

Abstract

Human tissues are an important link between organ-specific spatial molecular information, patient pathology, and patient treatment options. However, patient tissues are uniquely obtained by time and location, and limited in their availability and size. Currently, little knowledge exists about appropriate and simplified protocols for routine MS-based analysis of the various types and sizes of tissues. Following standard procedures used in pathology, we selected small fresh frozen uterine tissue samples to investigate how the tissue preparation protocol affected the subsequent proteomics analysis. First, we observed that protein extraction with 0.1% SDS followed by extraction with a 30% ACN/urea resulted in a decrease in the number of identified proteins, when compared to extraction with 30% ACN/urea only. The decrease in the number of proteins was approximately 55% and 20%, for 10 and 16 μm thick tissue, respectively. Interestingly, the relative abundance of the proteins shared between the two methods was higher when SDS/ACN/urea was used, compared to the 30% ACN/urea extraction, indicating the role of SDS to be beneficial for protein solubility. Second, the influence of tissue thickness was investigated by comparing the results obtained for 10, 16, and 20 μm thick (1 mm2) tissue using urea/30% ACN. We observed an increase in the number of identified proteins and corresponding quantity with an increase in the tissue thickness. Finally, by analyzing very small amounts of tissues (∼0.2 mm2) of 10, 16, and 20 μm thickness, we observed that the increase in tissue thickness resulted in a higher number of protein identifications and corresponding quantitative values.
The increasing relevance of proteomics for clinical research looms as national and international funding schemes focus on bringing innovative technologies closer to patients and their treatment options. This has spurred widespread interest among researchers who now aim to provide detailed molecular information on patient material, with the ambition to assist clinicians directly and/or convert patient tissues to “digital information” for subsequent investigation. To be successful, proteomics requires biochemical procedures that routinely and simply convert tissues to MS measurable material. The methods must be reliable and robust with reproducibility as a key performance indicator.
Human organs can contain the pathological condition of the patient, and therefore, the analysis of tissues, including subsections of tissues, can be of high relevance for clinicians aiming to treat or investigate patients based on their molecular profile. However, proteomics of tissues is not routinely employed. Clinical tissue samples are still mostly stored for future research and are archived as formalin-fixed paraffin embedded (FFPE) tissues and to a much lesser extent as fresh frozen (FF) tissues. FFPE is a routine method for tissue preservation and has existed for more than a century. Long-term storage of FFPE tissues at room temperature makes them readily available for use from repositories compared to FF tissues stored in freezers. However, during formalin fixation, proteins in FFPE tissues undergo chemical modifications and extensive cross-linking. Studies aimed at measuring differences from paired FFPE and FF tissues have revealed that a larger number of proteins can be identified from FF tissues and that 40–90% of the identified proteins are observed in both FF and FFPE tissues.1 Therefore, preferred samples for clinical proteomics are still FF tissues, when available.
Sample preparation is a key step in proteomics and several methodologies such as single-pot solid-phase-enhanced sample preparation (SP3),2 in-stage tip technology,3,4 or FASP5−11 have been reported for preparing tissue for subsequent MS analysis. We and others have also investigated mechanical disruption methods to extract proteins from tissues for subsequent MS analysis. Some of these include ultrasonication,12,13 picosecond infrared lasers (PIRL),14 and pressure cycling technology (PCT).15
A challenge to efficiently extract proteins from cells using chemical buffers is to ensure the proteins remain in-solution for subsequent digestion. To achieve this, the addition of components such as chaotropes, detergents, salts, and/or organic solvents are often used in the extraction buffer.16 Chaotropes as urea and guanidine hydrochloride facilitate protein denaturation and unfolding in-solution by influencing protein structure and stability. Urea-based buffers are widely used in tissue proteomics;17−21 however, the presence of high concentrations of urea can interfere with the later digestion step, and therefore, it needs to be diluted before the addition of trypsin.22 Moreover, it has been shown that presence of the urea can lead to carbamylation of amino and sulfhydryl groups and further modify lysine and arginine residues.23−25 Therefore, cyanate scavengers such as ammonium bicarbonate, methylamine, and TrisHCl are sometimes added to extraction buffers.23,26 The addition of organic solvents such as ACN and MeOH can alter protein conformation and facilitates their denaturation and subsequent digestion. The optimal concentration of the ACN for protein extraction varies between methods and has been reported in the range of 20–80%.27−31 Detergents are often added to the extraction buffers to improve extraction efficiency and protein solubilization. The most commonly used detergent is sodium dodecylsulfonate (SDS).32−34 SDS has been used for a long time as a component of buffers for protein extraction, however, as it is not compatible with MS due to ionization suppression, it must be removed from the sample using variety of methods including precipitation with ethyl acetate, trifluoroethanol (TFE), acetone, KCl, or spin columns.38−41 Besides SDS, nonionic (Triton-X100, NP-40, Brij) or zwitterionic (CHAPS) surfactants have also been used for processing of tissue samples. However, to avoid possible interferences from the surfactants and enable direct sample processing for MS, acid-labile detergents have been developed. The advantage of these compounds, which include Rapigest, MasDes, or ProteaseMax,42−45 is that they contain an acid-labile functional group and upon hydrolysis break down into innocuous MS compounds. However, sometimes these compounds are not as effective in protein extraction as SDS. Another disadvantage limiting their widespread uptake is their cost. A summary of some protocols reported for protein extraction from the biological materials using urea, SDS, MS-compatible detergents and other components present in extraction buffers is presented in Table 1. From this table, we see that different sample preparation protocols result in a diversity of protein recoveries, whereas the lytic strength (urea, detergent) or procedures for removal of noncompatible MS compounds showed to be most critical part in evaluation of procedures.
Table 1. Summary of the Protocols for Protein Extraction Using Urea and/or SDS-Based Buffers and Their Comparison with Other Procedures.
| sample | sample amount | buffer(s) composition | identified proteins | reference |
|---|---|---|---|---|
| brain | 0.5–10 mm2, 8 μm thick | 1. 100 mM NH4HCO3/20% ACN | 1665 | Drummond 201535 |
| 2. 0.2% Rapigest/50 mM NH4HCO3 | 1773 | |||
| 3. RIPA (50 mM Tris-HCl pH 7.4, 1% Triton X-100, 0.5% SDC, 0.1% SDS, 150 mM NaCl, 2 mM EDTA) | 1598 | |||
| liver | 5 μm thick | 1. 8 M urea (FASP) | 1693 | Tanca 201436 |
| 2. 0.2% SDS | 1358 | |||
| 3. 50 mM NH4HCO3 (direct tissue digestion) | 1015 | |||
| endometrial cancer | 8 μm thick, 63 nL | 100 mM NH4HCO3/20% ACN | 705a | Alkhas 201127 |
| sarcoma tumor | 140 mm2, 4 μm thick | 1. 7 M urea, 1 M NH4HCO3, 2 M thiourea | 572 | Luebker 201524 |
| 2. EXB Plus Extraction Buffer, 6% β-mercaptoethanol | 759 | |||
| kidney, brain, heart, lung, liver | 6 μm thick | 1. 0.2% Zwittergent, 10 mM Tris, 1 mM EDTA | 169b | Shen 20158 |
| 2. UPX universal extraction buffer | 180b | |||
| 3. 100 mM Tris, 100 mM DTT, 4% SDS (pH = 8) | 184b | |||
| 4. 0.5% PEG20000, 100 mM Tris, 100 mM DTT, 4% SDS (pH= 8) | 167b | |||
| 5. 8 M urea, 2 M thiourea, 65 mM DTT, 83 mM Tris, 4% CHAPS | 110b | |||
| X. laevis embryo | / | 1. NP-40 (FASP) | 2201 | Peuchen 201637 |
| 2. NP-40 (urea) | 1210c | |||
| 3. NP-40 (NH4HCO3) | 1080c | |||
| 4. SDS/Freon (FASP) | 907 | |||
| 5. SDS/Freon (urea) | 739c | |||
| 6. SDS/Freon (NH4HCO3) | 782c |
In-solution digestion.
Data shown for liver tissue.
Acetone precipitation.
The composition of the extraction buffer has a critical effect on which proteins may be detected. Due to the complexity of biological samples, methods evaluated as successful on model proteins or cell lines may be unsuccessful when analyzing tissues. The aim of our study was to examine several simple nonmechanical protocols for extraction and digestion of proteins from fresh frozen human uterus tissue. Protocols varied in their extraction and digestion procedure, use of detergent, and composition of the extraction solvent. We report the development and evaluation of relevant questions for proteomics analysis of uterus tissue: (1) Is SDS required for optimal and reproducible solubilization of proteins from tissues? (2) How does the size and thickness of a tissue relate to protein recovery? In this context, we analyzed small amounts of 10, 16, and 20 μm thick FF uterus tissues. We examined the influence of the sample preparation procedure on overall information and results were related to the different sizes of the tissues.
Materials and Methods
Materials and Reagents
Water (ULC/MS), acetonitrile (ACN, LC-MS grade), and formic acid (FA) were purchased from Biosolve (Valkenswaard, The Netherlands) and NH4HCO3 was obtained by Fluka (Zwijndrecht, The Netherlands). Acetone, urea, sodium dodecyl sulfate (SDS, ≥ 98.5%), dithiothreitol (DTT, ≥ 99%), iodoacetamide (IAA, ≥ 99%), trifluoroacetic acid (TFA, ≥ 99%), and trypsin (European Pharmacopoeia reference standard) were delivered by Sigma-Aldrich (Zwijndrecht, The Netherlands).
Fresh Frozen Tissue Samples
Cryosectioned fresh frozen (FF) tissues of smooth muscle from uterus were provided by Leiden University Medical Centre (LUMC, The Netherlands). Tissue sections of 10, 16, and 20 μm were cut from the tissue block, placed on the glass slide, and stored at −80 °C until used.
Fresh Frozen Uterus Tissue Preparation for LC-MS
Regions of tissue were cut with a scalpel (Martor KG, Germany), and tissue sections were then scraped from the glass slide into the extraction buffer. Area of each individual tissue section was measured using ImageJ software (version 1.49). All experiments were performed in triplicate.
Following removal of tissues from the glass slide, proteins were extracted using the different extraction buffers: (1) solution containing 50 mM Tris-HCl (pH = 8), 150 mM NaCl, and 0.1% SDS was added, and samples were incubated at 37 °C for 30 min. After incubation 400 μL of 80% acetone was added, samples were vortexed for 1 min and incubated for 1 h. After incubation, samples were centrifuged at 12 000g for 20 min, and the supernatant was discarded. Further, proteins were solubilized by using 100 μL of solution containing of 30% ACN, 100 mM NH4HCO3, 8 M urea, and 19.6 mM DTT. Samples were further incubated for 30 min at 37 °C; (2) 30% ACN, 100 mM NH4HCO3, 8 M urea, and 19.6 mM DTT, and samples were incubated for 30 min at 37 °C; (3) 60% ACN, 100 mM NH4HCO3, 8 M urea, and 19.6 mM DTT, and samples were incubated for 30 min at 37 °C. Following incubation, in each sample, 9.2 μL of 700 mM IAA was added, and the samples were alkylated for 1 h in the dark at room temperature.
Following alkylation, 120 μL of 1M NH4HCO3 and 880 μL of H2O were added to the samples. Samples were vortexed, and trypsin was added at 5 ng/mm246 of the tissue, whereupon the samples were left to digest. In Protocols 1, 2, and 4, proteins were digested overnight (18 h) at 37 °C with constant shaking. In Protocols 3 and 5, following 18 h of digestion, a second portion of trypsin was added, and samples were incubated for an additional 4 h (Figure 1).
Figure 1.

Schematic overview of the current study. (a) 10, 16, and 20 μm thick FF human uterus tissue were used in the study. (b) Proteins from the tissues were extracted using 3 different solutions varied by detergent (SDS) and organic solvent (ACN). (c) Protein digestion was performed over 18 h using a single portion of trypsin (Protocols 1, 2, and 4) or for an additional 4 h after the addition of second portion of trypsin (Protocols 3 and 5).
After digestion, samples were centrifuged for 45 min at 14 000g, and the supernatant was collected. Digestion was quenched by the addition of 50 μL of 5% TFA, into the sample solution. Peptides were desalted using Empore C18 solid phase extraction (SPE) cartridges (4 mm/1 mL, Sigma-Aldrich), and the remaining liquid was evaporated in a SpeedVac (miVac DNA concentrator GeneVac, SP Scientific). Dried samples were stored at −20 °C until analysis. Prior to LC-MS, samples were reconstituted in 3% ACN and 1% formic acid.
NanoLC-ESI-MS/MS
Samples were analyzed on an Eksigent Ekspert nanoLC 425 system (Sciex) coupled to the nanoelectrospray interface (nanoESI) of a TripleTOF 5600+. After injection, samples were loaded onto an Eksigent trap column (nano-LC trap set, ChromXP C18, 120 Å, 350 μm, 0.5 mm) and desalted with 3% ACN and 0.1% FA at 2 μL/min.
The resulting peptides were analyzed using an in-house packed analytical column (Magic C18 resin, 100 Å pore size, 5 μm particles, 75 μm i.d., 10 cm column length) at 300 nL/min. The elution gradient was composed of 5–40% B (0.1% FA in ACN) for 45 min, 40–100% B for 5 min, 100% B for 9 min, and then the gradient was changed from 100 to 5% B in 1 min. Mobile phase A was composed of 0.1% FA in H2O.
Samples were analyzed in intensity dependent acquisition (IDA) mode, and survey scans were acquired in 500 ms, with m/z from 400 to 1250 Da. In each duty cycle, 30 product ion scans were collected for 100 ms in the m/z range from 100 to 1800 Da, if exceeding 100 counts per seconds and if charge state was 2+ to 4+. Dynamic exclusion was used for half of the peak width, and rolling collision energy was used.
Data Processing Parameters
Peak lists from raw data files (.wiff) were created and converted into .mgf format using Protein Pilot Software 5.0 (Sciex, Singapore). Files were imported into SearchGui software47 and searched with X!Tandem against the Uniprot human database (downloaded November 22, 2016) with the search parameters as follows: enzyme trypsin, fixed modification carbamidomethylation of C; variable modification oxidation of methionin; maximum number of missed cleavages 2. Results were analyzed using Peptideshaker,48 and proteins at 1% FDR were filtered. Quantitative evaluation of relative protein abundances and their comparison for different extraction buffers and tissue thickness was performed using normalized spectral abundance factor (NSAF) reported by Peptideshaker. Protein sequences were obtained from the UniprotKB Web site (www.uniprot.org), and GRAVY indexes were obtained using the GRAVY calculator (http://www.gravy-calculator.de).
Results and Discussion
Influence of the MS-Related Instrumental Parameters
The peptide mixtures obtained from proteolysis of protein extracts of tissues are complex samples, and interaction between different compounds in complex mixtures can influence their detection. Therefore, sample-specific optimization of MS acquisition related parameters can lead to more effective detection and fragmentation of the peptides. In our study, variations in the TOF-MS and TOF-MS/MS accumulation time showed most distinct influence on the number of identified proteins and peptides (Figure S-1, Supporting Information). The optimal TOF-MS and TOF-MS/MS accumulation times were found to be 500 and 100 ms respectively, and these parameters were used for the rest of the study, as described in the Materials and Methods.
Protein Extraction Buffers: Evaluation of Efficiency and Reproducibility
Different methods for sample preparation were evaluated for 10, 16, and 20 μm thick FF human uterus tissues. The protocols used 3 extraction buffers (SDS/30% ACN/urea, 30% ACN/urea and 60% ACN/urea) and were also investigated for different digestion procedures (Protocols 1 to 5). The overview of the study workflow is schematically presented in the Figure 1.
Qualitative variability was established by determining the overlap between the 3 biological replicates of the 10 μm thick tissues, and results are shown in Figure 2. A comparison of the results from Protocol 1 and Protocol 2 for 10 μm tissue showed (Figure S-2, Supporting Information) an overlap of 30.7% of proteins between the protocols, while more unique proteins were identified using Protocol 2 (60.3%). Results were also evaluated at the quantitative level, and reproducibility of the protocols was assessed with their normalized spectral abundance factor (NSAF) values, as reported by the Peptideshaker software.48 Pearson correlation coefficients demonstrated for 10 μm thick tissue (Figure 2) that the protocols showed good reproducibility of the NSAF values among the replicates (Pearson r ≥ 0.78). Reproducibility was also evaluated for 16 and 20 μm thick tissue samples, and the results are shown in Figure S-3, Supporting Information.
Figure 2.

Summary of the results showing qualitative and quantitative comparison of five protocols for protein retrieval from 10 μm thick FF human uterus tissue. Venn diagrams show the distribution of identified proteins in biological replicates using Protocols 1 to 5, and correlation of the NSAF values between replicates is shown using Pearson r coefficient.
Protein extraction using 30% ACN/urea (Protocol 2) were found to lead to 55% (10 μm tissue) and 20% (16 μm tissue) increase in the number of identified proteins compared to extraction with SDS/30% ACN/urea (Protocol 1) (Figure 3a, Figure S-4, Supporting Information). The lower number of proteins identified using Protocol 1 may be related to sample loss during the precipitation procedure or incomplete SDS removal. While both ACN and methanol are commonly used at a range of concentrations,27−29 preliminary results in this study (data not shown) revealed that methanol did not contribute to protein or peptide identifications; therefore, ACN in the range 30–60% was used for optimization of the extraction buffer. Variations in the concentration of ACN showed comparable numbers of identified proteins in Protocols 2, 3, 4, and 5 (Figure S-4, Supporting Information).
Figure 3.

Overview of protein identifications and their quantitative values using various protocols and tissue amounts. (a) Relative comparison of identified proteins from 10 μm thick tissue after extraction and digestion using Protocols 1 to 5. Addition of SDS into the protein extraction buffer revealed a decrease in the number of identified proteins (Protocol 1 as compared to Protocols 2–5). Similar results were obtained for 16 μm thick tissue (Figure S-4, Supporting Information). (b) Quantitative comparison of shared-protein abundance by NSAF values after extraction and digestion using Protocol 2 for quantitation in the 10, 16, and 20 μm thick tissue. Abundance of the proteins after tissue processing using Protocol 1 and Protocol 2 are shown for: (c) 10 μm and (d) 16 μm thick FF human uterus tissue. Results are shown as mean ± SD.
The additional digestion (an extra addition of trypsin) performed in Protocols 3 and 5 did not increase the number of identified proteins; however, more shared proteins between 3 biological replicates for 16 and 20 μm thick tissue were found (Figure S-2 (a–c), Supporting Information).
Quantitative Evaluation of Proteins for Different FF Uterus Tissue Thicknesses
The thickness of a tissue section is an important factor as it relates to the amount of analyzed material. To examine the effect of the tissue thickness on protein recovery, we quantified proteins from 10, 16, and 20 μm microdissected tissues. The protocols were evaluated on 3 replicates, and our results demonstrated that an increase in tissue thickness led to an increased number of identified proteins. After protein extraction and digestion using Protocol 2 the number of identified proteins for 10, 16, and 20 μm thick tissue was 444 ± 31, 465 ± 11, and 535 ± 74 (mean ± SD), respectively (Figure S-4, Supporting Information). To examine protein abundance in relation to tissue thickness, proteins were assigned NSAF values. Our results also showed (Figure 3b) that increasing tissue thickness also led to an increased NSAF values, for matched areas of analyzed tissue in the samples.
To further investigate possible differences in protein extraction from Protocols 1 and 2, intensities of proteins detected by both protocols (i.e., the overlap) were compared. Evaluation of the tissue showed that there are 2400 cells/mm2 present in smooth muscle of uterus. Although Protocol 1 led to fewer identified proteins, it was demonstrated for both 10 and 16 μm thick tissue that overlap proteins exhibited higher NSAF values for Protocol 1 than Protocol 2 (Figure 3c,d).
Further, we then evaluated the minimum amount of tissue required for quantitative analysis. For this purpose, 0.2, 0.6, 0.8, 1.2, 1.6, and 2 mm2 of 10, 16, and 20 μm FF uterus tissue were analyzed. The results showed (Figure 4a) that increasing the amount of injected tissue led to an increase of number of identified proteins, following the same trend for all examined tissue thicknesses. Also, an increase in tissue thickness correlated with an increase in the measured abundance of identified proteins, reported as NSAF values. Comparison of results for different tissue thicknesses (Figure 4b) showed that 10 μm thick tissues had the lowest NSAF values compared to corresponding samples between 16 and 20 μm thick tissues. Overall, our results showed sufficient sensitivity of these method in that they could measure proteins from extremely small amounts of tissue, such as 0.2 mm2, while we observed a plateau in the number of identified proteins between 1 and 2 mm2.
Figure 4.
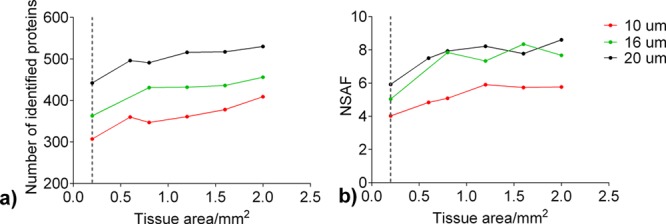
Quantitative comparison of protein abundance by NSAF values for 0.2, 0.6, 0.8, 1.2, 1.6, and 2.0 mm2 of 10, 16, and 20 μm thick FF uterus tissue. Results are obtained after extraction and digestion of the proteins using Protocol 2. (a) Number of identified proteins; (b) NSAF values compared across different samples.
Comparison of Physicochemical Properties of Identified Proteins
Qualitative evaluation of the identified proteins was performed in terms of sequence coverage, molecular weight, and GRAVY score, which is a simple method for calculating the hydropathic character of a protein.49 The interest in hydrophobic proteins is that these proteins may be membrane-associated and therefore difficult to solubilize. Proteins extracted and digested using Protocols 1 and 2 showed similar values of sequence coverage and NSAF values after their categorization into several bins, as a function of their molecular weight (Mw). The highest sequence coverage was found for proteins with Mw from 10 to 20 kDa (approximately 35%), followed by the proteins of 20–30 kDa (Figure 5a). Sequence coverage (Figure S-5, Supporting Information) and abundance of the proteins (Figure S-6, Supporting Information) as a function of their Mw were also evaluated for 16 and 20 μm thick FF uterus tissue.
Figure 5.
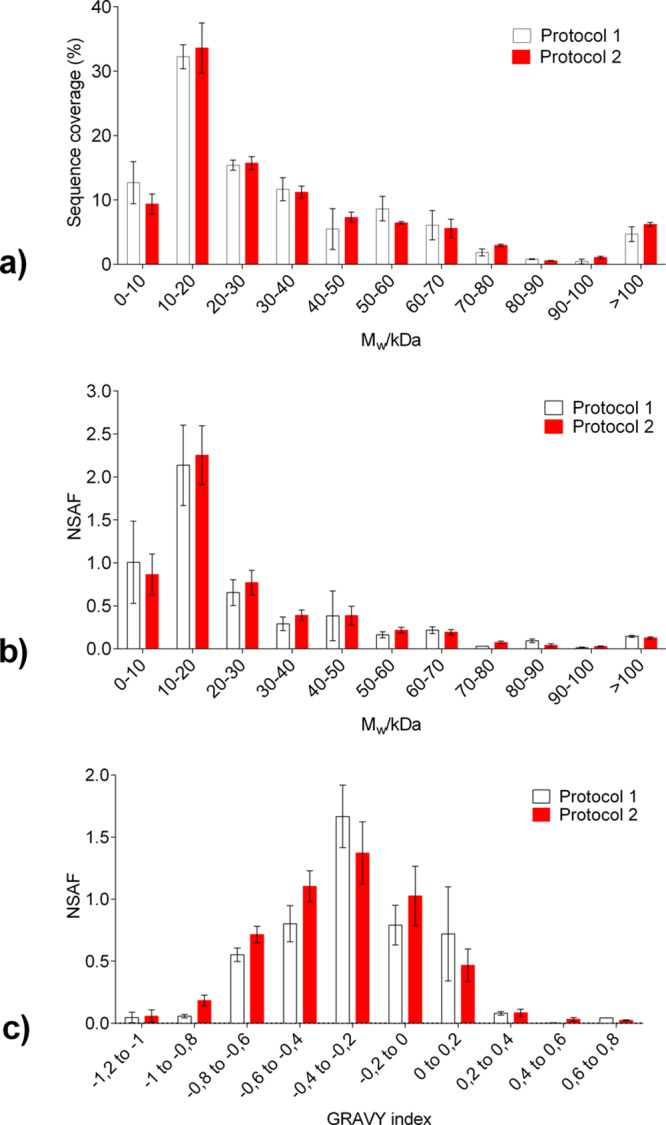
Distribution of the proteins identified from 3 biological replicates in 10 μm thick FF human uterus tissue after extraction and digestion using Protocols 1 and 2. (a) Sequence coverage; (b) NSAF values; and (c) GRAVY index. Results are shown as mean ± SD.
Figure 5c shows an analysis of the identified proteins in terms of their GRAVY. The results show that protein distribution is biased to a negative GRAVY value (in the region from −1.2 to 0), indicating that fewer proteins with hydrophobic character were detected. In both methods, the most abundant proteins were detected in the GRAVY index region from −0.4 to −0.2 (32% and 25% for Protocol 1 and 2, respectively) (Figure 5c). Comparison of the both methods shows that somewhat more proteins were detected in the region from −1 to −0.4 when Protocol 2 was used, while moving toward more positive values (regions −0.4 to −0.2 and 0 to 0.2) showed an increase in the abundance of the proteins extracted and digested using Protocol 1.
Conclusions
Retrieving protein information from small tissue amounts is important as patient samples are mostly limited in their number and sample size for each time point. The methods should be reproducible and effectively provide a synoptic overview of the aberrant protein expression, sequences and/or PTMs. Moreover, the development of analytical procedures that can effectively and reproducibly enable analysis of minute samples would spur on the development of less-invasive procedures for sampling of patient tissues. In this study, we evaluated several procedures in order to establish a simple yet fitting protocol for efficient and reproducible protein extraction and digestion. Our results show that using SDS, in combination with 30% ACN/urea results in 20% and 55% less identified proteins, when compared to a 30% ACN/urea extraction only. However, the protein abundance scores, expressed as NSAF values, from the SDS/30% ACN/urea mixture were higher when compared to 30% ACN/urea alone. Thus, SDS effectively solubilizes a subset of the proteome, but appears not to perform equally well on solubilizing all proteins, with the methods we tested.
Furthermore, we report that changes in the composition of the organic solvent did not contribute significantly to the number of identified proteins. However, an additional digestion with trypsin led to greater overlap between replicates. A method using 30% ACN/urea buffer was tested on very small amounts of the tissue, and these results show that a small increment in the amount of tissue leads to an asymptotic increase in the number of identified proteins and their abundances, which plateaus in the range of 1–2 mm2.
Acknowledgments
This work was supported by the Dutch National Sectorplan Natuur- en Scheikunde (SNS - 10.0119/D). The authors thank Inge H. Briaire- de Bruijn for assistance with microdissection of the uterus tissue samples and dr Marc Vaudel and Florian L. Lucas for their bioinformatics help. Also, the authors are grateful to Dr Jesper Kers for cell counting of uterus tissues.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b01937.
Instrumental settings comparison, additional qualitative and quantitative reproducibility of the methods, and lists of identified proteins (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Giusti L.; Lucacchini A. Expert Rev. Proteomics 2013, 10 (2), 165–177. 10.1586/epr.13.3. [DOI] [PubMed] [Google Scholar]
- De Graaf E. L.; Pellegrini D.; McDonnell L. A. J. Proteome Res. 2016, 15 (12), 4722–4730. 10.1021/acs.jproteome.6b00889. [DOI] [PubMed] [Google Scholar]
- Kulak N. A.; Pichler G.; Paron I.; Nagaraj N.; Mann M. Nat. Methods 2014, 11 (3), 319–324. 10.1038/nmeth.2834. [DOI] [PubMed] [Google Scholar]
- Murgia M.; Nagaraj N.; Deshmukh A. S.; Zeiler M.; Cancellara P.; Moretti I.; Reggiani C.; Schiaffino S.; Mann M. EMBO Rep. 2015, 16 (3), 387–395. 10.15252/embr.201439757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J. R.; Zougman A.; Nagaraj N.; Mann M. Nat. Methods 2009, 6 (5), 359–362. 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Duś-Szachniewicz K.; Ostasiewicz P.; Ziółkowski P.; Rakus D.; Mann M. J. Proteome Res. 2015, 14 (9), 4005–4018. 10.1021/acs.jproteome.5b00523. [DOI] [PubMed] [Google Scholar]
- Wisniewski J. R.; Ostasiewicz P.; Mann M. J. Proteome Res. 2011, 10 (7), 3040–3049. 10.1021/pr200019m. [DOI] [PubMed] [Google Scholar]
- Shen K.; Sun J.; Cao X.; Zhou D.; Li J. PLoS One 2015, 10 (11), e0142650. 10.1371/journal.pone.0142650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostasiewicz P.; Zielinska D. F.; Mann M.; Wiśniewski J. R. J. Proteome Res. 2010, 9 (7), 3688–3700. 10.1021/pr100234w. [DOI] [PubMed] [Google Scholar]
- Atrih A.; Mudaliar M. A. V; Zakikhani P.; Lamont D. J.; Huang J. T.-J.; Bray S. E.; Barton G.; Fleming S.; Nabi G. Br. J. Cancer 2014, 110 (6), 1622–1633. 10.1038/bjc.2014.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski J. R.; Mann M. Anal. Chem. 2012, 84, 2631–2637. 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
- Santos H. M.; Kouvonen P.; Capelo J.-L.; Corthals G. L. Proteomics 2013, 13 (9), 1423–1427. 10.1002/pmic.201200241. [DOI] [PubMed] [Google Scholar]
- Hansen K. C.; Kiemele L.; Maller O.; O’Brien J.; Shankar A.; Fornetti J.; Schedin P. Mol. Cell. Proteomics 2009, 8 (7), 1648–1657. 10.1074/mcp.M900039-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski M.; Wurlitzer M.; Omidi M.; Ren L.; Kruber S.; Nimer R.; Robertson W. D.; Horst A.; Miller R. J. D.; Schlüter H. Angew. Chem., Int. Ed. 2015, 54 (1), 285–288. 10.1002/anie.201407669. [DOI] [PubMed] [Google Scholar]
- Fowler C. B.; O’Leary T. J.; Mason J. T. J. Proteomics Bioinform. 2014, 7 (6), 151–157. 10.4172/jpb.1000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corthals G. L.; Wasinger V. C.; Hochstrasser D. F.; Sanchez J.-C. Electrophoresis 2000, 21 (6), 1104–1115. . [DOI] [PubMed] [Google Scholar]
- Guo T.; Wang W.; Rudnick P. A.; Song T.; Li J.; Zhuang Z.; Weil R. J.; DeVoe D. L.; Lee C. S.; Balgley B. M. J. Histochem. Cytochem. 2007, 55 (7), 763–772. 10.1369/jhc.7A7177.2007. [DOI] [PubMed] [Google Scholar]
- Bell L. N.; Saxena R.; Mattar S. G.; You J.; Wang M.; Chalasani N. Proteomics: Clin. Appl. 2011, 5 (7–8), 397–404. 10.1002/prca.201000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronci M.; Bonanno E.; Colantoni A.; Pieroni L.; Di Ilio C.; Spagnoli L. G.; Federici G.; Urbani A. Proteomics 2008, 8, 3702–3714. 10.1002/pmic.200701143. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Rigueiro T.; Valladares-Ayerbes M.; Haz-Conde M.; Blanco M.; Aparicio G.; Fernández-Puente P.; Blanco F. J.; Lorenzo M. J.; Aparicio L. A.; Figueroa A. Proteomics 2011, 11 (12), 2555–2559. 10.1002/pmic.201000809. [DOI] [PubMed] [Google Scholar]
- Guo T.; Kouvonen P.; Koh C. C.; Gillet L. C.; Wolski W. E.; Rost H. L.; Rosenberger G.; Collins B. C.; Blum L. C.; Gillessen S.; Joerger M.; Jochum W.; Aebersold R. Nat. Med. 2015, 21 (4), 407–413. 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownridge P.; Beynon R. J. Methods 2011, 54 (4), 351–360. 10.1016/j.ymeth.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Sun S.; Zhou J. Y.; Yang W.; Zhang H. Anal. Biochem. 2014, 446 (1), 76–81. 10.1016/j.ab.2013.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebker S. A.; Wojtkiewicz M.; Koepsell S. A. Proteomics 2015, 15 (21), 3744–3753. 10.1002/pmic.201500147. [DOI] [PubMed] [Google Scholar]
- McCarthy J.; Hopwood F.; Oxley D.; Laver M.; Castagna A.; Righetti P. G.; Williams K.; Herbert B. J. Proteome Res. 2003, 2 (3), 239–242. 10.1021/pr025564b. [DOI] [PubMed] [Google Scholar]
- Lin M.-F.; Williams C.; Murray M. V.; Conn G.; Ropp P. A. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2004, 803, 353–362. 10.1016/j.jchromb.2004.01.017. [DOI] [PubMed] [Google Scholar]
- Alkhas A.; Hood B. L.; Oliver K.; Teng P. N.; Oliver J.; Mitchell D.; Hamilton C. A.; Maxwell G. L.; Conrads T. P. J. Proteome Res. 2011, 10 (11), 5264–5271. 10.1021/pr2007736. [DOI] [PubMed] [Google Scholar]
- Russell W. K.; Park Z.-Y.; Russell D. H. Anal. Chem. 2001, 73 (11), 2682–2685. 10.1021/ac001332p. [DOI] [PubMed] [Google Scholar]
- Longuespée R.; Alberts D.; Pottier C.; Smargiasso N.; Mazzucchelli G.; Baiwir D.; Kriegsmann M.; Herfs M.; Kriegsmann J.; Delvenne P.; De Pauw E. Methods 2016, 104, 154–162. 10.1016/j.ymeth.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Hervey W. J.; Strader M. B.; Hurst G. B. J. Proteome Res. 2007, 6, 3054–3061. 10.1021/pr070159b. [DOI] [PubMed] [Google Scholar]
- Wall M. J.; Crowell A. M. J.; Simms G. A.; Liu F.; Doucette A. A. Anal. Chim. Acta 2011, 703 (2), 194–203. 10.1016/j.aca.2011.07.025. [DOI] [PubMed] [Google Scholar]
- Craven R. A.; Cairns D. A.; Zougman A.; Harnden P.; Selby P. J.; Banks R. E. Proteomics: Clin. Appl. 2013, 7 (3–4), 273–282. 10.1002/prca.201200065. [DOI] [PubMed] [Google Scholar]
- Nirmalan N. J.; Hughes C.; Peng J.; McKenna T.; Langridge J.; Cairns D. A.; Harnden P.; Selby P. J.; Banks R. E. J. Proteome Res. 2011, 10 (2), 896–906. 10.1021/pr100812d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoka L. C. Proteome Sci. 2008, 6, 30. 10.1186/1477-5956-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond E. S.; Nayak S.; Ueberheide B.; Wisniewski T. Sci. Rep. 2015, 5, 15456. 10.1038/srep15456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanca A.; Abbondio M.; Pisanu S.; Pagnozzi D.; Uzzau S.; Addis M. Clin. Proteomics 2014, 11 (1), 28. 10.1186/1559-0275-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peuchen E. H.; Sun L.; Dovichi N. J. Anal. Bioanal. Chem. 2016, 408 (17), 4743–4749. 10.1007/s00216-016-9564-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell A. M. J.; Wall M. J.; Doucette A. A. Anal. Chim. Acta 2013, 796, 48–54. 10.1016/j.aca.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Kachuk C.; Stephen K.; Doucette A. J. Chromatogr. A 2015, 1418, 158–166. 10.1016/j.chroma.2015.09.042. [DOI] [PubMed] [Google Scholar]
- Leon I. R.; Schwammle V.; Jensen O. N.; Sprenger R. R. Mol. Cell. Proteomics 2013, 12 (10), 2992–3005. 10.1074/mcp.M112.025585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. Y.; Dann G. P.; Shi T.; Wang L.; Gao X.; Su D.; Nicora C. D.; Shukla A. K.; Moore R. J.; Liu T.; Camp D. G. II; Smith R. D.; Qian W. J. Anal. Chem. 2012, 84 (6), 2862–2867. 10.1021/ac203394r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.-H.; Gregorich Z. R.; Chen A. J.; Hwang L.; Guner H.; Yu D.; Zhang J.; Ge Y. J. Proteome Res. 2015, 14 (3), 1587–1599. 10.1021/pr5012679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveliev S. V.; Woodroofe C. C.; Sabat G.; Adams C. M.; Klaubert D.; Wood K.; Urh M. Anal. Chem. 2013, 85 (2), 907–914. 10.1021/ac302423t. [DOI] [PubMed] [Google Scholar]
- Chen E. I.; Cociorva D.; Norris J. L.; Yates J. R. J. Proteome Res. 2007, 6 (7), 2529–2538. 10.1021/pr060682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waas M.; Bhattacharya S.; Chuppa S.; Wu X.; Jensen D. R.; Omasits U.; Wollscheid B.; Volkman B. F.; Noon K. R.; Gundry R. L. Anal. Chem. 2014, 86 (3), 1551–1559. 10.1021/ac403185a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehmas A. P.; Muth-Pawlak D.; Huhtinen K.; Saloniemi-Heinonen T.; Jaakkola K.; Laajala T. D.; Kaprio H.; Suvitie P. a; Aittokallio T.; Siitari H.; Perheentupa A.; Poutanen M.; Corthals G. L. J. Proteome Res. 2014, 13 (11), 4983–4994. 10.1021/pr500384n. [DOI] [PubMed] [Google Scholar]
- Vaudel M.; Barsnes H.; Berven F. S.; Sickmann A.; Martens L. Proteomics 2011, 11 (5), 996–999. 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- Vaudel M.; Burkhart J. M.; Zahedi R. P.; Oveland E.; Berven F. S.; Sickmann A.; Martens L.; Barsnes H. Nat. Biotechnol. 2015, 33 (1), 22–24. 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- Kyte J.; Doolittle R. F. J. Mol. Biol. 1982, 157 (1), 105–132. 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.