

Abstract
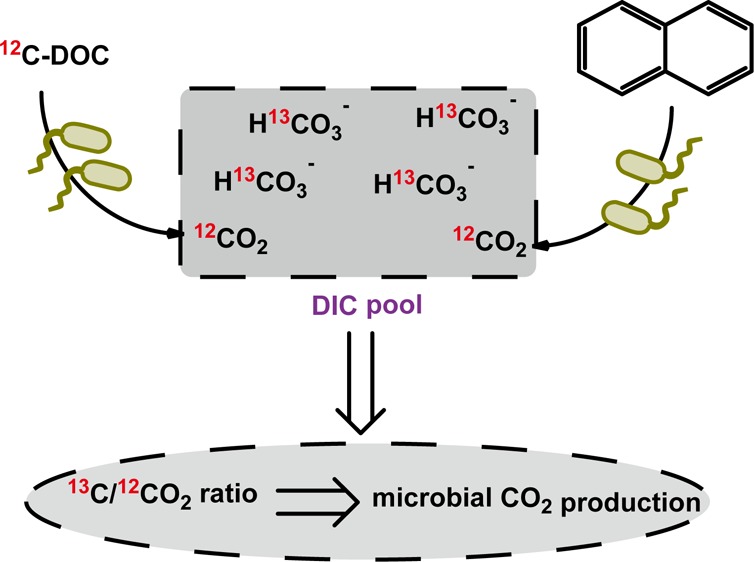
Assessing the biodegradation of organic compounds is a frequent question in environmental science. Here, we present a sensitive, inexpensive, and simple approach to monitor microbial mineralization using reverse stable isotope labeling analysis (RIL) of dissolved inorganic carbon (DIC). The medium for the biodegradation assay contains regular organic compounds and 13C-labeled DIC with 13C atom fractions (x(13C)DIC) higher than natural abundance (typically 2–50%). The produced CO2 (x(13C) ≈ 1.11%) gradually dilutes the initial x(13C)DIC allowing to quantify microbial mineralization using mass-balance calculations. For 13C-enriched CO2 samples, a newly developed isotope ratio mid-infrared spectrometer was introduced with a precision of x(13C) < 0.006%. As an example for extremely difficult and slowly degradable compounds, CO2 production was close to the theoretical stoichiometry for anaerobic naphthalene degradation by a sulfate-reducing enrichment culture. Furthermore, we could measure the aerobic degradation of dissolved organic carbon (DOC) adsorbed to granular activated carbon in a drinking water production plant, which cannot be labeled with 13C. Thus, the RIL approach can be applied to sensitively monitor biodegradation of various organic compounds under anoxic or oxic conditions.
Introduction
A crucial step in studying microbial degradation of organic compounds is using microcosm experiments to prove the degradability and to obtain degradation rates. Of special interest is to assess if the compounds are fully mineralized to CO2 or if they are just transformed to metabolites which might be even more toxic in some cases.1 A preferred method is to prove mineralization of the compound of interest by the development of labeled CO2 in airtight containers. To this end, target compounds are either labeled with stable (13C) or radioactive (14C) carbon isotopes and the evolution of 13CO2 or 14CO2 is measured over time, providing a very sensitive way for detection of low substrate turnover compared to residual substrate concentration measurements.1−4 Stable isotope methods are a preferred alternative in recent years, because the compounds are easier to handle and less expensive compared to radioactive labeling. However, the stable isotope labeling method is less applicable for higher-molecular weight compounds, because the 13C-labeled substrates usually require special synthesis, and are thus very expensive or even not commercially available. Furthermore, complex substrates such as dissolved organic carbon (DOC) or crude oil cannot be synthesized or labeled. Therefore, we aimed to develop a new method for quantifying biodegradation of organic compounds to CO2, which we call reverse stable isotope labeling (RIL).
In the classical stable isotope labeling experiments, fully or partially 13C-labeled compounds are added to the cultivation medium which is buffered with bicarbonate at natural 13C/12C isotopic abundance (i.e., with a 13C fraction x(13C) ≈ 1.1%). The change of the 13C/12C isotope ratio of CO2 in the headspace is then followed over time and used to calculate the quantity of evolved 13CO2. In our reverse stable isotope labeling approach, we simply reverse this set up by adding 13C-labeled bicarbonate with x(13C) values ranging from 2 to 50%. Then, a regular substrate or mixture for microbial degradation is supplied which has a normal stable isotope ratio at natural abundance (x(13C) ≈ 1.11%). Consequently, CO2 with a stable isotope ratio at natural abundance is released during mineralization. As the stable isotope ratio of the released CO2 differs significantly from the isotope ratio of the labeled background dissolved inorganic carbon (DIC), one can measure the changes in the 13C/12C isotope ratio of the total DIC (i.e., the sum of inorganic carbon species in the medium) over time which can be used to calculate CO2 production.
To demonstrate the feasibility and usefulness of the RIL method, we studied anaerobic degradation of aromatic hydrocarbons, which belong to the most recalcitrant priority pollutants in contaminated aquifers. The broader environmental relevance of this method was further shown by aerobic mineralization of DOC adsorbed to granular activated carbon (GAC) during drinking water production.
Experimental Section
Cultivation of Anaerobic Cultures
Thauera aromatica strain K172 (DSM 6984) and Geobacter metallireducens strain GS-15 (ATCC 53774/DSM 7210) were purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Both strains were cultivated as described before under anoxic conditions in a carbonate-buffered freshwater mineral medium at pH of 7.2–7.4 spiked with ∼0.8 mM pure toluene (Sigma-Aldrich, Steinheim, Germany),5 and amended with 5 mM nitrate or 50 mM ferric citrate as electron acceptors, respectively. The sulfate-reducing enrichment culture N47 was cultivated as described previously,6 with addition of 1 mL of 1.5% 2-methylnaphthalene or 3 mL of 1.5% naphthalene both dissolved in 2,2,4,4,6,8,8-heptamethylnonane (Sigma-Aldrich, Steinheim, Germany) to each cultivation bottle after autoclaving.
The uncharacterized iron-reducing, naphthalene-degrading enrichment culture SN was enriched from Pitch Lake in Trinidad and Tobago7 with ferrihydrite as electron acceptor according to previously described methods.8 It was transferred to freshwater mineral medium every 3–4 months when significant production of ferrous iron was indicated by the presence of black iron oxides. The culture was maintained in the lab for more than three years before our experiment, leading to a stable and sediment-free, iron-reducing culture (Supporting Information (SI) Figure S1). For the RIL experiment, 3 mL of 1.5% naphthalene dissolved in 2,2,4,4,6,8,8-heptamethylnonane were added to each cultivation bottle after autoclaving.
For every culture, RIL experiments were performed using five separate 250 mL serum bottles first filled with 135 mL of fresh medium without bicarbonate buffer and flushed with CO2/N2 (20:80, v/v). The NaH13CO3 (Sigma-Aldrich Co.) and NaH12CO3 (Sigma-Aldrich Co.) were injected separately through the stopper from filter-sterilized stock solutions (1 M each, headspace briefly flushed with N2) to produce a mixture of 30 mM bicarbonate in the medium. Considering the linearity range of IRMS/IRIS, we shifted the added labeled bicarbonate ratios from 50% to 10%. Afterward, substrates were injected through the stoppers. Then, three bottles were inoculated with 15 mL parent cultures whereas the other two were inoculated with autoclaved cultures (three times) serving as controls. All culture bottles were incubated at 30 °C in the dark.
Aerobic Mineralization of DOC on Granular Activated Carbon
GAC samples were taken from a testing facility (Mülheim, Germany) treating raw water from River Ruhr for drinking water production. First, the river water is filtered by slow sand filtration followed by ozonation. After ozonation, the water is filtered by a multilayer filter which removes particulate impurities. Afterward the water enters the down-flow GAC filter (30 cm diameter, 180 cm length) for adsorption and biological removal of DOC by attached biofilms.
For RIL experiments, the GAC particles (3 g each) were transferred to three separate 250 mL serum bottles filled with 50 mL influent water (after multilayer filter) under a laminar flow box. A large air headspace (200 mL) ensured oxic conditions in the bottles. The following three different incubations served as control: (1) fresh activated carbon + ozonized water; (2) ozonized water; (3) water after multilayer filter. For each microcosm, NaH13CO3 and NaH12CO3 at a ratio of 10:90 were injected separately through the stopper to produce a final concentration of 10 mM bicarbonate in the medium. DOC in each microcosm originated from DOC adsorbed to the activated carbon. All bottles were incubated at 17 °C in the dark to mimic the temperature conditions of the GAC filters.
Analytical Methods
Toluene concentrations were determined by headspace analyses with GC/MS (GC, Trace-DSQ; MS, Thermo Finnigan, San Jose, CA) in selective ion monitoring mode with a fused-silica capillary column DB-5 as previously described.9 Total concentrations of toluene in the cultures were calculated from concentrations in the liquid phase and in the headspace using the dimensionless air–water partitioning constant Kaw at 25 °C of 0.235.10 Adding naphthalene and 2-methylnaphthalene in 2,2,4,4,6,8,8-heptamethylnonane as carrier phase prohibited a reliable direct quantification of the substrate concentrations.8 Therefore, sulfate consumption was monitored using the barium-gelatin method to indirectly quantify naphthalene and 2-methylnaphthalene degradation.11 Iron(II) production was measured with the ferrozine assay.12 Cell growth was monitored by microscopically counting cell numbers.
Sample Preparation and Expression of Carbon Isotope Ratios
We adopted a procedure used to analyze carbon isotope ratios in natural water samples by taking liquid samples and liberating CO2 using phosphoric acid as reported previously (details in SI).13 Carbon isotope ratios (13C/12C, referred to as R) are reported in conventional delta notation (δ13C) as per mil (‰) values, relative to the Vienna Pee Dee Belemnite (VPDB) standard (eq 1).
| 1 |
where 0.0111802 is the ratio of 13C/12C in VPDB standard. To facilitate the comparison of results which are obtained from samples highly enriched in 13C compared to natural samples, the carbon isotope ratios were converted into 13C atom fractions (x(13C)) (%) from δ13C values according to Coplen.14 The conversions of x(13C) changes to CO2 production were based on isotope mass balance equations (details in the SI).
Isotope Ratio Mass Spectrometry (IRMS)
CO2 samples (100 μL) were taken from the headspace of 15 mL serum bottles with a gastight syringe and manually injected into a GC-IRMS system consisting of a TRACE GC Ultra gas chromatograph with split/splitless injector (GC) (both Thermo Fisher Scientific Corporation, Milan, Italy) coupled to a Finnigan MAT 253 isotope ratio mass spectrometer (IRMS) connected via a Finnigan GC combustion III interface (Thermo Fisher Scientific Corporation, Bremen, Germany). The temperature of the injector was held at 150 °C for 360 s. Helium 5.0 (AirLiquide, Oberhausen, Germany) was used as carrier gas with a constant flow rate of 1.6 mL min–1. The split flow was fixed at 160 mL min–1. CO2 was separated from other gases using a 30 m Rtx-Qbond column with an inner diameter of 0.32 mm and a film thickness of 10 μm (Restek GmbH, Bad Homburg, Germany). Certified pure CO2 gas (δ13C = −35.28 ‰) (AirLiquide, Oberhausen, Germany) was used as working reference gas. All samples were measured in triplicate. When compared to isotope ratio mid-infrared spectroscopy (IRIS), a secondary standard of 13CO2 gas (x(13C) = 10%, Sigma-Aldrich, Taufkirchen, Germany) was analyzed to correct data using a two-point calibration approach.
Isotope Ratio Mid-Infrared Spectroscopy (IRIS)
Samples were analyzed using a Thermo Fisher Delta Ray CO2 Isotope Ratio Infrared Spectrometer with Universal Reference Interface (URI) Connect.15−18 CO2-free synthetic air (Air Liquide, Oberhausen, Germany) was used as the carrier gas. CO2 in synthetic air at 414.2 ppm (Air Liquide, Oberhausen, Germany) was used for CO2 concentration calibration. Before measurements, two pure CO2 reference gases were used for calibration of carbon isotope ratios: one with δ13C values of −9.7‰ ((x(13C) = 1.10%, Thermo Fisher, Bremen, Germany) and the other with x(13C) = 10% (Sigma-Aldrich, Taufkirchen, Germany). For sample measurements, pure CO2 gas with x(13C) = 10% was used as working reference gas and samples were bracketed with working reference gas. The CO2 concentration for the working reference gas and the sample gas coming into the analyzer was set to 380 ppm to achieve optimal precision. The δ13C values were reported as average of 5 min’ measurements. The long measurement time under constant gas flows implies that IRIS needs a relatively larger gas sample size compared to IRMS.
Results and Discussion
Verification of the RIL Method
The proof of principle for the reverse stable isotope labeling (RIL) method was first verified with two reference cultures: (1) anaerobic toluene degradation by G. metallireducens and (2) anaerobic 2-methylnaphthalene degradation by the enrichment culture N47 (Figure 1). The medium was supplemented with unlabeled and 13C-labeled sodium bicarbonate at a ratio of 50:50 and carbon stable isotope ratios of DIC samples were measured with IRMS. During anaerobic toluene degradation by G. metallireducens, the initial 13C fractions of DIC samples (x(13C)initial = 33.16%) decreased by Δx(13C) = 13.03%, which corresponded to a consumption of total toluene in the bottle from 0.75 to 0.18 mM (Figure 1a). No significant changes could be observed for toluene concentrations or 13C fractions in control bottles. During anaerobic degradation of 2-methylnaphthalene by the enrichment culture N47, the initial 13C fractions of DIC samples (x(13C)initial = 34.01%) decreased by 13.62% concomitant with a measured reduction of sulfate from 9.97 to 8.01 mM (equal to theoretical consumption of 0.29 mM of 2-methylnaphthalene) (Figure 1b). No significant changes could be observed for sulfate concentrations or 13C fractions in control bottles. These two experiments provided proof-of-principle that measuring carbon stable isotope ratios of DIC by the RIL method can be used for demonstrating microbial degradation.
Figure 1.

Anaerobic degradation of (a) toluene by G. metallireducens, (b) 2-methylnaphthalene by the enrichment culture N47, (c) naphthalene by an uncharacterized iron-reducing enrichment culture SN, and (d) toluene by T. aromatica. The added bicarbonate buffer (30 mM) consisted of nonlabeled and 13C-labeled sodium bicarbonate at a ratio of 50:50 for G. metallireducens (a) and the enrichment culture N47 (b), 80:20 for the enrichment culture SN (c), and 90:10 for T. aromatica (d). Sulfate or iron reduction as surrogate measurements indicated methylnaphthalene or naphthalene degradation. Carbon isotope ratio changes were expressed as a percentage compared to the initial x(13C) value: Δx(13C) = (x(13C)t – x(13C)initial)/x(13C)initial × 100. The calculated CO2 production can be found in SI Figure S2. The x(13C) values of DIC samples for (a)–(c) were only measured by IRMS. Both IRMS and IRIS were performed for (d). Data points depict means of two or three parallel incubations measured three times each. Error bars represent standard deviation of the biological replicates.
We then used the RIL method to test if an uncharacterized enrichment culture can mineralize naphthalene under iron-reducing conditions (Figure 1c). In contrast to previous experiments, unlabeled and 13C-labeled sodium bicarbonate were added at a ratio of 80:20, making the initial x(13C) values lower than in the proof-of-principle study. Carbon stable isotope ratios of DIC samples were measured with IRMS. Naphthalene was the sole organic carbon source added. In control bottles inoculated with autoclaved cultures, the 13C fractions of DIC samples remained stable at ∼15.46%. In the bottles inoculated with active cultures, a considerable lag phase of about 90 days took place before a decrease in x(13C) values and an increase of ferrous iron could be observed. Then, the initial 13C fraction of DIC samples (x(13C)initial = 15.26%) dropped by 10.86% coupled to a concomitant ferrous iron production of 31 mM. This data provided unambiguous evidence that naphthalene mineralization to CO2 was coupled to iron reduction. But due to the nonlinearity of IRMS measurement at high isotope ratios (see below), the calculation of CO2 production and the electron balance were not accurate (SI Figure S2 and Table S1).
Evaluation of highly enriched x(13C) values measured by IRMS
For analysis of carbon stable isotope ratios by IRMS, CO2 samples are first ionized to different ions mass-to-charge ratios (m/z) at 44, 45, and 46. The resulting ions are subsequently separated in a magnetic field. Then, the Faraday cup collectors are used to specifically collect signals of each ion beam. In the case of samples at natural abundance, the 13C atom fractions are at x(13C) = ∼ 1.11%, making signals (voltage outputs) of m/z 45 and 46 much weaker compared to m/z 44 (SI Figure S3a). To get comparative intensities for the highest quality results, each Faraday cup is designed to connect its own amplification ohmic resistor, at a ratio of 3:300:1000 for m/z 44, 45, and 46, respectively (SI Figure S3a). When measuring samples highly enriched in 13C (e.g. x(13C) = 15–35%) by using the same standard setting, the amplified signals will not be in the same range, but with m/z 45 dominating the peak (SI Figure S3b), even though the absolute ratio of 13C/12C is smaller than 1. Consequently, the IRMS response could be imprecise. Aware of this, we preformed two linearity tests on an IRMS using two different reference gases to investigate if reported x(13C) values could significantly alter with changes in CO2 concentrations. Results showed a good linearity when we measured the natural abundance reference gas (x(13C) = 1.067%) (Figure 2a). For highly enriched samples (x(13C) = 10.3%), however, the measured x(13C) values increased with elevated gas partial pressures showing a larger positive slope of 0.02 (significantly linearly dependent on the CO2 concentration) (Figure 2a). This indicated that the results shown in Figures 1a-c could be inaccurate, as we could not apply reference gases at the same concentration. This was further confirmed by the stoichiometric calculations which showed great differences between the measured and the theoretical ratios between the reduction of electron acceptors or donors and CO2 production (SI Figure S2 and Table S1). Nevertheless, we could obtain a semiquantitative assessment of the degradation activities.
Figure 2.

Performance comparison of IRIS and IRMS. (a) Effect of CO2 concentrations (proportional to Peak Area All, that is, sum of peak areas of m/z 44, 45, and 46 of CO2) on the IRMS-measured 13C atom fractions for 13C-enriched gas (x(13C) = 11.073%; black squares) and natural abundance gas (x(13C) = 1.0671%; black triangles), n = 3. (b) Effect of CO2 concentrations on the IRIS-measured 13C atom fractions (x(13C) = 10.3%; blue circles) and the linear regression line showing IRIS-measured enriched 13C samples plotted against theoretical x(13C) values (n = 5; black triangles). No error bars are shown as they lie within the dimensions of the symbol.
One possible solution to alleviate this problem is to switch the feedback resistor of the preamplifier to adjust m/z 44 signal to an appropriate range.19 Unfortunately, this cannot be achieved due to the limitations of the used IRMS. Reinsch and Ambus adjusted resistor settings of the mass spectrometer to obtain higher measurement precision, but still they found that x(13C) values measured by IRMS for 13C-enriched samples (x(13C) = 50%) were also significantly dependent on CO2 concentrations.20 They subsequently used an empirical relationship to correct data but the measured x(13C) values never showed the expected values. Using IRMS is therefore only semiquantitative for determination of x(13C) values in highly enriched samples, which motivated us to search for a better alternative.
Using IRIS As an Alternative Method for Analyzing Highly Enriched 13C Samples
In contrast to IRMS, IRIS is capable of measuring enriched samples up to x(13C) = 25%.21 Some other early designed 13CO2/12CO2 infrared gas analyzer using tunable diode laser absorption (TDLA) techniques have been shown to be able to measure values of x(13C) as high as 5% or 10%, as long as the 13CO2 concentration is within the 13CO2 range of the calibration gas.22,23 Therefore, infrared gas analyzers can be a good alternative for measuring highly enriched samples in 13CO2-labeling experiments. The TDLA technique employed by the Delta Ray IRIS follows the Beer–Lambert law.24 While a laser beam scans through absorption lines of 13CO2 and 12CO2 of samples, the decrease of intensity of the laser can be directly linked to the concentration of the respective molecular species and hence carbon isotope ratios. For quantifications, absorptions within the scan range can be fitted by an algorithm adapted to certain environmental conditions within the laser cell (100 mbar pressure). For natural abundance samples, the peak areas for 13CO2 are relatively small compared to that for 12CO2 (SI Figure S4a).18 For 13C-enriched samples (e.g., x(13C) = 10.0%), the peak areas for 13CO2 increase strongly (SI Figure S4b). However, as long as the 13CO2 band does not reach the saturation level which we never observed, it can be used to determine carbon isotope ratios. Besides, IRIS can automatically determine CO2 concentrations of samples and adjust them to an optimal and constant concentration (380 ppm) going into the optical cell, which could address the issue of isotope ratio drifts caused by CO2 concentrations.18
To further confirm that IRIS can measure highly enriched 13C samples, we measured bicarbonate mixtures with different x(13C) values. The measured x(13C) versus expected x(13C) values showed an excellent linear correspondence (correlation coefficient R2 = 0.9996) over a wide range from x(13C) = 6% to x(13C) = 12%, giving a trendline of y = 1.0315x - 0.1177 (Figure 2b), demonstrating its capability to measure high 13C enrichments. The standard deviations of x(13C) values varied but were all below 0.006%. The precision is more than adequate for biological studies, where sample variation is considerably large than this analytical error (see below). We subsequently sampled different aliquots (0.2 to 0.6 mL) from a 10% 13C-labeled bicarbonate solution and added these into the vials to produce samples with different CO2 concentrations. Results illustrated that even though the CO2 concentrations of samples changed significantly the resultant variation was very low (11.073% ± 0.001%) and remained within the analytical precision (Figure 2b), demonstrating the efficiency of the automatic concentration adjustment by the analyzer.
For a direct comparison of IRMS and IRIS using the RIL method, nonlabeled and 13C-labeled sodium bicarbonate were added at a molar ratio of 90:10 to an anaerobic toluene degradation experiment with T. aromatica. IRIS and IRMS measurements produced a similar decreasing trend of x(13C) values, both indicating toluene mineralization (Figure 1d). Despite identical drops of x(13C) values, IRIS data showed a systematical offset from IRMS data at x(13C) = 0.145% on average. The difference could be related to the failure to produce the same concentration of injected CO2 samples as working reference gases for IRMS.
Sensitivity of CO2 Quantification under Anoxic Conditions Based on IRIS Measurements
The enrichment culture N47 was cultivated in the presence of 10% 13C-labeled sodium bicarbonate as an example to investigate if the RIL method can be used to sensitively quantify CO2 production based on IRIS measurement. Naphthalene was added as the sole organic carbon source while sulfate was supplied as the electron acceptor. In bottles inoculated with active cultures, we observed a reduction of 6.98 mM sulfate, concomitant with a production of 11.87 mM CO2 (Figure 3). The ratio for sulfate consumption to CO2 production (5.9:10) was very close to the theoretical stoichiometry (a ratio of 6:10).25 The closed electron balance indicates that the RIL method is reliable to quantify the extent of microbial mineralization.
Figure 3.
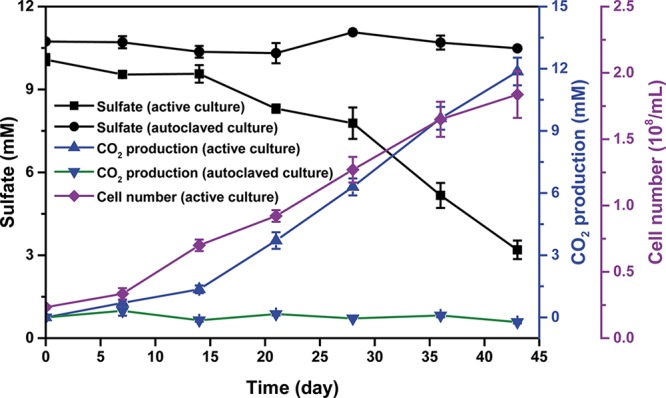
Anaerobic degradation of naphthalene by the sulfate-reducing enrichment culture N47. The added bicarbonate buffer (30 mM) consisted of nonlabeled and 13C-labeled sodium bicarbonate at a ratio of 90:10. The x(13C) values of DIC samples were measured by IRIS. Data points depict means of two or three parallel incubations. Error bars represent standard deviation of the biological replicates.
In control bottles with autoclaved cultures, a maximum variability of x(13C) = 0.05% was determined during the whole incubation time, which depicts the experimental error due to the sampling process and analytical uncertainty. To conclude that there is distinct evidence of biodegradation in bottles with active cultures, depletion in 13C-enrichment of CO2 (x(13C)background – x(13C)total) should be at least two times larger than the experimental error, meaning a production of 0.38 mM CO2 would be sufficient to provide evidence of biodegradation. This corresponds to 38 μM of naphthalene or 1.6% of the 2.34 mM of added naphthalene and clearly demonstrates the sensitivity of the method. This small extent of biodegradation would be impossible to assess using other methods, except for methods using direct stable or radioactive isotope labeled substrates. However, further theoretical consideration of the precision of the analysis predicts that the sensitivity of the method could be improved by a factor of 5–10 concerning the developed CO2. For example, optimization of measurements by IRIS showed that contamination of ambient air in the autosampler system could greatly contribute to analytical uncertainties. In fact, adding a preflushing step to the autosampler system with CO2-free synthetic air reduced the standard deviations of measured x(13C) from 0.028% down to 0.008% (n = 12, samples were taken one by one from the same culture bottle).
Applying the RIL Method to Determine Aerobic Mineralization of DOC on Granular Activated Carbon
Biological GAC filters are widely used in drinking water production systems for removal of undesired biodegradable DOC.26,27 During the steady-state of GAC filters when biomass concentration of the biofilm and effluent DOC concentration remain constant, the removal process is predominately performed by the indigenous microbial communities attached to the surface of activated carbon (i.e., BAC biofilms). It is of interest to characterize microbial activities (i.e., DOC removal rates) of such GAC filters. Most current studies focus on quantifying active biomass densities by, for example, adenosine triphosphate (ATP) analyses.28−30 Here we show that the RIL method can be used to directly assess the aerobic mineralization potential of DOC by biomass attached on the GAC. GAC particles were incubated in lab microcosms filled with influent water and 10% 13C-labeled sodium bicarbonate. DIC samples were taken regularly and shifts in isotopic composition of DIC samples were measured by IRIS to calculate CO2 production. In 22 days, no observable CO2 production could be found for incubations of only ozonized river water or multilayer filtered water (Figure 4). Insignificant amounts of CO2 production were observed in incubations of fresh GAC, which was most likely caused by carbonate adsorbed to the surface, in agreement with the decrease of pH values. For active microcosms with incubations of GAC particles + influent water, however, we could observe significant CO2 production. During the first 2 days, the degradation rate decreased and stayed almost linear afterward (Figure 4). Thus, these data indicated that the RIL method can be used to monitor microbial DOC mineralization rates performed by the biofilm matrix attached to the solid surface of GAC.
Figure 4.
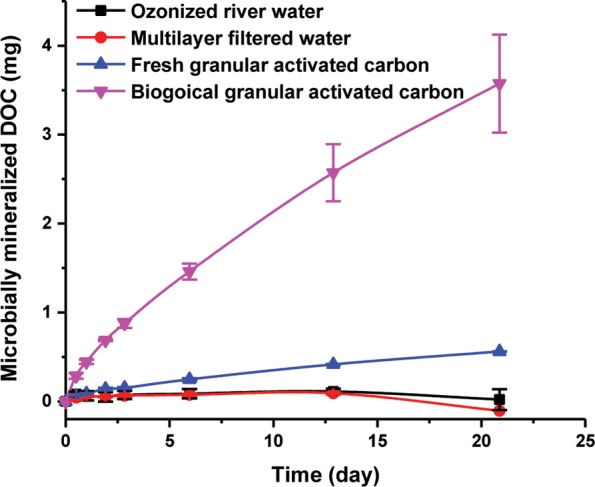
Aerobic mineralization of DOC adsorbed on the GAC filters by attached biofilms. The added bicarbonate (10 mM) consisted of nonlabeled and 13C-labeled sodium bicarbonate at a ratio of 90:10. The x(13C) values of DIC samples were measured by IRIS. Data points depict means of three parallel incubations. Error bars represent standard deviation of the biological replicates.
Environmental Significance
A typical question for research or practice would be to analyze if microorganisms in environmental samples or enrichment cultures can degrade specific organic compounds to CO2, similar to hydrocarbon degradation experiments presented here. Compared to other methods such as following substrate consumption, the RIL can directly prove the degradation of compounds to the end product CO2. Additionally, it does not come along with problems of substrate adsorption to matrix or stoppers, evaporation or other unspecific losses. However, the method runs into limitations if other easily degradable organic carbon is present in the sample (e.g., sediment or water), which can lead to additional release of nonlabeled CO2. This problem also exists in microcosm experiments when, for example, the consumption of electron acceptors such as molecular oxygen (in respiration test) or production of sulfide or ferrous iron are employed to follow biodegradation activity. In many cases, this problem is solved by appropriate controls without substrate if the organic carbon background is not too high.
However, this also turns into another application field of RIL: the very sensitive detection of biodegradation of complex organic substrates such as DOC, as demonstrated in our experiments with granular activated carbon. Measuring DOC degradation is notoriously difficult and needs severe experimental efforts.31 Due to its high sensitivity, RIL can detect even tiny degradation activities that are difficult to assess, otherwise. Only using labeled organic compounds could compete with this sensitivity, but organics such as DOC cannot be labeled. RIL has the advantage that it can analyze degradation of natural organic matter in such environmental samples to microcosms by only adding a little 13C-labeled DIC. Another big advantage is that degradation of compounds can be analyzed that are adsorbed to solid matrices, which is very difficult or almost impossible to measure with other methods at this precision and sensitivity. This is clearly shown by the DOC degradation experiment here. In the case of, for example, wood or plastics degradation, even the degradation of a solid itself could be analyzed.
As demonstrated here, RIL performs under different biodegradation scenarios. Extremely slow biodegradation experiments with very difficult, anaerobic degradation of polycyclic aromatic hydrocarbons can be analyzed as well as faster, aerobic degradation of DOC. The precise analysis of carbon stable isotope ratios by IRMS or IRIS allows for the detection of very small CO2 production. This makes RIL much more sensitive compared to directly measuring DIC concentrations or respiration tests (e.g., ISO 9408 and OECD 301B). If the production of DIC/CO2 during microbial degradation is large, for example, by fast-growing aerobic microorganisms and higher amounts of organic substrates, conventional GC–MS can be used for analyzing degradation of stable isotope-labeled compounds.32 For tiny or slow biodegradation, however, GC–MS cannot compete with tools for analyzing stable isotope ratios of CO2 such as IRMS and IRIS. IRMS does not produce accurate results if the carbon isotope ratios are too high and out of the linearity range. Thus, the Delta Ray IRIS provides the easiest and cheapest as well as the most precise analysis for 13C/12C ratios much higher than natural abundance.
One limitation for RIL is the need of closed systems. For example, aerobic incubations with continuous oxygen supply or open microcosms cannot be assessed by this method. In most cases, however, one can perform the same experiment in closed, stoppered vials when providing sufficient headspace.4 As the major advantage of RIL is assessing small amounts of biodegradation, aerobic degradation of big amounts of substrates will anyway not be a typical field of application.
It is noted that CO2 fixation by photosynthetic organisms, other autotrophs, or heterotrophic CO2 fixation does not affect RIL experiments. The release of CO2 during biodegradation changes the stable isotope ratio of the DIC because of the additional amount of 12CO2. CO2 fixation, however, does not change the carbon isotope ratio of the DIC because both isotopomers are consumed with the same rate. Stable isotope fractionation effects by discrimination of 13CO2 during fixation are orders of magnitude smaller than the changes in the isotope ratios observed here and do not have to be accounted for. This even applies to systems such as methanogenesis where CO2 is both released and consumed. While the release of CO2 during substrate degradation by fermenting organisms changes the stable isotope ratio of the DIC indicating mineralization similar to the sulfate-reducing culture shown above, the consumption of CO2 by methanogen does only affect the isotope ratios to a negligible extent. Methanogenesis produces one of the strongest stable isotope fractionation effects reported for environmental processes.33,34 Thus, we calculated the maximal CO2 carbon isotope fractionation of a theoretical methanogenic degradation experiment assuming the same conditions as in the sulfate-reducing experiment shown in Figure 3. The theoretical 13C isotope shift due to methanogenesis was only x(13C) ≈ 0.01% and much less than the isotope shift of 2.44% caused by fermentative mineralization (see SI for detailed calculations). The RIL method can thus also be applied for systems where both release and consumption of CO2 take place.
Acknowledgments
We thank Marcel Schulte and Nenad Stojanovic for help with Isotope Ratio Mass Spectrometry. We also thank Nils Stoebener and Magda Mandic for help with operating the Isotope Ratio Infrared Spectrometer. X.D. acknowledges a fellowship from the China Scholarship Council (File No. 201306060075). R.U.M. acknowledges funding from the ERC advanced grant EcOILogy (No. 666952).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.est.7b02909.
Details about sample preparation, Isotope Ratio Mid-Infrared Spectroscopy (IRIS), calculations and additional tables and figures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fischer A.; Manefield M.; Bombach P. Application of stable isotope tools for evaluating natural and stimulated biodegradation of organic pollutants in field studies. Curr. Opin. Biotechnol. 2016, 41, 99–107. 10.1016/j.copbio.2016.04.026. [DOI] [PubMed] [Google Scholar]
- Johnsen A. R.; Binning P. J.; Aamand J.; Badawi N.; Rosenbom A. E. The Gompertz function can coherently describe microbial mineralization of growth-sustaining pesticides. Environ. Sci. Technol. 2013, 47 (15), 8508–8514. 10.1021/es400861v. [DOI] [PubMed] [Google Scholar]
- Berry T. D.; Filley T. R.; Clavijo A. P.; Bischoff Gray M.; Turco R. Degradation and microbial uptake of C60 fullerols in contrasting agricultural soils. Environ. Sci. Technol. 2017, 51 (3), 1387–1394. 10.1021/acs.est.6b04637. [DOI] [PubMed] [Google Scholar]
- Bahr A.; Fischer A.; Vogt C.; Bombach P. Evidence of polycyclic aromatic hydrocarbon biodegradation in a contaminated aquifer by combined application of in situ and laboratory microcosms using 13C-labelled target compounds. Water Res. 2015, 69 (0), 100–109. 10.1016/j.watres.2014.10.045. [DOI] [PubMed] [Google Scholar]
- Widdel F.; Bak F., Gram-negative mesophilic sulfate-reducing bacteria. In The Prokaryotes; Springer, 1992; pp 3352–3378. [Google Scholar]
- Selesi D.; Jehmlich N.; von Bergen M.; Schmidt F.; Rattei T.; Tischler P.; Lueders T.; Meckenstock R. U. Combined genomic and proteomic approaches identify gene clusters involved in anaerobic 2-methylnaphthalene degradation in the sulfate-reducing enrichment culture N47. J. Bacteriol. 2010, 192 (1), 295–306. 10.1128/JB.00874-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meckenstock R. U.; von Netzer F.; Stumpp C.; Lueders T.; Himmelberg A. M.; Hertkorn N.; Schmitt-Kopplin P.; Harir M.; Hosein R.; Haque S.; Schulze-Makuch D. Water droplets in oil are microhabitats for microbial life. Science 2014, 345 (6197), 673–676. 10.1126/science.1252215. [DOI] [PubMed] [Google Scholar]
- Kleemann R.; Meckenstock R. U. Anaerobic naphthalene degradation by Gram-positive, iron-reducing bacteria. FEMS Microbiol. Ecol. 2011, 78 (3), 488–496. 10.1111/j.1574-6941.2011.01193.x. [DOI] [PubMed] [Google Scholar]
- Abu Laban N.; Selesi D.; Jobelius C.; Meckenstock R. U. Anaerobic benzene degradation by Gram-positive sulfate-reducing bacteria. FEMS Microbiol. Ecol. 2009, 68 (3), 300–311. 10.1111/j.1574-6941.2009.00672.x. [DOI] [PubMed] [Google Scholar]
- Staudinger J.; Roberts P. V. A critical review of Henry’s law constants for environmental applications. Crit. Rev. Environ. Sci. Technol. 1996, 26 (3), 205–297. 10.1080/10643389609388492. [DOI] [Google Scholar]
- Tabatabai M. A. A rapid method for determination of sulfate in water samples. Environ. Lett. 1974, 7 (3), 237–243. 10.1080/00139307409437403. [DOI] [Google Scholar]
- Stookey L. L. Ferrozine - a new spectrophotometric reagent for iron. Anal. Chem. 1970, 42 (7), 779–781. 10.1021/ac60289a016. [DOI] [Google Scholar]
- Assayag N.; Rive K.; Ader M.; Jezequel D.; Agrinier P. Improved method for isotopic and quantitative analysis of dissolved inorganic carbon in natural water samples. Rapid Commun. Mass Spectrom. 2006, 20 (15), 2243–2251. 10.1002/rcm.2585. [DOI] [PubMed] [Google Scholar]
- Coplen T. B. Guidelines and recommended terms for expression of stable-isotope-ratio and gas-ratio measurement results. Rapid Commun. Mass Spectrom. 2011, 25 (17), 2538–2560. 10.1002/rcm.5129. [DOI] [PubMed] [Google Scholar]
- Di Martino R. M. R.; Capasso G.; Camarda M. Spatial domain analysis of carbon dioxide from soils on Vulcano Island: Implications for CO2 output evaluation. Chem. Geol. 2016, 444, 59–70. 10.1016/j.chemgeo.2016.09.037. [DOI] [Google Scholar]
- Fischer T. P.; Lopez T. M. First airborne samples of a volcanic plume for δ13C of CO2 determinations. Geophys. Res. Lett. 2016, 43 (7), 3272–3279. 10.1002/2016GL068499. [DOI] [Google Scholar]
- Rizzo A. L.; Liuzzo M.; Ancellin M. A.; Jost H. J. Real-time measurements of δ13C, CO2 concentration, and CO2/SO2 in volcanic plume gases at Mount Etna, Italy, over 5 consecutive days. Chem. Geol. 2015, 411, 182–191. 10.1016/j.chemgeo.2015.07.007. [DOI] [Google Scholar]
- van Geldern R.; Nowak M. E.; Zimmer M.; Szizybalski A.; Myrttinen A.; Barth J. A.; Jost H. J. Field-based stable isotope analysis of carbon dioxide by mid-infrared laser spectroscopy for carbon capture and storage monitoring. Anal. Chem. 2014, 86 (24), 12191–12198. 10.1021/ac5031732. [DOI] [PubMed] [Google Scholar]
- Goodman K. J.; Brenna J. T. High sensitivity tracer detection using high-precision gas chromatography-combustion isotope ratio mass spectrometry and highly enriched uniformly carbon-13 labeled precursors. Anal. Chem. 1992, 64 (10), 1088–1095. 10.1021/ac00034a004. [DOI] [PubMed] [Google Scholar]
- Reinsch S.; Ambus P. In situ 13CO2 pulse-labeling in a temperate heathland-development of a mobile multi-plot field setup. Rapid Commun. Mass Spectrom. 2013, 27 (13), 1417–1428. 10.1002/rcm.6584. [DOI] [PubMed] [Google Scholar]
- Specification Sheet: Delta Ray – CO2 Isotope Ratio Infrared Spectrometer with URI Connect. https://tools.thermofisher.com/content/sfs/brochures/PS-30309-IR-MS-Delta-Ray-URI-Connect-PS30309-EN.pdf.
- Dannoura M.; Maillard P.; Fresneau C.; Plain C.; Berveiller D.; Gerant D.; Chipeaux C.; Bosc A.; Ngao J.; Damesin C.; Loustau D.; Epron D. In situ assessment of the velocity of carbon transfer by tracing 13C in trunk CO2 efflux after pulse labelling: variations among tree species and seasons. New Phytol. 2011, 190 (1), 181–192. 10.1111/j.1469-8137.2010.03599.x. [DOI] [PubMed] [Google Scholar]
- Plain C.; Gerant D.; Maillard P.; Dannoura M.; Dong Y.; Zeller B.; Priault P.; Parent F.; Epron D. Tracing of recently assimilated carbon in respiration at high temporal resolution in the field with a tuneable diode laser absorption spectrometer after in situ 13CO2 pulse labelling of 20-year-old beech trees. Tree Physiol. 2009, 29 (11), 1433–1445. 10.1093/treephys/tpp072. [DOI] [PubMed] [Google Scholar]
- Bowling D. R.; Sargent S. D.; Tanner B. D.; Ehleringer J. R. Tunable diode laser absorption spectroscopy for stable isotope studies of ecosystem-atmosphere CO2 exchange. Agr. Forest Meteorol. 2003, 118 (1–2), 1–19. 10.1016/S0168-1923(03)00074-1. [DOI] [Google Scholar]
- Meckenstock R. U.; Boll M.; Mouttaki H.; Koelschbach J. S.; Cunha Tarouco P.; Weyrauch P.; Dong X.; Himmelberg A. M. Anaerobic degradation of benzene and polycyclic aromatic hydrocarbons. J. Mol. Microbiol. Biotechnol. 2016, 26 (1–3), 92–118. 10.1159/000441358. [DOI] [PubMed] [Google Scholar]
- Simpson D. R. Biofilm processes in biologically active carbon water purification. Water Res. 2008, 42 (12), 2839–2848. 10.1016/j.watres.2008.02.025. [DOI] [PubMed] [Google Scholar]
- Velten S.; Boller M.; Köster O.; Helbing J.; Weilenmann H.-U.; Hammes F. Development of biomass in a drinking water granular active carbon (GAC) filter. Water Res. 2011, 45 (19), 6347–6354. 10.1016/j.watres.2011.09.017. [DOI] [PubMed] [Google Scholar]
- Velten S.; Hammes F.; Boller M.; Egli T. Rapid and direct estimation of active biomass on granular activated carbon through adenosine tri-phosphate (ATP) determination. Water Res. 2007, 41 (9), 1973–1983. 10.1016/j.watres.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Gibert O.; Lefèvre B.; Fernández M.; Bernat X.; Paraira M.; Calderer M.; Martínez-Lladó X. Characterising biofilm development on granular activated carbon used for drinking water production. Water Res. 2013, 47 (3), 1101–1110. 10.1016/j.watres.2012.11.026. [DOI] [PubMed] [Google Scholar]
- Wen G.; Kötzsch S.; Vital M.; Egli T.; Ma J. BioMig—a method to evaluate the potential release of compounds from and the formation of biofilms on polymeric materials in contact with drinking water. Environ. Sci. Technol. 2015, 49 (19), 11659–11669. 10.1021/acs.est.5b02539. [DOI] [PubMed] [Google Scholar]
- Panneer Selvam B.; Lapierre J.-F.; Guillemette F.; Voigt C.; Lamprecht R. E.; Biasi C.; Christensen T. R.; Martikainen P. J.; Berggren M. Degradation potentials of dissolved organic carbon (DOC) from thawed permafrost peat. Sci. Rep. 2017, 7, 45811. 10.1038/srep45811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J. S. C.; DeRito C. M.; Madsen E. L. Field-based and laboratory stable isotope probing surveys of the identities of both aerobic and anaerobic benzene-metabolizing microorganisms in freshwater sediment. Environ. Microbiol. 2008, 10 (8), 1964–1977. 10.1111/j.1462-2920.2008.01612.x. [DOI] [PubMed] [Google Scholar]
- Conrad R. Quantification of methanogenic pathways using stable carbon isotopic signatures: a review and a proposal. Org. Geochem. 2005, 36 (5), 739–752. 10.1016/j.orggeochem.2004.09.006. [DOI] [Google Scholar]
- Penger J.; Conrad R.; Blaser M. Stable carbon isotope fractionation by methylotrophic methanogenic Archaea. Appl. Environ. Microbiol. 2012, 78 (21), 7596–7602. 10.1128/AEM.01773-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.