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J Amer Statist Assoc. 2004;99:909–917. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.