

Abstract
Messenger RNA (mRNA) represents a promising class of nucleic acid drugs. Although numerous carriers have been developed for mRNA delivery, the inefficient mRNA expression inside cells remains a major challenge. Inspired by the dependence of mRNA on 3′ terminal poly adenosine nucleotides (poly A) and poly A binding proteins (PABPs) for optimal expression, we complex synthetic mRNA containing a poly A tail with PABPs in a stoichiometric manner and stabilize the ribonucleoproteins (RNPs) via a family of polypeptides bearing different arrangements of cationic side groups. We find that the molecular structure of these polypeptides modulates the degree of PABP-mediated enhancement of mRNA expression. This strategy elicits an up to 20-fold increase in mRNA expression in vitro and ~4-fold increase in mice. These findings suggest a set of new design principles for gene delivery via synergistic co-assembly of mRNA with helper proteins.
Keywords: polyamine, poly A binding protein, polycation, mRNA, gene delivery
Graphical Abstract
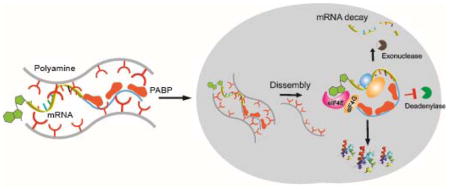
Co-assembly of mRNA/PABP nanoplexes with polyamines enhances mRNA translation via improvement of both mRNA stability and protein translation.
mRNA-based therapeutics have been extensively investigated recently and harbor great potential towards clinical translation.[1–4] Unlike protein-based biologics, mRNA co-opts natural biological processes to express proteins and thereby achieve a desired therapeutic effect.[5–6] In contrast to DNA, however, mRNA does not need to enter the nucleus to be functional, which allows for transfection of non-dividing cells with potentially high efficiency.[7] Additionally, mRNA does not integrate into the genome and hence has little risk of insertional mutagenesis.[8–9] These advantages enable the potential treatment of a broad spectrum of diseases, many of which cannot be addressed with current technologies.[1–2] Nevertheless, inefficient transfection of exogenous mRNA remains a key barrier to broad applications of mRNA-based drugs.[10]
Endogenous mRNA associates with specific proteins at different stages when trafficking from the nucleus to cytoplasmic ribosomes for maximal protein production. In contrast, the existing approach towards transfecting exogenous mRNA remains direct complexation of mRNA with cationic carriers.[11–13] These carriers are designed to protect mRNA from degradation, enable uptake by cells, and facilitate endosomal escape.[10] However, it was recently reported that certain polycation-based delivery vehicles can partially block mRNA from effectively recognizing the complementary protein responsible for mRNA translation.[14] To circumvent this problem, we recently demonstrated that preloading the 5′ end of mRNA with recombinant cap-binding protein, eIF4E, increases the formation of the mRNA translation initiation complex and leads to enhanced mRNA transfection via improved mRNA stability and expression inside cells.[15] Nevertheless, the 3′ poly A tail of mRNA is susceptible to de-adenylation (an enzyme-mediated hydrolysis of poly A), which can trigger RNA degradation by 3′–5′ RNA exonucleases in the cytoplasm.[16] Additionally, fundamental biology studies have shown that the poly A tail of endogenous mRNA is able to bind multiple PABPs in a tandem manner, which not only improves mRNA stability, but also stimulates mRNA translation in eukaryotic cells. [16–18]
In light of these observations, we speculated that existing gene delivery approaches of encapsulating mRNA directly in a cationic nanoplex could potentially prevent transfected mRNA from easily accessing and binding key translational proteins such as intracellular PABP. This lowered access could lead to de-adenylation, mRNA degradation, and reduced mRNA translation (Figure 1A). In contrast, co-encapsulation of the mRNA with a recombinant form of PABP may alleviate such steric hindrance, thereby improving mRNA expression (Figure 1B). To deliver the mRNA/PBAP complexes into cells, we designed a library of cationic polypeptides derived from N-carboxyanhydride polymerization of L-benzyl aspartate, followed by exhaustive amination of the ester groups to achieve varied architectures and numbers of charged groups in the side chains. These side chains can be divided into two major classes: linear and branched oligoalkylamines, which are composed of ethylene- or propane-diamine units (Supplemental Scheme 1). This well-defined library can then be used to determine the role of polyamine structure in mRNA transfection in co-delivery with PABP.
Figure 1. Overview of bio-inspired assembly for enhanced mRNA delivery.

(A) The state-of-the-art approach of directly complexing mRNA with cationic carriers for mRNA delivery. When mRNA associates with cationic carriers, the poly-A tail may not be readily accessible to endogenous PABP due to steric hindrance. Upon release from the complex, however, mRNA is susceptible to degradation (deadenylase and exonucleases), resulting in poor expression of the desired protein. (B) Preassembly of mRNA/PABP nanoplexes with polyamines enhance mRNA translation via improvement of both mRNA stability and protein translation. (C) Molecular structures of polycation carriers used in this study. Note that amination of the backbone likely induces intramolecular isomerization of the repeating unit, aspartamide, generating two isomers.[19]
Following purification from E.coli, recombinant human PABP retained its poly A-binding ability and spontaneously assembled with poly A-tailed mRNA in vitro to form RNPs, as confirmed by a gel shift assay (Supplementary Figs. 1A and B). On the other hand, the synthetic mRNA utilized in this study contains pseudouridine- and 5-methylcytidine-substituted nucleotides along with a poly A tail at the 3′ end (120 adenosine residues) for optimal mRNA expression, which has been demonstrated in previous studies.[1–2, 20] Since each PABP protein is known to bind a continuous stretch of 12 adenosine residues, each mRNA molecule is theoretically able to bind a maximum of ten PABP proteins.[21] Therefore, we first tested three different molar ratios of PABP to mRNA (2:1, 10:1 and 50:1) and subsequently encapsulated them with seven different polyamines. A range of N/P ratios (protonable amines in polyamines relative to phosphates in mRNA) was previously screened, and an N/P ratio of 50 was chosen for maximal mRNA transfection without noticeable cytotoxicity. At this ratio, both luciferase mRNA and luciferase mRNA/PABP formed stable nanoplexes (~100 nm) with all seven polyamines, with positive zeta potentials (Supplementary Figs. 2A and B). As shown in Figures 2B and C, when the ratio of PABP to mRNA reached 10:1, co-delivery of PABP resulted in enhanced luciferase expression relative to mRNA transfection alone by the same polyamine in HEK293T cells. Interestingly, five out of the seven polyamines led to enhanced mRNA transfection via co-delivery of PABP, with “2-2-2-2” yielding up to a 20-fold increase in luciferase mRNA expression. Since sufficient nanoplex uptake is a prerequisite for high transfection efficiency, we evaluated whether PABP increases luciferase mRNA uptake in HEK293T cells. At 24 hours post-transfection, FITC-mRNAs were present in 90–95% of total cells, independent of the PABP protein and polyamine (Supplementary Fig. 3A). Quantification of intracellular FITC-mRNA amounts via mean fluorescence intensity (MFI) showed that PABP slightly enhanced mRNA uptake compared to transfection of mRNA alone (Supplementary Fig. 3B). Nevertheless, there was little correlation between the dramatic enhancement of luciferase expression and the slight increase in mRNA uptake via co-delivery of PABP for each polyamine (Figure 2C and Supplementary Fig. 3B). Considering the fact that all polyamines were derived from the same polyaspartamide backbone, these results further imply that the increase in mRNA expression through stabilized preassembly with PABP is dependent on the side chain.
Figure 2. mRNA/PABP nanoplex enhances luciferase expression in a polyamine architecture-dependent manner.

(A) Schematic of synthetic luciferase mRNA with a 120 (A) tail binding to multiple PABP proteins in a tandem manner. One PABP protein binds to 12 continuous adenosine nucleotides. (B) Luciferase mRNA (100 ng) was preassembled with PABP at the indicated molar ratios and transfected with polyamines at a 50:1 N/P ratio. Luciferase expression was quantified 24 hours after transfection into HEK293T cells. The polyamine structures are shown in Supplemental Scheme 1. (C) Fold change in luciferase expression. For each type of polyamine, the increased luciferase expression via delivery of mRNA/PABP was normalized to the expression from delivery of mRNA alone and presented as the fold change. All experiments were performed twice in quadruplicates. Data represent the mean ± SEM (n=4). **, P<0.01; ***, P<0.001; ns, no significance.
Endogenous mRNA species with long poly A sequences are abundant inside cells and can potentially compete with synthetic mRNA for co-transfected PABP proteins. To further investigate the fundamental structure–activity relationships for co-delivery, we hypothesized that physical stabilization of mRNA and PABP in the nanoplexes is critical for enhanced mRNA translation during intracellular trafficking to ribosomes (Figure 3A). To directly confirm whether polyamines differentially modulate the physical interactions between mRNA and PABP inside the nanoplexes, luciferase mRNA and PABP protein were labeled with fluorescein isothiocyanate (FITC) and cyanine 5 (Cy5), respectively. In principle, when these two dyes are in close vicinity of each other (<10 nm) within cells, Förster resonance energy transfer (FRET) can be detected by flow cytometry. Consequently, an intracellular FRET assay can quantitatively evaluate the proximity between recombinant PABP and mRNA in a biologically relevant environment following transfection into cells. Mean FRET intensity measurements showed that only triethylenetetramine (“2-2-2”), tetraethylenepentamine (“2-2-2-2”), pentaethylenehexamine (“2-2-2-2-2”), tris(3-aminopropyl)amine (“branched 3-3-3”), and N,N′-Bis(3-aminopropyl)-1,3-propanediamine (“3-3-3”) resulted in relatively high degrees of co-localization of protein and mRNA within the complexes at a 10:1 (Cy5-PABP/FITC-mRNA) molar ratio 24 hours after transfection into HEK293T cells (Figure 3B and C). These observations correlated with the ability of the polyamines to enhance PABP-mediated mRNA expression (Figure 2B and C). Our intracellular FRET assay results therefore suggest that in addition to facilitating the internalization of nanoplexes into cells, polyamine carriers also stabilize the association between mRNA and PABP during cytosolic delivery, which is critical for PABP-mediated enhancement of mRNA transfection.
Figure 3. Side chains of polyamine carriers modulate nano-scale distances that define the functional assembly of mRNA and PABP.

(A) Schematic of stabilized mRNA/PABP complex via polyamines. (B) Representative histograms from the intracellular FRET assay measuring the degree of co-localization between FITC-mRNA and Cy5-PABP. At 24 hours after transfection of Cy5-PABP or FITC-mRNA/Cy5-PABP into HEK293T cells with each polyamine, cells were analyzed by flow cytometry using a 488 nm laser and 695 nm detector with 40 nm bandwidth. The polyamine structures are shown in Supplemental Scheme 1. (C) Mean FRET intensity, calculated by subtracting the fluorescence signal of Cy5-PABP-transfected cells from that of FITC-mRNA/Cy5-PABP-transfected cells. Data represent the mean ± SEM (n=2).
To demonstrate potential applications for gene therapy, we next investigated whether PABP could enhance mRNA delivery in vivo. As a proof-of-concept, luciferase mRNA or luciferase mRNA/PABP was packaged with the polyamine-”2-2-2-2”, which exhibited maximal enhancement of mRNA expression in vitro (Figure 2B and C). The transgene expression peaked at 6 hours following intravenous injection into Balb/c mice and was primarily found in the lungs due to the cationic nature and sizes of these complexes (Figure 4A). The signals decayed exponentially over 48 hours in both mRNA/polyamine and mRNA/PABP/polyamine groups, indicating the nature of transient gene expression in mRNA-mediated gene delivery. However, preassembly with PABP increased luciferase expression by ~4 fold relative to mRNA/polyamine alone at 6 hours (Figure 4A–D). Nevertheless, the degree of enhancement was lower than that observed in vitro. This difference may reflect challenges in transfecting primary cells and the lower stability of mRNA/PABP nanoplexes in blood plasma when delivered systemically in mice. Future studies investigating strategies to improve the plasma stability of nanoplexes and/or attach a targeting moiety can potentially allow for efficient, tissue-specific gene delivery in vivo.
Figure 4. Delivery of mRNA/PABP nanoplexes enhances mRNA expression in mice.

Bioluminescence imaging (BLI) at (A) 6, (B) 24, and (C) 48 hours after tail vein injection of luciferase mRNA or mRNA/PABP packaged with polyamine-(2-2-2-2). A third mouse from each treatment is not shown. (D) Quantification of luciferase expression in the lungs of Balb/c mice via BLI. Data represent the mean ± SEM (n=3); *, P<0.05.
To enable efficient cellular transfection, we previously investigated polyamines with linear aminoethylene repeat units in clustering mRNA/G cap-binding protein (eIF4E) complexes and observed differences in enhancement of eIF4E-mediated mRNA expression in cells. In the current study, however, we first engineered synthetic mRNA with a long poly A tail that allows for recognition of multiple PABP proteins in a tandem fashion for optimal mRNA expression. By modulating the stoichiometry between the mRNA strand and the poly A-binding protein, PABP, we achieved a substantial increase of mRNA expression inside cells. Moreover, we expanded the polyamine library to include additional side chain structures such as a propanediamine unit, ring structure, and branched conformations. As a result, we discovered additional polycation structures for improved mRNA transfection in co-delivery with PABP. By comparing a series of polyamines with the same backbone but distinct side chain amines, we discovered that the ability of PABP to enhance mRNA expression is dependent on the side chain structure. Among the seven polyamines examined, a clear correlation between PABP-mediated increase in mRNA expression and the stability of mRNA/PABP association was observed. Furthermore, comparison with a previous study demonstrated that the functional assembly of mRNA/eIF4E and mRNA/PABP complexes displayed preferences towards different polyamine structures for maximal enhancement of mRNA expression in vitro and in vivo. In particular, the ethylenediamine repeat appears to be very effective, particularly in linear form. This dependence on the polyamine structure may reflect differences in the mRNA-associated protein itself (e.g. surface charge and protein size) and mRNA/protein interactions. Future work will examine how different polyamines coordinate the interactions between mRNA and different proteins (eIF4E and PABP) at the atomic scale and compare these to the natural conformation of ribonucleoprotein complexes in protein structure databases.
This work highlights how leveraging mRNA biology and materials chemistry can inspire new approaches to engaging mRNA in active protein expression inside cells. This co-delivery strategy of nucleic acid and associated translational protein can be applied to other nucleic acid-based drugs such as small interference RNA (siRNA), microRNA (miRNA), aptamers, and DNA plasmids, by identifying complementary proteins that can potentially facilitate the functions of these nucleic acids inside cells. With respect to the theoretical significance, a variety of mRNA carriers have been developed to enable robust mRNA delivery and expression in vitro and in vivo. However, studies on the detailed mechanisms are usually focused on the material properties related to buffering effects and charge distribution, often independent of the specific type of nucleic acid under investigation. Our work suggests that characterization of the dynamic interactions between the synthetic delivery vehicle, mRNA, and the cellular proteins responsible for mRNA expression may offer a unique avenue towards the development of new mRNA vehicles with significantly greater therapeutic efficacy.
Supplementary Material
Acknowledgments
This work was supported by the Department of Defense (DoD) Ovarian Cancer Research Program Teal Innovator Award (PTH), the DoD Peer Reviewed Orthopaedic Research Program (PRORP) Idea Development Award (PTH) and the Koch Institute Quinquennial Postdoctoral Fellowship (JL). We thank: Glenn Paradis at the David H. Koch Institute Flow Cytometry Core at MIT for providing assistance in the intracellular FRET experiment; Department of Comparative Medicine at MIT for assistance of animal study. CW acknowledges the National Science Foundation Graduate Research Fellowship.
Footnotes
Lab webpage: https://hammondlab.mit.edu/
References
- 1.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. Nature. 2016;534:396–401. doi: 10.1038/nature18300. [DOI] [PubMed] [Google Scholar]
- 2.Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, et al. Nature. 2017;543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM, Spater D, Xu H, Tabebordbar M, Gorbatov R, et al. Nature biotechnology. 2013;31:898–907. doi: 10.1038/nbt.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, Huppmann M, Mays LE, Illenyi M, Schams A, et al. Nature biotechnology. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
- 5.Kreiter S, Selmi A, Diken M, Koslowski M, Britten CM, Huber C, Tureci O, Sahin U. Cancer research. 2010;70:9031–9040. doi: 10.1158/0008-5472.CAN-10-0699. [DOI] [PubMed] [Google Scholar]
- 6.Boczkowski D, Nair SK, Nam JH, Lyerly HK, Gilboa E. Cancer research. 2000;60:1028–1034. [PubMed] [Google Scholar]
- 7.Zou S, Scarfo K, Nantz MH, Hecker JG. Int J Pharm. 2010;389:232–243. doi: 10.1016/j.ijpharm.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sahin U, Kariko K, Tureci O. Nat Rev Drug Discovery. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 9.Schlake T, Thess A, Fotin-Mleczek M, Kallen KJ. Rna Biol. 2012;9:1319–1330. doi: 10.4161/rna.22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirschman JL, Bhosle S, Vanover D, Blanchard EL, Loomis KH, Zurla C, Murray K, Lam BC, Santangelo PJ. Nucleic acids research. 2017 doi: 10.1093/nar/gkx290. [DOI] [PMC free article] [PubMed]
- 11.Aini H, Itaka K, Fujisawa A, Uchida H, Uchida S, Fukushima S, Kataoka K, Saito T, Chung UI, Ohba S. Scientific reports. 2016;6:18743. doi: 10.1038/srep18743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jekhmane S, de Haas R, Paulino da Silva Filho O, van Asbeck AH, Favretto ME, Hernandez Garcia A, Brock R, de Vries R. Nucleic acid therapeutics. 2017;27:159–167. doi: 10.1089/nat.2016.0660. [DOI] [PubMed] [Google Scholar]
- 13.Udhayakumar VK, De Beuckelaer A, McCaffrey J, McCrudden CM, Kirschman JL, Vanover D, Van Hoecke L, Roose K, Deswarte K, De Geest BG, et al. Advanced healthcare materials. 2017;6 doi: 10.1002/adhm.201601412. [DOI] [PubMed] [Google Scholar]
- 14.Uchida H, Itaka K, Uchida S, Ishii T, Suma T, Miyata K, Oba M, Nishiyama N, Kataoka K. Journal of the American Chemical Society. 2016;138:1478–1481. doi: 10.1021/jacs.5b11726. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Wang W, He Y, Li Y, Yan EZ, Zhang K, Irvine DJ, Hammond PT. ACS nano. 2017 doi: 10.1021/acsnano.6b08447. [DOI] [PMC free article] [PubMed]
- 16.Wang Z, Day N, Trifillis P, Kiledjian M. Molecular and cellular biology. 1999;19:4552–4560. doi: 10.1128/mcb.19.7.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wakiyama M, Imataka H, Sonenberg N. Current biology : CB. 2000;10:1147–1150. doi: 10.1016/s0960-9822(00)00701-6. [DOI] [PubMed] [Google Scholar]
- 18.Gray NK, Coller JM, Dickson KS, Wickens M. The EMBO journal. 2000;19:4723–4733. doi: 10.1093/emboj/19.17.4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakanishi M, Park JS, Jang WD, Oba M, Kataoka K. React Funct Polym. 2007;67:1361–1372. [Google Scholar]
- 20.Pardi N, Tuyishime S, Muramatsu H, Kariko K, Mui BL, Tam YK, Madden TD, Hope MJ, Weissman D. Journal of controlled release : official journal of the Controlled Release Society. 2015;217:345–351. doi: 10.1016/j.jconrel.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deo RC, Bonanno JB, Sonenberg N, Burley SK. Cell. 1999;98:835–845. doi: 10.1016/s0092-8674(00)81517-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.