

Abstract
Native electron capture dissociation (NECD) is a process during which proteins undergo fragmentation similar to that from radical dissociation methods, but without the addition of exogenous electrons. However, after three initial reports of NECD from the cytochrome c dimer complex, no further evidence of the effect has been published. Here, we report NECD behavior from horse spleen ferritin, a ∼490 kDa protein complex ∼20-fold larger than the previously studied cytochrome c dimer. Application of front-end infrared excitation (FIRE) in conjunction with low- and high-m/z quadrupole isolation and collisionally activated dissociation (CAD) provides new insights into the NECD mechanism. Additionally, activation of the intact complex in either the electrospray droplet or the gas phase produced c-type fragment ions. Similar to the previously reported results on cytochrome c, these fragment ions form near residues known to interact with iron atoms in solution. By mapping the location of backbone cleavages associated with c-type ions onto the crystal structure, we are able to characterize two distinct iron binding channels that facilitate iron ion transport into the core of the complex. The resulting pathways are in good agreement with previously reported results for iron binding sites in mammalian ferritin.
Interest in native mass spectrometry for characterization of biologically relevant macromolecular assemblies has increased in recent years,1 with great insights into composition and function provided for soluble2,3 and integral membrane complexes.4,5 Gas-phase fragmentation of these complexes by electron-based and collision-based dissociation techniques allows for unprecedented characterization of primary and higher-order structures.6−10 Of these, native electron capture dissociation (NECD), first introduced in 2003 by Breuker and McLafferty, proved both unexpected and useful for the elucidation of the heme-binding and gas-phase unfolding of the dimeric species of cytochrome c.(11,12) However, finding examples of this effect on other macromolecular complexes has proven elusive, and the underlying mechanism has not been investigated further.
NECD is a fragmentation process that occurs during ion transfer through a heated capillary in native electrospray ionization (nESI) of an iron-containing protein complex. Covalent bond cleavage near residues in contact with the heme results in apparent complementary c- and y-fragment ions.13 Changes in higher-order complex structure (e.g. unfolding) can perturb the heme-protein interactions, causing variation in the formation of NECD products. By analyzing changes in these fragment ion yields, it was possible to study the early gas-phase unfolding of cytochrome c,(12) and even the early partitioning of protons onto the dimer ions.14 Unfortunately, despite the promise of NECD as a technique for protein characterization, no further studies of the effect, either theoretical or experimental, have since been reported.
Here, we report the NECD phenomenon on the ∼490 kDa 24-mer ferritin, a protein complex known to store up to 4,500 Fe atoms.15 Unlike the previously studied cytochrome c dimer, ferritin does not contain a covalently bound heme group, instead using a di-iron site to catalytically convert soluble Fe(II) to an inorganic mineral similar to ferrihydrate (composed of Fe(III), oxygen, and small amounts of phosphate) which is stored in the core of the protein complex.16−19 Iron ions are channeled through subunit helices17 and protein nanocage pores20,21 and stored in the central cavity for future cellular use.22,23 Ferritin from mammalian tissues is made up of different proportions of the L (liver)- and H (heart)-chain; in horse they have masses of 19.9 and 21.2 kDa, respectively. The H-chain provides catalytic activity, and the L-chain is implicated in structural stability and cavity mineralization.24−26 The present work analyzed horse spleen holo-ferritin, which is made up of ∼90% L-chain.
Experimental Section
Apo- and holoferritin from horse spleen and cytochrome c from horse heart (Sigma) were desalted with 100 kDa and 10 kDa molecular weight cutoff filters, respectively, into 150 mM ammonium acetate at slightly acidic pH (∼5.6–7.0). Samples were diluted to a final concentration of 2 μM (ferritin) and 100 μM (cytochrome c) for the intact complex and sprayed using a custom-built nanospray source as described previously.27,28
All mass spectrometry measurements were performed on a Q-Exactive HF (Thermo Scientific) modified to allow for efficient transfer and quadrupole isolation of ions up to ∼10,000 m/z.6 Spectra were acquired at a resolving power of 120,000 (at 200 m/z), with the exception of the ferritin MS1, which was acquired at 7,500 resolving power (at 200 m/z), and smoothed for more accurate mass determination. The mass error presented here represents the standard deviation of the masses determined from the apex of each charge state peak in a single spectrum and, therefore, indicates the maximum precision of the measurement. NECD fragment ions from ferritin were collected as two separate spectra, one at higher energy in the collision cell for fragments from sites 6–79, and one at lower energy for fragments from sites 114–130. Data analysis was performed with Xtract (Thermo Scientific) or mMass29 software, and graphical fragment maps were created with Prosight Lite.30 All intensity values used for the determination of weighted-average charges were normalized by charge to account for the detection bias in the Orbitrap mass analyzer.
Front-end infrared excitation (FIRE) experiments used a 20 W continuous-wave CO2 laser (Synrad Firestar V20). The laser was attenuated with a 1.0 optical density (O.D.) nickel-coated zinc selenide neutral density filter and then aligned unfocused to the inlet capillary with protected gold mirrors. An average power of 1.2 W was used for FIRE experiments.
Safety Considerations
Vigilance and proper protective equipment should be used when handling ferritin, cytochrome c, and any other sample. Additionally, the high-voltage in the electrospray source can cause dangerous electrical shocks when not properly shielded. Finally, proper eye protection should be worn when using a CO2 or any other laser, especially as pointing it at the highly reflective front end of an instrument may cause unpredictable scattering.
Results and Discussion
Horse spleen ferritin was desalted and characterized by native top down mass spectrometry (nTDMS)6,31 (Figure 1). The MS1 spectrum (Figure 1a) indicated the presence of the intact complex with a mass of 490,380 ± 59 Da, consistent with the L22H2 complex and a ∼9500 Da iron oxide mineral. Broad spectral peaks (full width at half max of ∼3000 Da) were likely caused by hetereogeneity in the core mass but may also indicate small molecule adduction common with native MS.32 Collisional activation in the electrospray source produced peaks at lower m/z (Figure 1b, isotopically resolved) corresponding in mass to the ejected ferritin L-chain, with the previously reported31 cysteine methyl-disulfide modification at near complete stoichiometry. The observed signal is at a much (∼6-fold) higher charge state than would be predicted from a symmetric charge partitioning during monomer ejection, indicating that it undergoes asymmetric charge partitioning.33,34
Figure 1.
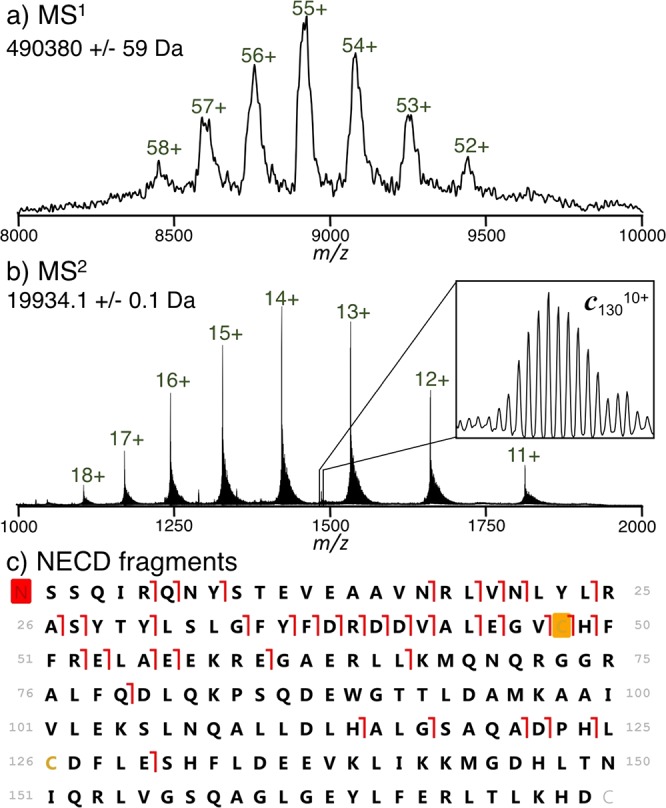
Analysis of ferritin by native top-down mass spectrometry. The MS1 spectrum (a) exhibits broad peaks corresponding to the mass of the intact 24-mer. After gas-phase activation in the source region, the intact L-chain is ejected along with c-fragment ions (MS2, b). Mapping the fragment ions onto the protein sequence (c) shows peptide bond cleavage at 37 unique sites, with cleavages from sites 5–79 determined using higher-energy collisions in the HCD cell, and those from sites >113 determined using lower-energy collisions. The red box on the N-terminus corresponds to an acetylation, and the orange box to a cysteine methyl disulfide modification.
In addition to ejected monomer, several fragment ions were observed and clearly resolved in the MS2 spectrum (Figure 1b, inset). Closer examination of these species revealed that they did not correspond to the traditional b- and y-fragments generated from threshold dissociation,35 but were instead consistent with the mass of c-fragment ions. In conjunction with fragment ions observed from a spectrum acquired at higher collision energy, products from cleaving 37 L-chain backbone sites were characterized (Figure 1c, matching fragment masses for this and other fragment maps are listed in Table S-1, Supporting Information). On the other hand, isolation and collisional fragmentation of a single charge state of the ejected monomer produced only b- and y-fragments (Figure S-1, Supporting Information). No evidence for the intact or fragmented H-chain was observed.
The observation of c-type fragment ions is characteristic of radical fragmentation processes such as electron capture or electron transfer dissociation (ETD).36,37 However, here no exogenous electrons were added or transferred. Instead, the formation of these product ions recalls the heme-mediated intramolecular electron transfer and fragmentation of NECD.11,12,14 Unlike the original reports of NECD on the cytochrome c dimer, the ∼20-fold larger ferritin complex does not contain a heme group, and no complementary y-fragment ions were observed. Therefore, in order to confirm that the c-type cleavage products were from NECD, it was necessary to better characterize the mechanism driving fragment ion formation in ferritin.
First, we confirmed the identity of the observed c-fragments by quadrupole-isolating and using collisionally activated dissociation (CAD) to characterize the 5+ charge state of c44, the most abundant NECD product (Figure 2). CAD produced b- and y-fragments originating from 50% of all peptide bonds present in the c44 precursor, including seven y-fragments corresponding (<5 ppm) to the mass of the sequence with a C-terminal amide. The y-fragments in this MS3 experiment contain the mass of the “ragged end” of the original NECD cleavage, which is characteristic of a c-fragment. Other NECD fragments were insufficiently abundant to perform a similar characterization, but were consistent in mass with c-fragments. Thus, we were able to exclude other potential factors (e.g., unexpected adducts and modifications) that may have confounded the mass values and confirm the presence of c-fragments in the spectrum with high confidence.
Figure 2.

Isolation and CAD fragments provide unambiguous characterization of the NECD-produced c44 5+ fragment ion. The C-terminal amide corresponds to the “ragged end” of the original NECD cleavage (after tautomerization), which is characteristic of a c-fragment ion and not hydrolysis.
However, while NECD products from cytochrome c were shown to form primarily in the source region of the instrument,11 the location where NECD of ferritin occurs remained more ambiguous. Isolation and collisional activation of the intact ferritin complex gave clear evidence of NECD (Figure S-2, Supporting Information), albeit at a lower efficiency. This is in contrast with isolation and fragmentation of the ejected monomer (Figure S-1, Supporting Information), which produced no c-fragments. Therefore, both cytochrome c and ferritin require the intact complex to fragment. However, unlike cytochrome c, ferritin can evidently form NECD fragments in the gas phase, after quadrupole isolation. The apparent discrepancy between NECD of these two systems could be due to significant water solvation of the ferritin ions even after transfer to the gas phase. Additionally, it is possible that fragment ions are being formed in the source region but are only separated after activation in the gas phase.38
In order to examine whether ferritin NECD fragmentation can also occur in the source region, we used a 20 W CO2 continuous-wave laser aligned with the inlet capillary of the mass spectrometer in a method termed front-end infrared excitation (FIRE) (experimental setup, Figure S-3, Supporting Information). In order to better characterize the effects of FIRE, we used a 100 μM solution of cytochrome c, as its NECD behavior has been extensively characterized previously.11,12,14 Application of FIRE produced NECD (both c- and y-fragments) of cytochrome c dimer species at inlet capillary temperatures low enough not to produce any fragment ions without laser irradiation (Figure S-4, Supporting Information). Using the high precision afforded by varying the distance between the ESI tip and inlet in the FIRE experiment, we correlated the relative signal of the major NECD peak (y555+) with the distance between the laser and nESI spray tip (Figure S-5, Supporting Information). The intensity dropped drastically between 2 mm and 3 mm, indicating that FIRE has little effect on nESI droplets after they have been sufficiently desolvated. Misalignment of the laser so that it only irradiated the droplet pathway between the nESI tip and the inlet capillary also drove NECD (Figure S-6, Supporting Information), providing further evidence that FIRE affects the droplets most just after they leave the nESI nozzle. Because application of FIRE to ferritin also produced NECD fragments (Figure S-7, Supporting Information), we can conclude that this process occurs in the nESI droplet as well.
As reported previously for NECD14 and ETD39 fragments of cytochrome c dimer, ferritin c-fragments also undergo asymmetric charge partitioning, as is evident from the great decrease in m/z between the precursor and its product ions (from >8000 to 1000–2000, respectively, Figure 1a,b). Plotting the average charge of each NECD fragment with respect to cleavage site indicates a relatively high level of charge density in the fragment ions, with discrete steps corresponding to the locations of arginines (Figure 3a); residues known to be charged in nESI.40 In comparison, CAD fragments from the isolated 14+ ejected monomer (overall weighted-average charge of ejected monomers = 13.93) exhibited a similar distribution of charge states also repeating the discrete jumps in charge at arginines 18, 26, and 39 (Figure 3a). The overall similarity in charge between the NECD and CAD fragments indicates that the protons have partitioned to analogous locations after the fragments have been ejected from the intact complex.
Figure 3.

Charge distribution observed in ferritin fragment ions. (a) The observed charge states of c-type fragment ions from NECD fragmentation of the intact complex and the b- and y-type fragments from CAD of the 14+ ejected monomer exhibit many of the same discrete steps. In order to plot them on the same axis as the N-terminal fragments, the charge of y-fragments is displayed as the difference 14–observed charge. The locations of the 11 arginine residues in ferritin are marked, as they often correlate with increases in charge. (b) The asymmetric charge partitioning factor (ACPF) is displayed for each of the observed NECD fragments with two or more charges. While there is no noticeable change in the ACPF for larger fragment ions, all but two partition more asymmetrically than the ejected monomer (dotted line at ACPF = 6.1).
While the overall charge state of a fragment is a good measure of charge density, we use the asymmetric charge partitioning factor (ACPF) to determine the full magnitude of charge partitioning for products of different mass.39 The ACPF indicates the fold change between how much charge an ejected subunit has when compared to how much it would have upon symmetric charge partitioning. Thus, a symmetric charge partitioning would correspond to an ACPF of 1, whereas higher values indicate an increasing magnitude of the effect. Plotting the ACPF values of observed NECD fragments (with two or more charges) with respect to their cleavage sites shows that the magnitude of asymmetric charge partitioning remained relatively constant with respect to fragment size (Figure 3b). Surprisingly, only two fragments partitioned less than the ejected monomer (ACPF = 6.1); the overall average ACPF was 7.2. Therefore, the NECD fragments are not only undergoing asymmetric charge partitioning but are doing it more so than even the ejected monomer.
The c-fragments observed from ferritin have many of the same characteristics as those found previously for NECD from cytochrome c. Therefore, because previous work on NECD has shown that fragment formation only occurs near residues that interact with the iron-bearing heme group, the location of fragments in ferritin too should be dependent on iron-side chain interactions. However, cleavages occur between two residues, so any given cleavage site could indicate up to two individual iron-binding interactions. The assignment of residues to cleavage sites can be simplified because not all amino acids are likely to coordinate to iron and were therefore excluded from assignment.
Plotting the yields of c-fragment ions with respect to their location on the protein sequence (Figure 4a) exhibits two regions of major fragmentation: sites 34–49 and 117–130. High-intensity fragmentation from these regions strongly indicates iron-binding interactions. The first, 34–49, corresponds to a loop-pocket region (Figure 4b), which is a cavity in the protein cage hypothesized to be involved in the release of iron atoms into the cell.19,41 The second, located on a 3-fold axis in the cage structure (Figure 4c) has been implicated in the uptake of soluble Fe2+ prior to deposition in the inner iron core.19,41 Previous studies to characterize these regions have relied on overexpressed systems and alternate metals; NECD directly mapped iron interactions on a mammalian system from small amounts of an endogenous sample.
Figure 4.

Yields of c-type fragment ions plotted with respect to their cleavage site (a) with the X-ray crystal structure of ferritin (PDB: 1IER) comprised of all L-subunits (shown in different colors). Mapping the major fragment ions from NECD onto the crystal structure for sites 43–53 (b) indicates that the cleavages are centered in the helix-loop region. Fragment ions from sites 114–130 (c) instead indicate cleavages from the 3-fold axis pore.
Surprisingly, little fragmentation was observed around the catalytically active residues 53–63 and 135–140. However, only the H-chain is considered catalytically active.25 Thus, NECD fragments, which are observed exclusively from the L-chain, cannot be directly correlated to di-iron site oxidation of soluble Fe2+. Additionally, activation of apoferritin from horse spleen, which should contain no iron oxide mineral, produced many similar c-fragments (Figure S-8), suggesting that the effect cannot be attributed to the core. However, a recent study by Carmona et al. has shown that the light chain of horse ferritin is critical for the transfer of an electron across the boundary of the ferritin cage.42 The apparent dependence of ferritin NECD on electron transfer brings it in line with the electron-transport protein cytochrome c and could explain why NECD is not observed from other systems.
Despite the strong correlation between ferritin NECD fragment ions and iron-binding channels, how the iron ions can generate radical-type fragment ions is not completely clear. However, with the improved fragment ion characterization presented here we propose a potential mechanism for NECD in ferritin. In the first step, a subunit partially unfolds, driving a similar proton transfer process to that predicted for monomer ejection.43,44 This step explains the “stepped” appearance of charge distribution in NECD fragment ions (Figure 3a), as the protons are no longer mobilized and have already been distributed prior to cleavage. While most of the subunit monomer has unfolded, the stronger iron-mediated protein–protein interactions are maintained. With increasing energy, these interactions are finally broken, transferring an electron from complexed iron and cleaving the monomer’s peptide bond.
Conclusions
Native electron capture dissociation displayed promise as a new technique for elucidating iron binding interactions when it was first presented in 2003. However, no examples beyond the original cytochrome c dimer have since been described.11,12,14 Here, we demonstrate a second system displaying the NECD phenomenon, the ∼490 kDa protein complex ferritin. Leveraging information from FIRE-assisted dissociation and gas-phase isolation/dissociation, we show that the resulting fragments are predominantly NECD type c-ions, undergo asymmetric charge partitioning, and can be formed in the electrospray droplet or in the gas phase upon collisional or infrared activation. The formation of NECD products, postulated here to be liberated from previously unfolded monomers, could be further probed with future experiments using electron capture dissociation, which has been used to monitor structural changes in other gaseous protein complexes like the hemoglobin tetramer.45
When mapped on the crystal structure of the all L-chain ferritin, prevalent NECD fragments indicated significant iron binding at the loop-pocket and 3-fold axis regions. These residues are not implicated in the catalytic process of ferritin but instead correspond to channels which mediate iron ion transport into the protein cage. Future applications of NECD could therefore probe the migration of iron and potentially other metals through the ferritin iron-binding channels and would require orders-of-magnitude less sample than crystallography experiments. While the applicability of NECD remains limited to two protein complexes, its readout of iron-binding channels provides a high potential for the targeted analysis of perturbations in these and potentially other systems.
Acknowledgments
The authors acknowledge generous support from the W. M. Keck Foundation (DT061512). OSS and MOM are supported by National Science Foundation Graduate Research Fellowships (2014171659 and DGE-0824162, respectively). KB is supported by FWF projects Y372 and P27347. The authors would also like to acknowledge helpful insight from Eva-Maria Schneeberger and Nicole Haverland.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01581.
Author Contributions
§ OSS and MOM contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Loo J. A. Mass Spectrom. Rev. 1997, 16, 1–23. . [DOI] [PubMed] [Google Scholar]
- Heck A. J.; Van Den Heuvel R. H. Mass Spectrom. Rev. 2004, 23, 368–389. 10.1002/mas.10081. [DOI] [PubMed] [Google Scholar]
- Schneeberger E. M.; Breuker K. Angew. Chem., Int. Ed. 2017, 56, 1254–1258. 10.1002/anie.201610836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrera N. P.; Di Bartolo N.; Booth P. J.; Robinson C. V. Science 2008, 321, 243–246. 10.1126/science.1159292. [DOI] [PubMed] [Google Scholar]
- Hopper J. T.; Yu Y. T.; Li D.; Raymond A.; Bostock M.; Liko I.; Mikhailov V.; Laganowsky A.; Benesch J. L.; Caffrey M.; Nietlispach D.; Robinson C. V. Nat. Methods 2013, 10, 1206–1208. 10.1038/nmeth.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov M. E.; Damoc E.; Denisov E.; Compton P. D.; Horning S.; Makarov A. A.; Kelleher N. L. Anal. Chem. 2013, 85, 11163–11173. 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Wen J.; Blankenship R. E.; Gross M. L. J. Am. Soc. Mass Spectrom. 2010, 21, 1966–1968. 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell A. E.; Dodds E. D.; Bandarian V.; Wysocki V. H. Anal. Chem. 2011, 83, 2862–2865. 10.1021/ac200452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Nelp M. T.; Bandarian V.; Wysocki V. H. ACS Cent. Sci. 2015, 1, 477–487. 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Wongkongkathep P.; Van Orden S. L.; Ogorzalek Loo R. R.; Loo J. A. J. Am. Soc. Mass Spectrom. 2014, 25, 2060–2068. 10.1007/s13361-014-0928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuker K.; McLafferty F. W. Angew. Chem., Int. Ed. 2003, 42, 4900–4904. 10.1002/anie.200351705. [DOI] [PubMed] [Google Scholar]
- Breuker K.; McLafferty F. W. Angew. Chem., Int. Ed. 2005, 44, 4911–4914. 10.1002/anie.200500668. [DOI] [PubMed] [Google Scholar]
- Roepstorff P.; Fohlman J. Biomed. Mass Spectrom. 1984, 11, 601. 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- Breuker K. Int. J. Mass Spectrom. 2006, 253, 249–255. 10.1016/j.ijms.2006.04.004. [DOI] [Google Scholar]
- Mann S.; Bannister J. V.; Williams R. J. J. Mol. Biol. 1986, 188, 225–232. 10.1016/0022-2836(86)90307-4. [DOI] [PubMed] [Google Scholar]
- Theil E. C. Curr. Opin. Chem. Biol. 2011, 15, 304–311. 10.1016/j.cbpa.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldar S.; Bevers L. E.; Tosha T.; Theil E. C. J. Biol. Chem. 2011, 286, 25620–25627. 10.1074/jbc.M110.205278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstead P. D.; Hudson A. J.; Artymiuk P. J.; Andrews S. C.; Banfield M. J.; Guest J. R.; Harrison P. M. FEBS Lett. 1994, 350, 258–262. 10.1016/0014-5793(94)00781-0. [DOI] [PubMed] [Google Scholar]
- Theil E. C.; Tosha T.; Behera R. K. Acc. Chem. Res. 2016, 49, 784–791. 10.1021/ar500469e. [DOI] [PubMed] [Google Scholar]
- Behera R. K.; Theil E. C. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 7925–7930. 10.1073/pnas.1318417111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernacchioni C.; Pozzi C.; Di Pisa F.; Mangani S.; Turano P. Chem. - Eur. J. 2016, 22, 16213–16219. 10.1002/chem.201602842. [DOI] [PubMed] [Google Scholar]
- Theil E. C. Annu. Rev. Biochem. 1987, 56, 289–315. 10.1146/annurev.bi.56.070187.001445. [DOI] [PubMed] [Google Scholar]
- Chasteen N. D.; Harrison P. M. J. Struct. Biol. 1999, 126, 182–194. 10.1006/jsbi.1999.4118. [DOI] [PubMed] [Google Scholar]
- Grace J. E. Jr.; Van Eden M. E.; Aust S. D. Arch. Biochem. Biophys. 2000, 384, 116–122. 10.1006/abbi.2000.2068. [DOI] [PubMed] [Google Scholar]
- Levi S.; Santambrogio P.; Cozzi A.; Rovida E.; Corsi B.; Tamborini E.; Spada S.; Albertini A.; Arosio P. J. Mol. Biol. 1994, 238, 649–654. 10.1006/jmbi.1994.1325. [DOI] [PubMed] [Google Scholar]
- Carmona F.; Poli M.; Bertuzzi M.; Gianoncelli A.; Gangemi F.; Arosio P. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 522–532. 10.1016/j.bbagen.2016.12.011. [DOI] [PubMed] [Google Scholar]
- Wojcik R.; Dada O. O.; Sadilek M.; Dovichi N. J. Rapid Commun. Mass Spectrom. 2010, 24, 2554–2560. 10.1002/rcm.4672. [DOI] [PubMed] [Google Scholar]
- Skinner O. S.; Do Vale L. H.; Catherman A. D.; Havugimana P. C.; de Sousa M. V.; Compton P. D.; Kelleher N. L. Anal. Chem. 2015, 87, 3032–3038. 10.1021/ac504678d. [DOI] [PubMed] [Google Scholar]
- Strohalm M.; Kavan D.; Novak P.; Volny M.; Havlicek V. Anal. Chem. 2010, 82, 4648–4651. 10.1021/ac100818g. [DOI] [PubMed] [Google Scholar]
- Fellers R. T.; Greer J. B.; Early B. P.; Yu X.; LeDuc R. D.; Kelleher N. L.; Thomas P. M. Proteomics 2015, 15, 1235–1238. 10.1002/pmic.201400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner O. S.; Havugimana P. C.; Haverland N. A.; Fornelli L.; Early B. P.; Greer J. B.; Fellers R. T.; Durbin K. R.; Do Vale L. H.; Melani R. D.; Seckler H. S.; Nelp M. T.; Belov M. E.; Horning S. R.; Makarov A. A.; LeDuc R. D.; Bandarian V.; Compton P. D.; Kelleher N. L. Nat. Methods 2016, 13, 237–240. 10.1038/nmeth.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassou C. A.; Williams E. R. Analyst 2014, 139, 4810–4819. 10.1039/C4AN01085J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light-Wahl K. J.; Schwartz B. L.; Smith R. D. J. Am. Chem. Soc. 1994, 116, 5271–5278. 10.1021/ja00091a035. [DOI] [Google Scholar]
- Jurchen J. C.; Williams E. R. J. Am. Chem. Soc. 2003, 125, 2817–2826. 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverland N. A.; Skinner O. S.; Fellers R. T.; Tariq A. A.; Early B. P.; LeDuc R. D.; Fornelli L.; Compton P. D.; Kelleher N. L. J. Am. Soc. Mass Spectrom. 2017, 28, 1203–1215. 10.1007/s13361-017-1635-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. J. Am. Chem. Soc. 1998, 120, 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Syka J. E.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schennach M.; Schneeberger E. M.; Breuker K. J. Am. Soc. Mass Spectrom. 2016, 27, 1079–1088. 10.1007/s13361-016-1363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton P. D.; Fornelli L.; Kelleher N. L.; Skinner O. S. Int. J. Mass Spectrom. 2015, 390, 132–136. 10.1016/j.ijms.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbeck J. D.; Severs J. C.; Gao J.; Wu Q.; Smith R. D.; Whitesides G. M. J. Phys. Chem. B 1998, 102, 10596–10601. 10.1021/jp980768u. [DOI] [Google Scholar]
- Turano P.; Lalli D.; Felli I. C.; Theil E. C.; Bertini I. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 545–550. 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona U.; Li L.; Zhang L.; Knez M. Chem. Commun. (Cambridge, U. K.) 2014, 50, 15358–15361. 10.1039/C4CC07996E. [DOI] [PubMed] [Google Scholar]
- Sciuto S. V.; Liu J.; Konermann L. J. Am. Soc. Mass Spectrom. 2011, 22, 1679–1689. 10.1007/s13361-011-0205-x. [DOI] [PubMed] [Google Scholar]
- Loo R. R.; Loo J. A. J. Am. Soc. Mass Spectrom. 2016, 27, 975–990. 10.1007/s13361-016-1375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W.; Zhang H.; Blankenship R. E.; Gross M. L. Protein Sci. 2015, 24, 1325–1332. 10.1002/pro.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.