

Abstract
Alzheimer’s disease (AD), a common neurodegenerative disease, is characterized by the aggregation of amyloid-β (Aβ) peptides. The interactions of Aβ with membranes cause changes in membrane morphology and ion permeation, which are responsible for its neurotoxicity and can accelerate fibril growth. However, the Aβ-lipid interactions and how these induce membrane perturbation and disruption at the atomic level and the consequences for the Aβ organization are not entirely understood. Here, we perform multiple atomistic molecular dynamics (MD) simulations on three protofibrillar Aβ9–40 trimers. Our simulations show that, regardless of the morphologies and the initial orientations of the three different protofibrillar Aβ9–40 trimers, the N-terminal β-sheet of all trimers preferentially binds to the membrane surface. The POPG lipid bilayers enhance the structural stability of protofibrillar Aβ trimers by stabilizing inter-peptide β-sheets and D23-K28 salt-bridges. The interaction causes local membrane thinning. We found that the trimer structure related to Alzheimer’s disease brain tissue (2M4J) is the most stable both in water solution and at membrane surface, and displays slightly stronger membrane perturbation capability. These results provide mechanistic insights into the membrane-enhanced structural stability of protofibrillar Aβ oligomers and the first step of Aβ-induced membrane disruption at the atomic level.
Introduction
Alzheimer’s disease (AD), which was first identified more than a hundred years ago, is a neurodegenerative brain disease recognized as the prevailing cause of dementia as well as a major cause of death1. Between 2000 and 2013, deaths resulting from stroke, heart disease and cancer decreased 23%, 14%, and 11%, respectively, whereas those from Alzheimer’s disease increased 71%2. The accumulation and aggregation of amyloid beta (Aβ) outside neurons (senile plaques) and abnormal tau inside neurons (neurofibrillary tangles) are considered as two biomarkers of Alzheimer’s disease and are also believed to contribute to the damage of neural tissues in human brains3, 4. Aβ is an intrinsically disordered protein (IDP) in solution. Compared to folded proteins, the free energy landscape of IDPs is rather shallow, consisting of many metastable states generally separated by lower energy barriers, which allows conformational transitions to occur readily5. Thus IDPs usually have higher aggregation propensity than folded proteins6.
Aβ peptide results from proteolysis of the amyloid precursor protein (APP) in the transmembrane region by β- and γ-secretase. Protease cleavage of APP at different positions by γ-secretase generates a variety of Aβ peptides such as Aβ1–37, Aβ1–40, Aβ1–42 and Aβ1–437. Previous solution NMR experiments reported that Aβ monomer is largely unstructured in aqueous solution8, 9, while it can also adopt a partially folded structure with a 310 helix from residues H13 to D23 (PDB ID: 2LFM)10. A recent NMR study showed that Aβ1–40 adopts a partially helical structure upon binding to zwitterionic lipid bilayers prior to amyloid formation11. Aβ monomers can readily undergoes aggregation by a nucleation–elongation process leading to the formation of various oligomers and amyloid fibrils12. Aβ1–42 aggregates faster in vitro13 and is identified as more cellular toxic than the other alloforms14, while Aβ1–40 levels in human brains are nearly 5 times higher than Aβ1–42 levels15. The main components of the senile plaques in the brains of AD patients are Aβ1–40 and Aβ1–42 peptides16, with a wide range of Aβ1–40/Aβ1–42 ratios17. Both Aβ1–40 and Aβ1–42 are good model systems in the study of aggregation mechanism and neuronal toxicity of Aβ peptides18. As demonstrated by different experimental techniques such as solid-state nuclear magnetic resonance (ssNMR) and electron microscopy, Aβ fibrils are structurally polymorphic and exhibit significant differences in the extent and locations of stable β-sheets19–22. Several ssNMR-derived Aβ1–40/Aβ1–42 fibril models were reported15, 23–28. Three of them have been extensively studied and their PDB IDs are 2BEG (residues 17–42 for synthetic Aβ1–42 fibrils)25, 2LMN (residues 9–40 for synthetic Aβ1–40 fibrils)26 and 2M4J (residues 1–40 for AD-brain-derived Aβ1–40 fibrils)15. More importantly, Aβ fibrils with variable structures display distinct levels of toxicity in neuronal cell cultures22, 29. Thus, studies of the interactions between different fibrils or fibril-like oligomers and membranes, especially the amyloid form found in Alzheimer’s disease brain tissue (2M4J)15 can illuminate important pathology-related occurrences.
Experimental studies reported that mature fibrils and intermediate aggregates of Aβ are cytotoxic30–32. Toxicity is related to their interactions with the neuronal lipid bilayer33. Membranes can serve as a site for accumulation/nucleation, greatly accelerating the rate of fibrillization33, 34; but they can be disrupted in this process33, 34. Membrane thinning, pore formation and fragmentation are three possible Aβ-induced membrane disruption mechanisms33–36. Aβ interacts preferentially with anionic phospholipids37, 38 and the disruption effect is more prominent in negatively charged membranes38–40. However, the molecular mechanism of amyloid neurotoxicity is still not fully understood.
To get mechanistic insights into membrane disruption by toxic amyloids in atomic detail, several computational groups have investigated the interactions of Aβ oligomers with lipid bilayers by performing molecular dynamics (MD) simulations41–47. Jang et al examined the structural properties of modeled Aβ9–42/Aβ17–42 channels in zwitterionic POPC lipid bilayers and obtained Aβ channel conformations with diameters consistent with AFM measurements41, 48–50. Zhao et al studied the trimerization of helical Aβ1–42 peptide inside cholesterol-rich DPPC lipid bilayers and observed formation of short parallel β-sheet structures43. MD simulations by Poojari et al. demonstrated that compared to transmembrane Aβ1–42 monomers, membrane-embedded β-sheet-rich tetramers increased the water flow through the POPC bilayers44. Brown et al. reported that the binding of disordered Aβ1–42 tetramers on POPC membranes resulted in a greater membrane perturbation than that on cholesterol-rich membranes47. Multiple 80-ns MD simulations by Yu et al showed that the interactions of protofibrillar Aβ17–42 pentamers with a mixed POPC and POPG membrane are stronger than those with POPC membranes45, indicative of the role of surface charge in Aβ-membrane interactions. Recently, Tofoleanu et al explored the influence of chemical compositions of POPC and POPE headgroups on the interactions of Aβ9–40 protofilaments with membranes by performing MD simulations ranging from 25–150 ns. They found that POPC membranes exhibited weaker interactions with Aβ9–40 protofilaments than POPE51. However, a systematic study of the interactions of protofibrillar Aβ oligomers with lipid bilayers that would evaluate and compare these is lacking.
In this study, we investigate how protofibrillar Aβ oligomers and membranes interact with and mutually influence each other by performing extensive all-atom molecular dynamics (MD) simulations in the absence and presence of a lipid bilayer. The structures of Aβ1–42 have special folds due to the K28-A42 salt bridge28, 52. We focus on the Aβ9–40 sequence which may capture the common features of Aβ40 and Aβ42. Three different Aβ9–40 trimers were constructed using the three extensively-studied ssNMR-derived fibril models (PDB ID: 2BEG, 2LMN and 2M4J) which have a U-shaped topology consisting of a β-sheet—turn—β-sheet conformation15, 25, 26. 2M4J structure is found in the brain tissue of patients with Alzheimer’s disease22. Using the ssNMR-derived fibrils to construct the Aβ9–40 trimers allows us to investigate the interactions between Aβ protofibrils and membranes. These trimers were used to model the protofibrillar Aβ oligomers. An anionic POPG bilayer was employed to model the phospholipid membrane. Our selection of trimer is motivated by two previous studies. A single-molecule study by Ding et al reported that trimers and tetramers may be the smallest Aβ1–40 oligomers in the anionic lipid bilayers and could be the origin of neurotoxicity53. A MD simulation work by Zhao et al. showed that protofibrillar Aβ trimers might be the smallest seeding nucleus on the self-assembled monolayer surfaces54. We carried out four independent 200-ns MD simulations for each protofibrillar Aβ9–40 trimer. The initial minimum distance of between the backbone of Aβ and the surface of the POPG lipid bilayer is ~0.8 nm. To avoid the bias of the initial orientation of Aβ9–40 trimer with respect to the POPG bilayer surface on the simulation results, we considered four different initial orientations for each protofibrillar trimer (see Table 1). Our simulations show that regardless of the conformations of the three different Aβ9–40 trimers and their initial orientations relative to the membrane surface, the N-terminal β-sheet region of Aβ preferentially binds to the membrane surface due to strong electrostatic interactions between the positively charged N-terminus (NH3+) and side chains of K16 residue and the negatively charged phosphate groups in the POPG head groups. The membrane-adsorbed Aβ9–40 trimers have higher β-sheet content and more salt-bridges than the trimers in water, demonstrating that the POPG membrane stabilizes the protofibrillar Aβ trimers. On the other hand, the Aβ trimers cause membrane perturbation by decreasing the local thickness of lipid bilayers.
Table 1.
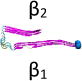
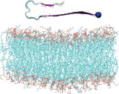
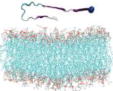
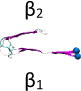
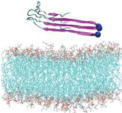
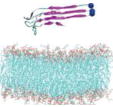
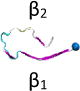
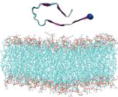
Name and initial state of Aβ and Aβ-POPG systems. The PDB ID of each fibril model denotes the three constructed protofibrillar Aβ9-40 trimers. In the four initial states of each Aβ-POPG system, the orientations of the protofibrillar Aβ trimers with respect to the POPG bilayer surface differ. According to the initial orientations of each trimer relative to the lipid bilayer surface, the four initial Aβ-POPG states are labelled as β1-to-lipid, β2-to-lipid, face-to-lipid and back-to-lipid. The β1-to-lipid and β2-to-lipid represent respectively the two systems where the N-terminal and C-terminal β-sheets face to the lipid bilayer surface. The face-to-lipid and back-to-lipid represent respectively the systems where the face and back of the protofibrillar Aβ trimer face to the lipid bilayer surface. The Aβ trimer is shown in cartoon representation, with the β-sheet in purple, the coil in white and the turn in cyan. The Cα atom of each G9 residue is denoted by a blue sphere. The lipids are shown in line representation, with oxygen, phosphorus, hydrogen and carbon atoms in red, tan, white and cyan, respectively. For clarity, counterions and water molecules are not shown.
| Aβ system | Aβ-POPG system | |||||
|---|---|---|---|---|---|---|
| Name | Initial state | Name | Initial states | |||
| β1-to-lipid | β2-to-lipid | face-to-lipid | back-to-lipid | |||
| 2BEG |
|
2BEG-POPG |
|
|

|

|
| 2LMN |
|
2LMN-POPG |

|

|
|
|
| 2M4J |
|
2M4J-POPG |
|

|
|
|
Materials and Methods
Protofibrillar Aβ9–40 trimers
The amino acid sequence of the 32-residue Aβ9–40 peptide is NH3+-GYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVV-COO−. To simulate the experimental neutral pH condition, the side chain of Lys (Lys+), Glu (Glu−) and Asp (Asp−) are charged. The N- and C-termini (NH3+, COO−) are also charged. The atomic coordinates of each Aβ9–40 peptide were extracted respectively from the three ssNMR-derived Aβ fibril models (PDB ID: 2BEG, 2LMN and 2M4J)15, 25, 26. Each peptide chain has a U-shaped conformation. 2BEG fibrils (residues 17–42) have β-strands at residues L17−S26 and I31−V40 and a turn region spanning residues N27-A3025, while 2LMN fibrils (residues 9–40) have β-strands at Y10−D23 and A30−G38 and a turn at residues V24-G2926. 2M4J Fibrils (residues 1–40) show a deformed U-shaped conformation, with β-strand-like regions at residues V12−F19 and I31-V36, a turn at F20-A30, a bend at G37−G38 and a more ordered N-terminal region15. In order to facilitate comparison of the structural properties of the three protofibrillar Aβ trimers, in this study, the same amino acid sequence (i.e. Aβ9–40) was considered for the three trimers. Thus, residues 9–16 (with sequence GYEVHHQK in β-strand conformation) were added to the originally published 2BEG structure, while residues 41 and 42 were removed. No modifications were done for the originally published 2LMN structure. Residues 1–8 were removed from the original 2M4J. Each protofibrillar Aβ9–40 trimer was constructed by in-register parallel stacking of the β-strands (Fig. S1). For convenience, we use the PDB ID (2BEG, 2LMN and 2M4J) of each NMR-derived fibril model to denote the three Aβ9–40 trimers (see Table 1). Note that the 2BEG and 2M4J systems used in this work were modified from the originally published pdb structures. In spite of modifications, the elongated 2BEG and truncated 2M4J still maintain respectively the structural feature of the original 2BEG and 2M4J fibrils: the U-shaped conformation stabilized respectively by inter- and intra-molecular D23-K28 salt-bridges.
POPG lipid bilayer
The POPG lipid bilayer is composed of 2×100 lipids (i.e., 100 lipids in each leaflet). The initial coordinates and force field parameters are obtained from lipidbook55, 56. The initial size of the POPG bilayer is ~8 nm×8 nm in the x-y plane. The molecular structure of a POPG lipid is given in Fig. S2. The properties of POPG membranes have been recently determined by MD simulations and small-angle neutron and X-ray scattering experiments, indicating that the molecular area is 66.0±1.3 Å2 and the bilayer thickness is 36.7±0.7 Å57. The POPG bilayer is fully anionic, and has been routinely used in experiments and simulations to probe the interaction of negatively charged membrane with various charged peptides. Beschiaschvili and Seelig studied the binding of cyclic somatostatin analogue peptides with neutral and negatively charged monolayer films58. They found that binding of the positively charged peptide and the negatively charged POPG surface was enhanced as compared to the binding to a neutral POPC membrane58. A study exploring membrane selectivity of the antimicrobial peptide KIGAKI using solid-state NMR spectroscopy found that strong electrostatic interaction between the cationic peptide KIGAKI and anionic POPG lipids is not the only factor determining the antimicrobial activity59. Nevertheless, the use of a fully anionic lipid bilayer allows us to observe Aβ-membrane interactions within relatively short simulation time scales as Aβ prefers to interact with anionic lipids37, 38, thus saving computational cost.
Aβ-POPG system
Each Aβ9–40 protofibrillar trimer was placed on the upper leaflet of the POPG, with a minimum distance of ~0.8/0.5 nm between the backbone/all-atoms of Aβ and the surface of the POPG lipid bilayer. To eliminate the bias of the initial orientation of Aβ9–40 trimers relative to the POPG surface on the simulation results, we chose four different initial orientations for each trimer (see Table 1): N-terminal β1-stand (β1 surface) or C-terminal β2-strand (β2 surface) facing the lipid surface; one side of the strand-turn-strand conformation (face) or the other side of the strand-turn-strand conformation (back) orienting towards the bilayer surface. The four surfaces (β1, β2, face and back) of the Aβ trimers are shown in Fig. S1. Each initial orientation corresponds to an initial state of MD simulations. Thus, there are four different initial states for each Aβ-POPG system. The four initial Aβ-POPG states are labelled as β1-to-lipid, β2-to-lipid, face-to-lipid and back-to-lipid (Table 1).
Simulation details
All MD simulations were performed in the isothermal-isobaric (NPT) ensemble using GROMACS-4.5.3 software package60. The Amber99SB force field61 was used for the Aβ9–40 trimers. The POPG lipids were described with the Jämbeck force field56, 62, 63. During the preparation stage for the MD simulations, the Aβ-POPG system was placed in the center of a rectangular box of 8.2×8.3×13.7 nm3, with the distance between Aβ trimer and the box wall being ~3.0 nm. Periodic boundary conditions were applied. Then the Aβ-POPG system was fully solvated in simple point charge (SPC) water. Na+ and Cl− ions were added to neutralize the system, which provides an additional 0.1 M salt concentration. Bond length of peptides and water molecules were constrained respectively using the LINCS64 and SETTLE65 algorithms, allowing an integration time step of 2 fs. The protein, POPG membrane and non-protein (water molecules and counterions) groups were separately coupled to an external heat bath with a relaxation time of 0.2 ps using a velocity rescaling coupling method66. The temperature of the system is kept close to 310 K, above the gel-liquid crystal phase transition temperature (271 K) of POPG bilayers67. The pressure was kept at 1 bar using the Parrinello-Rahman method68 with a coupling constant of 1.0 ps. Electrostatic interactions were treated with the particle mesh Ewald (PME) method with a real space cutoff of 1.0 nm. The van der Waals interactions were calculated using a cutoff of 1.4 nm. Each system was energy-minimized by steepest descent for 50000 steps. After minimization, the solvent was equilibrated in a 100-ps NVT MD run with position restraints on the protein. The solvent equilibration was followed by another 100-ps NVT MD run without position restraints on the protein. Then a 100-ps NPT MD run was performed on the full system. This was followed by production MD runs for each system. Four independent 200-ns production MD simulations were conducted for each of the three different protofibrillar Aβ trimers in the presence of POPG membrane (Aβ-POPG system) (12 simulations in total). For comparison, we also carried out a 200-ns MD simulation for each Aβ9–40 trimer in water without POPG membrane (Aβ system). The name and initial state of Aβ and Aβ-POPG systems are given in Table 1. A summary of the MD setup details is given in Table 2.
Table 2.
A summary of the MD setup details. For each system, we present the name of the system, the size of the simulation box, the total number of atoms, simulation time, the number of MD runs and the number of initial states.
| System | Box size (nm3) | Total number of atoms | Simulation time (ns) | Number of MD runs | Number of initial states |
|---|---|---|---|---|---|
| 2BEG | 7.5×7.5×9.0 | 49751 | 200 | 1 | 1 |
| 2LMN | 7.5×7.5×9.0 | 49769 | 200 | 1 | 1 |
| 2M4J | 7.5×7.5×9.0 | 49739 | 200 | 1 | 1 |
| 2BEG-POPG | 8.2×8.3×13.7 | 91001 | 200×4 | 4 | 4 |
| 2LMN-POPG | 8.2×8.3×13.7 | 91001 | 200×4 | 4 | 4 |
| 2M4J-POPG | 8.2×8.3×13.7 | 91001 | 200×4 | 4 | 4 |
Analysis
The β-sheet content was calculated using the DSSP program69. A salt bridge is formed if the minimum distance between the side chain COO− group of D23 and the Nζ atom of the side chain NH3+ group of K28 (i.e. the D23-K28 distance) is less than 0.4 nm. The thickness of the POPG bilayer was estimated by the local average of phosphor-to-phosphor distance70. All the snapshots are displayed using the VMD program71.
Results and Discussion
Adsorption dynamics of three protofibrillar Aβ trimers on POPG lipid bilayers
To explore the interaction mechanism of protofibrillar Aβ trimers with anionic lipid bilayers, we firstly investigate the adsorption behavior of each trimer on POPG bilayers. Figure 1 shows the time evolution of the minimum distance between each amino acid residue of the 2BEG trimer and the POPG bilayer and snapshots at five different time points starting from four different initial states. In the MD run with the initial state of β1-to-lipid (Fig. 1(a)), the turn region (residues G25-K28) is adsorbed on the bilayer first, followed by the N-terminal residues G9-K16, resulting in membrane binding of the β1 and turn regions. This observation is not surprising as the simulation started from the β1-to-lipid state. In the MD runs initiating from β2-to-lipid (Fig. 1(b)), face-to-lipid (Fig. 1(c)) and back-to-lipid (Fig. 1(d)), the N-terminal residues G9-H14 are adsorbed on the bilayer surface first, then residues Q15-K28 in the β1 region sequentially bind. This leads to the binding of the full β1 region and the turn region to the membrane surface. Of particular interest is the adsorption process observed in Fig. 1(b) that although C-terminal residues initially face towards the membrane surface, the Aβ trimer still rotates itself and the N-terminal residues bind to the lipid bilayer. Overall, irrespective of the initial orientations, it is the β1 region (containing positively-charged N-terminus NH3+ and residues K16) or the turn region (containing positively-charged residues K28) that initiate the adsorption, which results in the binding of β1+turn region of 2BEG trimer to the lipid bilayers (see the snapshots at t=200 ns in Fig. 1).
Fig. 1.
Time evolution of the minimum distance between all atoms of each residue in the three chains and the POPG lipid bilayer for the 2BEG-POPG system starting from four initial states: β1-to-lipid (a), β2-to-lipid (b), face-to-lipid (c) and back-to-lipid (d). Snapshots at five time points are given to visualize the adsorption behavior of 2BEG timers on the POPG bilayer surface.
The adsorption dynamics of the 2LMN trimer to the POPG bilayer is shown in Fig. 2. In the two MD runs starting from β1-to-lipid (Fig. 2(a)) and face-to-lipid (Fig. 2(c)), the N-terminal residues G9-H14 bind to the bilayer surface first, followed by residues Q15-K28, leading to membrane binding of the β1+turn region of the 2LMN trimer (snapshots in Fig. 2(a, c)). When starting from β2-to-lipid (Fig. 2(b)) and back-to-lipid (Fig. 2(d)), the adsorption to the POPG bilayer surface is initiated from the turn region (residues G25-K28). Only the turn region binds to the POPG surface within the 200 ns of these two MD runs, indicating that a long time is needed for the adsorption of β1 region on POPG bilayers. The detailed process can be seen from snapshots at five time points in Fig. 2.
Fig. 2.
Time evolution of the minimum distance between all atoms of each residue in the three peptide chains and the POPG lipid bilayer for the 2LMN-POPG system in MD runs starting from four states: β1-to-lipid (a), β2-to-lipid (b), face-to-lipid (c) and back-to-lipid (d). The snapshots at five time points are also given to visualize the adsorption process of 2LMN timers on the POPG bilayer surface.
Fig. 3 shows the adsorption behavior of the 2M4J trimer onto the POPG bilayer. In the two runs starting from β1-to-lipid (Fig. 3(a)) and back-to-lipid (Fig. 3(d)), the adsorption is initiated from the N-terminal residues G9-H14. At the beginning of the β2-to-lipid simulation (Fig. 3(b)), residues in β2 and turn regions are quite close to the membrane surface, and at t=18 ns N27 residues bind to the bilayer. Shortly afterwards, the trimer rotates and the turn region dissociates from the bilayer, and then N-terminal residues G9-Y10 anchor into the bilayer. This is followed by sequential membrane binding of other residues in β1 region and of the turn region, leading to the β1+turn region binding. This binding event indicates that the β1 region has a preference to bind to the POPG bilayer although initially β2 and turn regions have atomic contacts with the bilayer. For the face-to-lipid (Fig. 3(c)), it appears that the whole Aβ trimer comes close to the membrane surface at first. After that, N-terminal residues keep contacts with the bilayer, while C-terminal residues in the β2 region gradually dissociate. The detailed process can be seen from the snapshots at five time points in Fig. 3.
Fig. 3.
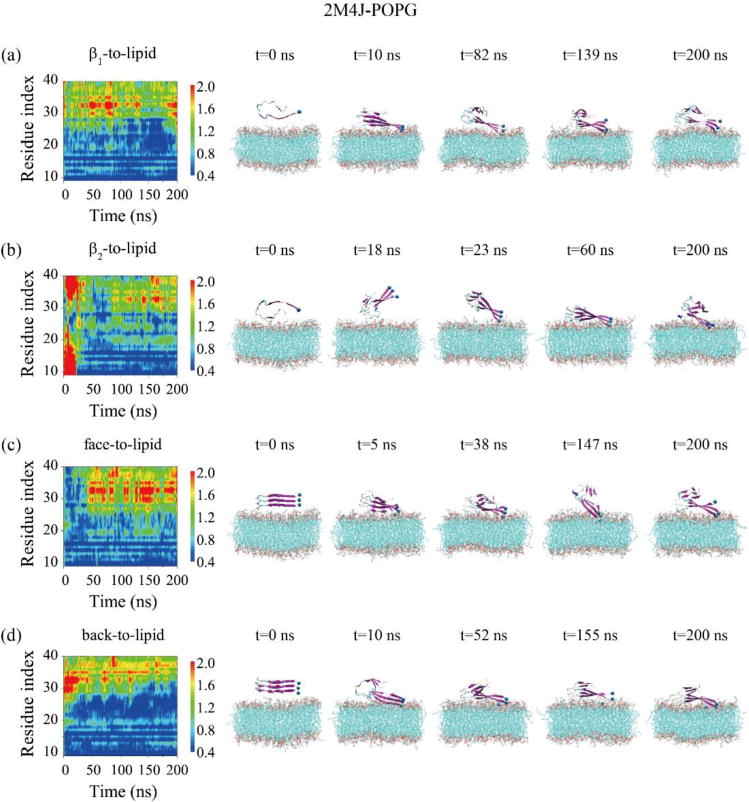
Time evolution of the minimum distances between all atoms of each residue in the three chains and the POPG lipid bilayer for 2M4J-POPG system in MD runs starting from four different initial states: β1-to-lipid (a), β2-to-lipid (b), face-to-lipid (c) and back-to-lipid (d). The snapshots at five different time points are given to visualize the adsorption dynamics of 2M4J timers on the POPG bilayer surface.
By comparing all the simulation results, we find that the three distinct trimers display similar adsorption behaviors, regardless of their structural morphologies and initial orientations. Namely, N-terminal residues G9-K16 in β1 region preferentially bind to the membrane surface, followed by other residues in the β1 and turn regions, leading to the binding of β1+turn region of Aβ trimer onto the lipid bilayers. This finding indicates that our simulation results do not depend on the initial states of Aβ-POPG systems. In term of absorption rate of the N-terminal β1 region, the three trimer forms are in the order of 2M4J > 2BEG > 2LMN.
To identify the key residues that bind to the POPG bilayer, we calculated the binding probability of each residue for the three trimers (Fig. 4). Our calculation shows that N-terminal residues G9-K16 (containing the positively charged N-terminal NH3+ and residues K16) in the β1 region exhibit the highest membrane binding probability and turn region G25-G29 (containing positively charged residues) display the second highest binding probability. These results indicate that electrostatic interaction is an important driving force to facilitate the adsorption on the anionic membrane. This finding is consistent with previous studies showing that electrostatic interactions with negatively charged lipids enhance peptide adsorption onto lipid monolayers/bilayers45, 72. The relative higher membrane binding probabilities of the residues in the N-terminal β1 region also suggest strong interactions between N-terminal residues of Aβ trimers and POPG bilayers.
Fig. 4.
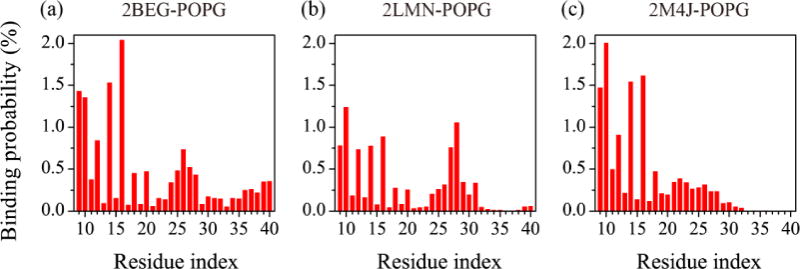
Membrane binding probability of each amino acid residue for the three protofibrillar Aβ trimers in the Aβ-POPG system. The binding probabilities were calculated using all heavy atoms over the last 50 ns for each Aβ-POPG system.
The influence of POPG bilayers on the structural stabilities of the Aβ trimmers
We conducted a 200-ns MD simulation for each trimer in water without the lipid bilayers. Fig. 5(a) and (b) show respectively the time evolution of the root-mean-square deviation (RMSD) of each trimer with respect to its initial conformation and the number of backbone hydrogen bonds. It can be seen from Fig. 5 that the RMSDs of the three trimers all reach a plateau after t=110 ns and their values are 0.8 nm for 2BEG, 1.1 nm for 2LMN and 0.5 nm for 2M4J. The RMSD values reflect different structural stabilities in water: the 2M4J trimer is the most stable, 2BEG is the less stable and 2LMN is the least stable. This finding is supported by the time evolution of the number of backbone hydrogen bonds (H-bonds) in Fig. 5(b). Namely, 2M4J has the largest number of H-bonds (note that it still increases with simulation time), 2BEG has the less and 2LMN has the least. The different structural stabilities in water can be clearly seen from the snapshots of Aβ trimer generated at t=0, 100 and 200 ns (Fig. 5(c, d, e)). The 2M4J trimer keeps its protofibrillar structure during the full process of the 200-ns MD simulation. The increase of its RMSD value with simulation time (blue curve in Fig. 5(a)) mainly results from the twisting of the three strands with respect to each other (see the snapshots at t=100 and 200 ns in Fig. 5(e)).
Fig. 5.
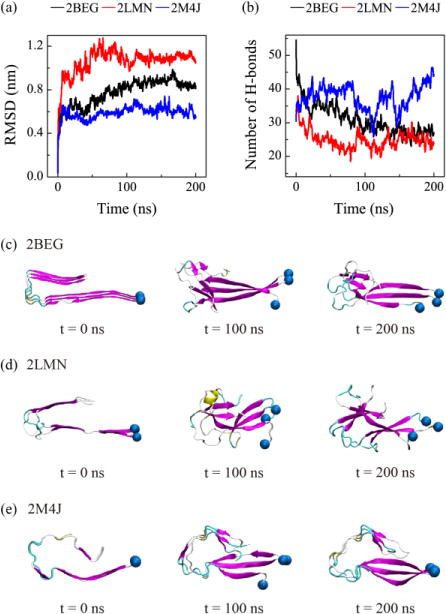
Structural stability analysis of the three protofibrillar Aβ9-40 trimers in water. Time evolution of the Cα-RMSD (a) and the number of backbone hydrogen bonds (b) of each trimer. Snapshots of Aβ trimers generated at 0, 100 and 200 ns for 2BEG (c), 2LMN (d) and 2M4J (e) systems are also shown. The snapshots at t=0 ns are the same as the initial states shown in Table 1 for the Aβ system.
The structural stability of each protofibrillar Aβ9–40 trimer in the presence of POPG lipid bilayers are investigated by monitoring the time evolution of the Cα-RMSD and the number of backbone H-bonds of Aβ trimer in MD runs starting from four different initial states (Fig. 6). For comparison, the time evolution of these two parameters for each Aβ trimer in water without POPG bilayers are also given (black curve). It can be seen from Fig. 6 that the Cα-RMSDs and backbone H-bond number of the three trimers reach a plateau after t=50 ns for 2BEG and after 100 ns for both 2LMN and 2M4J, indicating our simulations are reasonably converged. Figure 6(a, b, c) shows that 2BEG, 2LMN and 2M4J trimers have different RMSD values, implying their different structural stabilities on the POPG bilayer surface. 2M4J trimer is the most stable (with the smallest RMSD), 2BEG trimer is the less stable (larger RMSD) and 2LMN trimer is the least stable (with the largest RMSD value). The 2BEG and 2LMN trimers on the bilayer surface have smaller RMSD values and more backbone H-bonds than the trimers in water (Fig. 6(a, b, d, e)). The results reveal that membrane-bound protofibrillar 2BEG and 2LMN trimers are more stable than Aβ trimers in aqueous solution, highlighting the role of POPG bilayer in stabilizing the structures of Aβ trimers. In the MD run starting from β2-to-lipid (green curve in Fig. 6(a)), the RMSD value of the membrane-bound 2BEG trimer is almost the same as that of the isolated trimer in water, due to the slow adsorption of the trimer on the bilayer surface (see Fig. 1(b)). We also found that the RMSD values and the number of H-bonds of the membrane-bound 2M4J trimer resembles those of the trimer in water (Fig. 6(c, f)), implying that the 2M4J trimer on POPG has similar structural stability as in water. The small RMSD values (~0.5 nm) of 2M4J trimer in Fig. 6(c), together with the snapshots in Fig. 3 and Fig. 5(e), demonstrate that the 2M4J trimer is structurally stable both on the POPG membrane surface and in water. Thus, the POPG bilayer has minor influence on the structure of the 2M4J trimer. Overall, POPG bilayers can stabilize the protofibrillar Aβ trimers although the stabilization effect of the bilayer on 2M4J is not as obvious as that on 2BEG and 2LMN.
Fig. 6.
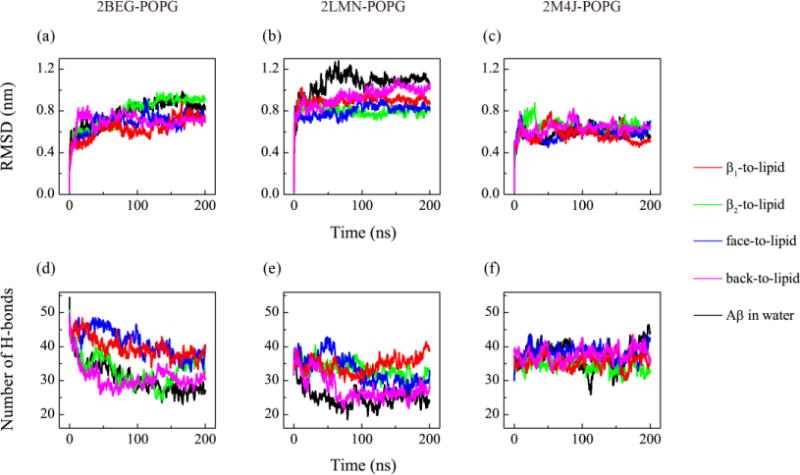
Structural stability analysis of the three protofibrillar Aβ9-40 trimers in the presence of POPG lipid bilayers. For each Aβ-POPG system (2BEG-POPG, 2LMN-POPG, 2M4J-POPG), we show the time evolution of the Cα-RMSD (a, b, c) and the number of backbone hydrogen bonds (d, e, f) of Aβ trimer in MD runs starting from four states: β1-to-lipid (red), β2-to-lipid (green), face-to-lipid (blue) and back-to-lipid (pink). For comparison, the results for each Aβ trimer in water without POPG bilayers are also given (black).
To identify the residues that contribute most to the structural stability of each trimer in the presence of POPG bilayers, we calculate the β-sheet propensity of each amino acid residue (red curves in Fig. 7). For each trimer, residue-based β-sheet propensity is obtained by averaging the β-sheet probability of each residue over the data in the four MD trajectories. For comparison, the β-sheet propensity for each trimer in water without POPG bilayers are also calculated (black curves in Fig. 7). We see from Fig. 7 that the β-sheet propensities of most residues in the β1 region of Aβ in the Aβ-POPG system are higher than those in water, while the β-sheet propensities of most of the residues in the β2 region are only slightly affected. This can also be seen from the time evolution of the secondary structure of each residue in the three MD runs for Aβ and in the 12 MD runs for Aβ-POPG system (Fig. S3). This result, together with the residue-based membrane binding probability in Fig. 4, indicates that membrane binding stabilizes the β-sheet structure of the N-terminal residues, which is favorable for the structural stability of Aβ trimers, especially for 2BEG and 2LMN trimers.
Fig. 7.
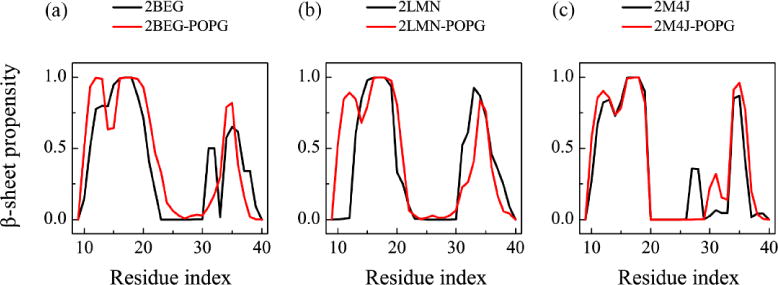
The residue-based β-sheet propensity of each Aβ9-40 trimer in the absence and presence of POPG lipid bilayers. The β-sheet propensity of each residue was calculated using the last 50 ns simulation data for both Aβ (black) and Aβ-POPG systems (red). For each Aβ-POPG system, the β-sheet propensity is the average of the four MD trajectories.
Previous experimental and computational studies suggested that D23-K28 salt-bridges enhance the fibrillization rate73, 74 and stabilize Aβ fibril structures15, 24–26. To examine whether POPG bilayers affect the D23-K28 salt-bridges, we calculate the PDF (probability density function) of the D23-K28 distance (including both intra- and inter-peptide D23-K28 distances) in isolated (black curves in Fig. 8) and in membrane-bound Aβ trimers (red curves in Fig. 8). The D23-K28 distance in Aβ-POPG system is the average of the four different MD runs. The PDF of the D23-K28 distance in each MD run for Aβ and Aβ-POPG systems are presented in Fig. S4. Two peaks (Fig. 8(a, b), black curves) are observed for the isolated 2BEG and 2LMN trimers: a small sharp peak centered at 0.28 nm and a broad peak around 0.6 nm. The probability density distribution of the two peaks implies that the D23-K28 salt-bridges in the isolated 2BEG and 2LMN trimers are almost lost. In the Aβ-POPG system, the peak at 0.28 nm becomes much larger (red curves in Fig. 8(a, b)), implying that most of the D23-K28 salt bridges are preserved in the membrane-bound trimers and the interaction of Aβ trimers with POPG bilayers protects the salt bridges. The peak at 0.28 nm in the membrane-bound 2M4J trimer also becomes larger than that in the isolated trimer (Fig. 8(c)), indicative of the stabilizing role of POPG bilayers on the D23-K28 salt-bridges in the 2M4J trimer. These results demonstrate that POPG bilayers stabilize the D23-K28 salt bridges that were reported to play a crucial role in Aβ fibrillization73, 74.
Fig. 8.
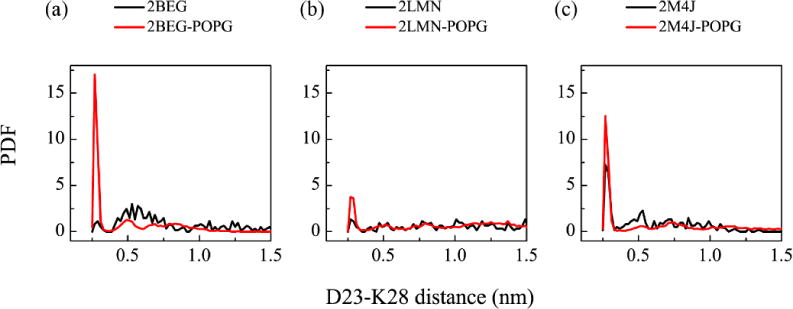
Effect of POPG bilayers on the D23-K28 salt-bridge. Probability density function of the D23-K28 distance in Aβ (black curve) and Aβ-POPG (red curve) systems. The D23-K28 distance in Aβ-POPG system is the average of the four MD runs. All calculations were based on the last 50 ns data of each MD run.
Altogether, our simulations demonstrate that protofibrillar Aβ9–40 trimers become much more stable upon binding to the POPG bilayer. It is expected that the membrane-stabilized trimers would enhance the fibril formation. Consistent with our prediction, recent studies reported that Aβ fibrillation can be accelerated in the presence of anionic lipid membranes33, 34.
The influence of protofibrillar Aβ trimers binding on POPG bilayers
Membrane integrity is essential for cellular activities. To investigate the perturbation of the POPG bilayers incurred by binding of the trimers, we calculated the bilayer thickness distribution in the x-y plane for each Aβ-POPG system in the MD runs starting from β1-to-lipid, β2-to-lipid, face-to-lipid and back-to-lipid. The bilayer thickness distribution map is presented in Fig. 9. For comparison, the distribution map of a pure POPG bilayer is also given (Fig. 9(a)). The color bar from red to white represents the bilayer thickness decreasing from 3.7 to 3.2 nm. We see from Fig. 9(a) that pure POPG bilayer is almost uniform with a thickness of 3.7 nm. In contrast, the Aβ-POPG system exhibits obvious local membrane thinning (the white region in Fig. 9(b–m)) except for the bilayer in Fig. 9(g) and (i). In the two MD runs corresponding to Fig. 9(g) and (i), only the turn region is adsorbed on the POPG bilayer surface (see the snapshots at t=200 ns in Fig. 2(b) and (d)), while both the N-terminal β1 region and the turn region bind to the bilayer surface in all other MD runs. This observation, together with the different bilayer thickness distribution seen in Fig. 9 reveals that the membrane binding of protofibrillar Aβ trimers results in bilayer thinning and that it is the β1-POPG interaction that contributes mostly to the membrane thinning. This finding is consistent with a recent study showing that the susceptibility of neuronal cells to different types of oligomeric assemblies is directly related to the extent of binding of such oligomers to the cellular membrane75.
Fig. 9.
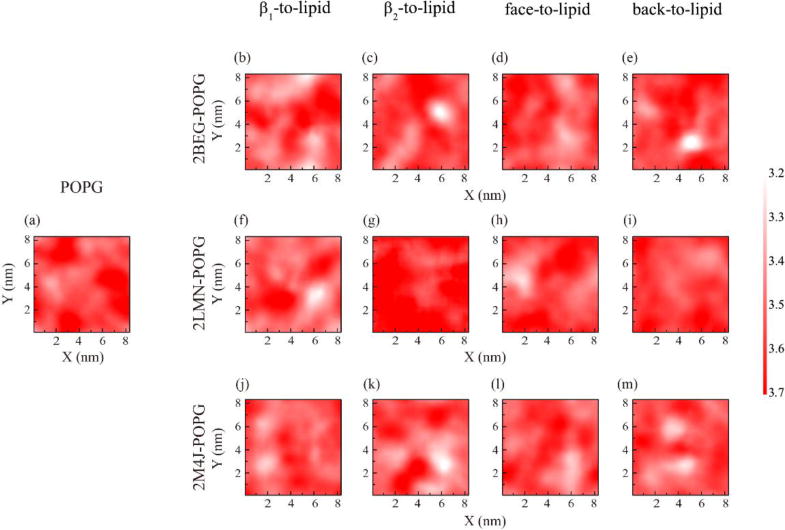
The POPG bilayer thickness distribution map over the x-y plane for each Aβ-POPG system in the MD runs starting from different initial states: β1-to-lipid, β2-to-lipid, face-to-lipid and back-to-lipid. For comparison, the bilayer thickness distribution map of a pure POPG bilayer is also presented. The bilayer thickness was calculated using the last 50-ns data of each MD run.
We then calculated the lipid tail order parameter SCD for lipids in the upper leaflet using the formula SCD=0.5 〈3cos2 θ−1〉, where θ is the angle between the bilayer normal and the C-H bond vector (in the simulations) or the C-D bond vector (in experiments)76. The angular brackets denote a time and lipid ensemble average76, 77. Figure 10(a, b) gives the absolute SCD values of acyl chain 1 and 2 (i.e. sn-1 and sn-2) of POPG lipids in each Aβ-POPG system. The sn-1 and sn-2 chains and the carbon atoms in the two chains are labelled in Fig. S2. For comparison, the SCD values of the two chains from a 200 ns MD run of a pure POPG lipid bilayer are also given in Fig. 10(a, b). We see from Fig. 10(a) that the SCD values of sn-1 chains in all Aβ-POPG systems are close to each other, but slightly smaller than those in a pure POPG bilayer. We noted that 2M4J (green line) leads to the lowest SCD values, indicating larger membrane perturbation. The differences among the SCD values of sn-2 chains in all systems are negligible. Consistent with the change of SCD values of sn-1 chains, the lipid area in each Aβ-POPG system becomes slightly larger than that in the pure POPG system, with 2M4J being the most obvious again (Fig. 10(c)). These results indicate that while membrane binding of protofibrillar Aβ trimers does not significantly impact the ordering of the POPG bilayer within our simulation time scale, structural polymorphism may still have different membrane perturbation effects. To examine whether the Aβ-induced membrane perturbation can cause water penetration into the POPG bilayer, we calculated the water density distribution along the membrane normal (i.e. the z-axis direction) for each Aβ-POPG system (Fig. S5). For comparison, the water density distribution along the membrane normal in a pure POPG bilayer is also given. The membrane center is at z=0 nm and the thickness of the hydrophobic tail region is ~2.8 nm (from z = −1.4 to 1.4 nm). The trimer binds on the upper surface (z = ~1.7 nm) of the POPG bilayer. As in the pure POPG system, the water density in the central hydrophobic tail region (from z = −1.0 to 1.0 nm) of POPG bilayer in the Aβ-POPG system is almost zero, indicating that membrane perturbation is not able to cause water penetration into the POPG bilayer within our simulation time scale. This finding suggests that binding alone is not sufficient for membrane permeabilization, consistent with a previous Forster resonance energy transfer (FRET) study showing that Aβ membrane binding and permeabilization are distinct processes influenced separately by membrane charge and fluidity38.
Fig. 10.
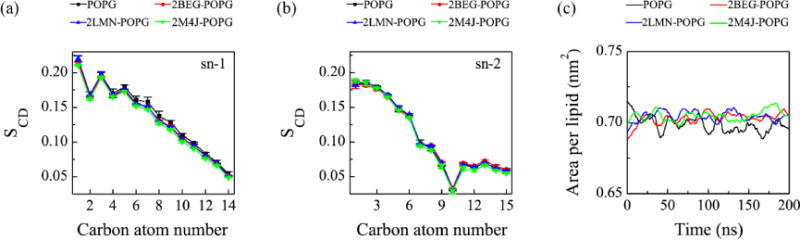
Influence of membrane binding of protofibrillar Aβ trimers on the order of the head and the tail atoms of POPG lipids in the upper leaflet of the bilayer. The order parameter SCD for the sn-1 chain (a) and sn-2 chain (b) of POPG as a function of carbon atom index. (c) Area per lipid as a function of simulation time. For each Aβ-POPG system, both the SCD values and the area per lipid are the average over the four different MD runs using the last 50 ns simulation data. The SCD and the area per lipid of a neat POPG lipid bilayer are also given for comparison.
Membrane thinning, pore formation and membrane fragmentation have been considered as three possible mechanisms of Aβ-induced membrane disruption33–36. Our simulations demonstrate that binding of protofibrillar Aβ trimers results in membrane thinning. Reduced thickness regulates the aggregation and cytotoxicity of Aβ78. As membrane binding is the initial step of Aβ-membrane interaction, we propose that membrane thinning might be the first step of Aβ-induced membrane disruption and this will result in pore formation or membrane fragmentation.
We note that there are some limitations in this work. For example, we used a fully anionic POPG lipid bilayer to model the membrane in order to observe Aβ-membrane interactions within relatively short time scale of simulations due to the large size of the simulated system. We used Aβ9–40 (an elongated 2BEG and a truncated 2M4J) trimers to model the full-length Aβ trimers in order to facilitate comparison of the structural properties of the three protofibrillar Aβ trimers in the absence and presence of phospholipid bilayers and to save computational cost. Finally, due to the short time scale of MD simulations, only the turn region of the 2LMN trimer binds to the POPG surface within the 200 ns of MD runs, while most of part of the trimers are exposed to water (Fig. 2(b, d)). Thus, it is desirable to study Aβ-protofibril-membrane interactions by employing a realistic model membrane (such as a mixed POPC and POPG bilayer) and full-length Aβ protofibrils and by performing long time scale MD simulations. This remains to be determined in a future study.
Conclusions
We have systematically investigated the interactions between anionic lipid bilayers and protofibrillar Aβ9–40 trimers constructed using the three NMR-derived fibril structures, 2BEG, 2LMN and 2M4J, by carrying out all-atom molecular dynamics simulations. We particularly focused on the adsorption dynamics of the trimers, the effect of POPG lipid bilayers on the stability of trimers, and the perturbation of the membrane induced by the interactions of Aβ with the lipid bilayers. Simulations starting from four states of each Aβ-POPG system reveal that irrespective of their structural details and initial orientations, the three distinct protofibrillar Aβ trimers display similar adsorption dynamics. N-terminal residues G9-K16 in β1 region preferentially bind to the membrane surface, followed by other residues in the β1 and turn regions, leading to the binding of β1+turn region of Aβ trimer to the lipid bilayers. This adsorption behavior is mostly dominated by the electrostatic interaction between positively charged residues and negatively charged POPG lipid bilayers. Lipid bilayers enhance the structural stability of Aβ trimers by stabilizing the β-sheet content and the D23-K28 salt-bridges. On the other hand, the POPG lipid bilayer is also influenced by the binding of protofibril Aβ trimers. Aβ binding decreases the local thickness, leading to membrane thinning which is related to aggregation and toxicity of Aβ peptides78. In particular, Aβ structure found in Alzheimer’s disease brain tissue (2M4J) is the most stable both in water solution and on membrane surface, and exhibits slightly stronger membrane perturbation ability. This investigation shows that peptide-membrane interaction can induce membrane thinning which is responsible for Aβ toxicity and suggests that the membrane may promote Aβ fibril growth by enhancing the structural stability of Aβ oligomers, ultimately providing insights into Aβ-lipid interaction and the mechanism of lipid perturbation by Aβ protofibrils at atomic level.
Supplementary Material
Acknowledgments
This work has been supported by the MOST of China (Grant No. 2016YFA0501702) and the NSF of China (Grant No. 11674065). It has also been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E. This research was supported [in part] by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
All simulations reported in this work had been performed using the high-performance computational facilities at the National High Performance Computing Center of Fudan University.
References
- 1.Wilson RS, Segawa E, Boyle PA, Anagnos SE, Hizel LP, Bennett DA. Psychology and aging. 2012;27:1008–1017. doi: 10.1037/a0029857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2016;12:459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 3.LaFerla FM, Green KN, Oddo S. Nature reviews Neuroscience. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 4.Ballatore C, Lee VM, Trojanowski JQ. Nature reviews Neuroscience. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 5.Wei G, Xi W, Nussinov R, Ma B. Chem Rev. 2016;116:6516–6551. doi: 10.1021/acs.chemrev.5b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uversky VN, Dave V, Iakoucheva LM, Malaney P, Metallo SJ, Pathak RR, Joerger AC. Chemical reviews. 2014;114:6844–6879. doi: 10.1021/cr400713r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 8.Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJ, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG. Journal of the American Chemical Society. 2004;126:1992–2005. doi: 10.1021/ja036813f. [DOI] [PubMed] [Google Scholar]
- 9.Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. Protein science : a publication of the Protein Society. 2005;14:1581–1596. doi: 10.1110/ps.041292205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vivekanandan S, Brender JR, Lee SY, Ramamoorthy A. Biochemical and biophysical research communications. 2011;411:312–316. doi: 10.1016/j.bbrc.2011.06.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korshavn KJ, Bhunia A, Lim MH, Ramamoorthy A. Chemical communications (Cambridge, England) 2016;52:882–885. doi: 10.1039/c5cc08634e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, Rigby AC, Teplow DB. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16522–16527. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Proc Natl Acad Sci U S A. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. The Journal of biological chemistry. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 15.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pitschke M, Prior R, Haupt M, Riesner D. Nature medicine. 1998;4:832–834. doi: 10.1038/nm0798-832. [DOI] [PubMed] [Google Scholar]
- 17.Gravina SA, Ho L, Eckman CB, Long KE, Otvos L, Jr, Younkin LH, Suzuki N, Younkin SG. The Journal of biological chemistry. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 18.Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete NV, Cote S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, Derreumaux P. Chemical reviews. 2015;115:3518–3563. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bertini I, Gonnelli L, Luchinat C, Mao J, Nesi A. Journal of the American Chemical Society. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 20.Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Fandrich M. Journal of molecular biology. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kodali R, Williams AD, Chemuru S, Wetzel R. J Mol Biol. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tycko R. Neuron. 2015;86:632–645. doi: 10.1016/j.neuron.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma B, Nussinov R. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:14126–14131. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. Proc Natl Acad Sci U S A. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. Proc Natl Acad Sci U S A. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petkova AT, Yau WM, Tycko R. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Nat Struct Mol Biol. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Science (New York, NY) 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Hong S, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benilova I, Karran E, De Strooper B. Nature neuroscience. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 32.Lesne SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain : a journal of neurology. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kotler SA, Walsh P, Brender JR, Ramamoorthy A. Chemical Society reviews. 2014;43:6692–6700. doi: 10.1039/c3cs60431d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Butterfield SM, Lashuel HA. Angewandte Chemie (International ed in English) 2010;49:5628–5654. doi: 10.1002/anie.200906670. [DOI] [PubMed] [Google Scholar]
- 35.Sciacca MF, Kotler SA, Brender JR, Chen J, Lee DK, Ramamoorthy A. Biophysical journal. 2012;103:702–710. doi: 10.1016/j.bpj.2012.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jang H, Arce FT, Ramachandran S, Kagan BL, Lal R, Nussinov R. Chemical Society reviews. 2014;43:6750–6764. doi: 10.1039/c3cs60459d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alarcon JM, Brito JA, Hermosilla T, Atwater I, Mears D, Rojas E. Peptides. 2006;27:95–104. doi: 10.1016/j.peptides.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Wong PT, Schauerte JA, Wisser KC, Ding H, Lee EL, Steel DG, Gafni A. Journal of molecular biology. 2009;386:81–96. doi: 10.1016/j.jmb.2008.11.060. [DOI] [PubMed] [Google Scholar]
- 39.Williams TL, Serpell LC. The FEBS journal. 2011;278:3905–3917. doi: 10.1111/j.1742-4658.2011.08228.x. [DOI] [PubMed] [Google Scholar]
- 40.Sabate R, Espargaro A, Barbosa-Barros L, Ventura S, Estelrich J. Biochimie. 2012;94:1730–1738. doi: 10.1016/j.biochi.2012.03.027. [DOI] [PubMed] [Google Scholar]
- 41.Jang H, Arce FT, Capone R, Ramachandran S, Lal R, Nussinov R. Biophysical journal. 2009;97:3029–3037. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang Z, Luo Y, Zhang Y, Wei G. The journal of physical chemistry B. 2011;115:1165–1174. doi: 10.1021/jp107558e. [DOI] [PubMed] [Google Scholar]
- 43.Zhao LN, Chiu SW, Benoit J, Chew LY, Mu Y. The journal of physical chemistry B. 2011;115:12247–12256. doi: 10.1021/jp2065985. [DOI] [PubMed] [Google Scholar]
- 44.Poojari C, Kukol A, Strodel B. Biochimica et biophysica acta. 2013;1828:327–339. doi: 10.1016/j.bbamem.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Yu X, Wang Q, Pan Q, Zhou F, Zheng J. Physical chemistry chemical physics : PCCP. 2013;15:8878–8889. doi: 10.1039/c3cp44448a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tofoleanu F, Buchete NV. Journal of molecular biology. 2012;421:572–586. doi: 10.1016/j.jmb.2011.12.063. [DOI] [PubMed] [Google Scholar]
- 47.Brown AM, Bevan DR. Biophysical journal. 2016;111:937–949. doi: 10.1016/j.bpj.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang H, Teran Arce F, Ramachandran S, Capone R, Lal R, Nussinov R. The journal of physical chemistry B. 2010;114:9445–9451. doi: 10.1021/jp104073k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. Journal of molecular biology. 2010;404:917–934. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang H, Zheng J, Lal R, Nussinov R. Trends Biochem Sci. 2008;33:91–100. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 51.Tofoleanu F, Brooks BR, Buchete NV. ACS chemical neuroscience. 2015;6:446–455. doi: 10.1021/cn500277f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG. Journal of the American Chemical Society. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding H, Schauerte JA, Steel DG, Gafni A. Biophysical journal. 2012;103:1500–1509. doi: 10.1016/j.bpj.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao J, Wang Q, Liang G, Zheng J. Langmuir : the ACS journal of surfaces and colloids. 2011;27:14876–14887. doi: 10.1021/la2027913. [DOI] [PubMed] [Google Scholar]
- 55.Domanski J, Stansfeld PJ, Sansom MS, Beckstein O. The Journal of membrane biology. 2010;236:255–258. doi: 10.1007/s00232-010-9296-8. [DOI] [PubMed] [Google Scholar]
- 56.Jambeck JP, Lyubartsev AP. Journal of chemical theory and computation. 2013;9:774–784. doi: 10.1021/ct300777p. [DOI] [PubMed] [Google Scholar]
- 57.Kucerka N, Holland BW, Gray CG, Tomberli B, Katsaras J. J Phys Chem B. 2012;116:232–239. doi: 10.1021/jp208920h. [DOI] [PubMed] [Google Scholar]
- 58.Beschiaschvili G, Seelig J. Biochemistry. 1990;29:10995–11000. doi: 10.1021/bi00501a018. [DOI] [PubMed] [Google Scholar]
- 59.Lu JX, Blazyk J, Lorigan GA. Biochim Biophys Acta. 2006;1758:1303–1313. doi: 10.1016/j.bbamem.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 60.Hess B, Kutzner C, van der Spoel D, Lindahl E. Journal of chemical theory and computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 61.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jambeck JP, Lyubartsev AP. The journal of physical chemistry B. 2012;116:3164–3179. doi: 10.1021/jp212503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jambeck JP, Lyubartsev AP. Journal of chemical theory and computation. 2012;8:2938–2948. doi: 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- 64.Hess B, Bekker H, Berendsen HJ, Fraaije JG. Journal of computational chemistry. 1997;18:1463–1472. [Google Scholar]
- 65.Miyamoto S, Kollman PA. Journal of computational chemistry. 1992;13:952–962. [Google Scholar]
- 66.Bussi G, Donadio D, Parrinello M. The Journal of chemical physics. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 67.Wiedmann T, Salmon A, Wong V. Biochimica et biophysica acta. 1993;1167:114–120. doi: 10.1016/0005-2760(93)90150-8. [DOI] [PubMed] [Google Scholar]
- 68.Parrinello M, Rahman A. Journal of Applied Physics. 1981;52:7182–7190. [Google Scholar]
- 69.Kabsch W, Sander C. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 70.Elmore DE. FEBS letters. 2006;580:144–148. doi: 10.1016/j.febslet.2005.11.064. [DOI] [PubMed] [Google Scholar]
- 71.Humphrey W, Dalke A, Schulten K. Journal of molecular graphics. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 72.Maltseva E, Kerth A, Blume A, Mohwald H, Brezesinski G. Chembiochem : a European journal of chemical biology. 2005;6:1817–1824. doi: 10.1002/cbic.200500116. [DOI] [PubMed] [Google Scholar]
- 73.Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Biochemistry. 2005;44:6003–6014. doi: 10.1021/bi0474867. [DOI] [PubMed] [Google Scholar]
- 74.Reddy G, Straub JE, Thirumalai D. The journal of physical chemistry B. 2009;113:1162–1172. doi: 10.1021/jp808914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Evangelisti E, Cascella R, Becatti M, Marrazza G, Dobson CM, Chiti F, Stefani M, Cecchi C. Scientific reports. 2016;6:32721. doi: 10.1038/srep32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vermeer LS, de Groot BL, Reat V, Milon A, Czaplicki J. European biophysics journal : EBJ. 2007;36:919–931. doi: 10.1007/s00249-007-0192-9. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Y, Luo Y, Deng Y, Mu Y, Wei G. PloS one. 2012;7:e38191. doi: 10.1371/journal.pone.0038191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH, Ramamoorthy A. The Journal of biological chemistry. 2017;292:4638–4650. doi: 10.1074/jbc.M116.764092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.