

Abstract
The present work reports on the development of photo-cross-linkable gelatins sufficiently versatile to overcome current biopolymer two-photon polymerization (2PP) processing limitations. To this end, both the primary amines as well as the carboxylic acids of gelatin type B were functionalized with photo-cross-linkable moieties (up to 1 mmol/g) resulting in superior and tunable mechanical properties (G′ from 5000 to 147000 Pa) enabling efficient 2PP processing. The materials were characterized in depth prior to and after photoinduced cross-linking using fully functionalized gelatin-methacrylamide (gel-MOD) as a benchmark to assess the effect of functionalization on the protein properties, cross-linking efficiency, and mechanical properties. In addition, preliminary experiments on hydrogel films indicated excellent in vitro biocompatibility (close to 100% viability) both in the presence of MC3T3 preosteoblasts and L929 fibroblasts. Moreover, 2PP processing of the novel derivative was superior in terms of applied laser power (≥40 vs ≥60 mW for gel-MOD at 100 mm/s) as well as post-production swelling (0–20% vs 75–100% for gel-MOD) compared to those of gel-MOD. The reported novel gelatin derivative (gel-MOD-AEMA) proves to be extremely suitable for direct laser writing as both superior mimicry of the applied computer-aided design (CAD) was obtained while maintaining the desired cellular interactivity of the biopolymer. It can be anticipated that the present work will also be applicable to alternative biopolymers mimicking the extracellular environment such as collagen, elastin, and glycosaminoglycans, thereby expanding current material-related processing limitations in the tissue engineering field.
Introduction
Gelatin and its derivatives are of specific interest in the field of biomaterials because they are characterized by high biocompatibility combined with excellent cell-interactive properties due to the presence of Arg-Gly-Asp (RGD) motifs in the gelatin backbone.1−3 In addition, gelatin is derived from collagen, which is a major component of the natural extracellular matrix (ECM), rendering it an ideal ECM mimic.4−7 Furthermore, it is a cost-effective,8,9 food and drug administration (FDA) approved,10 bioresorbable polymer that can be degraded enzymatically.5,6,9,11,12
To date, one of the most commonly applied hydrogel materials for biofabrication and tissue engineering purposes is methacrylamide-modified gelatin (gel-MOD) or gelatin-methacryloyl hydrogels (gel-MA), which can be obtained by functionalization of the primary amines of the (hydroxy)lysine and ornithine side groups present in gelatin with methacrylic anhydride.4,13−22 (Figure 1A) As a result, a photo-cross-linkable derivative is obtained that is suitable for laser-based rapid prototyping techniques including two photon polymerization (2PP).5,6,23−25 Important material limitations remain, however, in terms of mechanical and swelling properties after cross-linking. Consequently, several strategies were proposed to tackle this issue: varying the degree of substitution, grafting of other biomolecules,26 the formation of interpenetrating networks,27 or combining the gelatin with a second (synthetic) material (e.g., polyesters) to increase the overall stiffness of the final construct.28−30
Figure 1.

(A, B) Reaction scheme of the synthesis of gel-MOD (A) and gel-MOD-AEMA (B) with the corresponding equations to calculate the degree of substitution (DS). (C) 1H NMR spectrum of gel-MOD-AEMA with the characteristic methacrylamide signals at 5.75 and 5.51 ppm depicted in red, the methacrylate signals at 6.20 and 5.80 ppm depicted in green, and the reference signal corresponding to the -CH3 groups present in valine, leucine, and isoleucine at 1.01 ppm depicted in black. (D) Influence of gel-MOD concentration on the carboxylic acid DS of gel-MOD-AEMA.
2PP of naturally derived polymers has gained increasing interest for the development of porous constructs with (sub)micrometer-scaled features for tissue engineering purposes.5,6,23,31 Unfortunately, laser-based processing of natural polymers mimicking the ECM is often concomitant with limitations including swelling-related deformations and compromised spatial resolution. As an example, Ovsianikov et al. previously performed 2PP on gelatin-based hydrogels (gel-MOD/gel-MA) at relatively high concentrations (20 wt %) with and without cells to explore the tissue engineering potential of the developed scaffolds.5,6,23 However, the reported structures did not fully match the implemented CAD because of postprocessing aberrations as a consequence of swelling and inferior mechanical properties.5 Because these scaffolds were fabricated from precursor concentrations close to the gelatin solubility limit, no substantial improvement in CAD model reproducibility can be realized by increasing the gelatin concentration.
With the aim of overcoming the limitations described above to improve the two-photon polymerization potential of gelatin hydrogels, we report a novel photo-cross-linkable gelatin. In this respect, the two-photon polymerization potential refers to several aspects of 2PP including the minimally required spatiotemporal energy to obtain reproducible structures. This spatiotemporal energy is defined both by the applied laser power as well as the scan speed of the voxel. Additionally, the two-photon polymerization potential also refers to the mimicry between the applied CAD and the final structure obtained. This feature is determined both by postproduction swelling of the hydrogel during the development process as well as the mechanical properties of the material. Indeed, a higher stiffness results in superior load-bearing capabilities, rendering the support of smaller features possible. Additionally, reproducible structuring at lower concentrations can also be considered as part of the “2PP potential”. As a consequence, we report on a novel photo-cross-linkable gelatin possessing a higher number of cross-linkable functionalities compared to those of the gold standard gel-MOD. We anticipate that, by introducing additional cross-linkable functionalities, the hydrogels of this novel derivative will be characterized by a higher network density after cross-linking and outperform currently reported gelatin derivatives32−35 (e.g., gel-MOD/gel-MA, gel-SH, ...) in 2PP potential.
Materials and Methods
Materials
Gelatin type B (isolated from bovine hides by an alkaline process) was supplied by Rousselot (Ghent, Belgium). Methacrylic anhydride, 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC), and deuterium oxide were purchased at Sigma-Aldrich (Diegem, Belgium) and used as received. Dimethyl sulfoxide (DMSO) (99.85%) and N-hydroxysuccinimide (98%) (NHS) were purchased at Acros (Geel, Belgium). 2-Aminoethyl methacrylate hydrochloride (AEMA·HCl) was obtained from Polysciences (Conches, France), and Irgacure 2959 (1-[4-(2-hydroxyethoxy)-phenyl]-2-hydroxy-2-methyl-1-propane-1-one) was purchased at BASF (Antwerp, Belgium). The dialysis membranes Spectra/Por (MWCO 12.000–14.000 g/mol) were obtained from Polylab (Antwerp, Belgium).
Methods
Methacrylation of the Primary Amines in Gelatin B
The methacrylation of gelatin B, with the aim of obtaining gel-MOD, was performed as described in previous reports.36 Briefly, 100 g of gelatin B (38.5 mmol amines) was dissolved in 1 L of phosphate buffer (pH 7.8) at 40 °C under continuous mechanical stirring followed by the addition of 2.5 equiv of methacrylic anhydride (14.34 mL, 96.25 mmol) and reacted for 1 h. Next, the reaction mixture was diluted with 1L double distilled water (DDW) (ρ = 18.2 MΩ cm) and dialyzed against distilled water (MWCO 12000–14000 g/mol) over 24 h at 40 °C (water changed 5 times) followed by freezing and lyophilization (Christ freeze-dryer alpha I-5).
Methacrylation of the Carboxylic Acids Present in Gel-MOD
Fully functionalized gel-MOD (10 g, 10.980 mmol carboxylic acids) was dissolved in 300 mL of dry DMSO at 50 °C under an inert atmosphere at reflux conditions. After complete dissolution, 1.2 equiv of 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC) (2.525 g; 13.176 mmol) and 1.5 equiv N-hydroxysuccinimide (NHS) (1.895 g; 16.48 mmol) were added together with 50 mL of dry DMSO. After 30 min, 1.5 equiv of 2-aminoethyl methacrylate hydrochloride (AEMA·HCl) was added (4.546 g, 21.96 mmol) together with 0.01 equiv (18 mg, 0.1098 mmol) of 4-tert-butyl catechol (i.e., inhibitor) together with 50 mL of dry DMSO. The solution was shielded from light and stirred overnight at 50 °C followed by precipitation in a 10-fold excess of cold acetone and filtered on a glass filter no. 4. The precipitate was redissolved in DDW and dialyzed using distilled water (MWCO 12000–14000 g/mol) at 40 °C over 24 h (water changed 5 times) followed by freezing and lyophilization. The obtained gelatin derivative will be referred to as gel-MOD-AEMA throughout the rest of the paper.
NMR Spectroscopy
The modification of gelatin was quantified via 1H NMR spectroscopy (Bruker WH 500 MHz) using D2O as solvent at elevated temperature (40 °C). The integrations characteristic for methacrylamide (5.5 ppm (s,1H) and 5.51 ppm (s, 1H)) (gel-MOD) or methacrylates (6.20 ppm (s, 1H) and 5, 80 (s, 1H) (gel-MOD-AEMA) were compared to the integration corresponding with the inert hydrogens of Val, Leu, and Ile at 1.01 ppm (18H) according to the following formula (based on the amino-acid composition).
 |
1 |
Molecular Weight Determination via Gel Permeation Chromatography (GPC)
GPC measurements were performed on a Waters 610 fluid unit and a Waters 600 control unit equipped with a Waters 410 RI detector. Samples were prepared by dissolving approximately 10 mg of material in 1 mL of DMSO. The mobile phase consisted of DMSO in the presence of 0.2 M LiCl. All samples were measured at 40 °C. A five point calibration curve was prepared using Pullulan standards. The obtained results were analyzed using Waters Empower 2 software.
Physical Gelation Study via Differential Scanning Calorimetry
Hydrogel building block solutions (10 w/v %, 40 mg each) in double distilled water were placed into a hermetic Tzero pan (TA Instruments, Zellik, Belgium). As a reference, an empty hermetic Tzero pan was applied. The samples were subjected to a preparatory program as described by Prado et al.37 First, a temperature ramp of 20.00 °C/min was applied to reach a temperature of 60.00 °C. The sample was stabilized for 20 min. Next, a ramp of 10.00 °C/min was applied to cool the sample to a temperature of 15.00 °C followed by stabilizing the samples at 15 °C for 20 min. Then, a temperature ramp of 20.00 °C/min was applied until a temperature of −10.00 °C was reached, which was followed by a final ramp of 5.00 °C/min until a temperature of 60.00 °C was obtained. All measurements were performed on a TA Instruments Q 2000 with an RSC 500 cooler (Zellik, Belgium). The results were analyzed using Q series software. Ten w/v % solutions were applied for most characterization experiments as they enable straightforward sample manipulation where both physical and chemical cross-linking can clearly be observed.
Preparation of Gelatin Films via Film Casting
The obtained gelatin derivatives gel-MOD and gel-MOD-AEMA (0.250–0.750 g) were dissolved in 5 mL of phosphate buffered saline (PBS, pH 7.4) at 40 °C to obtain concentrations of 5, 10, and 15 w/v %. After complete dissolution, 2 mol % (relative to the amount of double bonds) of an 8 mg/mL stock solution of Irgacure 2959 in DDW was added to the mixture followed by degassing for at least 30 s. Next, the heated solution was injected between two parallel glass plates covered with a thin Teflon sheet and separated by a 1 mm thick silicone spacer. Next, the molds were stored in the fridge for 60 min to induce physical gelation. Finally, the hydrogel was irradiated from both sides with UV-A light (365 nm, 2 × 4 mW/cm2) for 30 min. From the obtained films, 3 samples with a diameter of 0.8 cm were punched to determine the gel fraction. The remaining films were incubated in 20 mL of DDW at 37 °C over 48 h to obtain equilibrium swelling.
Gel Fraction and Swelling Determination
The gel fraction was determined by freeze-drying films with a diameter of 0.8 cm immediately after cross-linking. Next, the dry mass of these films was determined (md,1), and the films were incubated in DDW at 37 °C for 24 h. After equilibrium swelling, the films were freeze-dried again, and the dry mass was determined again (md,2). The gel fraction was determined by comparing the final dry mass to the original one such that
| 2 |
All measurements were performed in triplicate, and the standard deviation was calculated.
The equilibrium swelling ratio was determined using circular films with a diameter of 0.8 cm punched from equilibrium swollen sheets. Before determining the swollen mass (ms) of the films, the excess water on the surface was gently removed using tissue paper. Afterward, the samples were freeze-dried to determine the dry mass (md). The swelling ratio was then calculated using the formula
| 3 |
Rheological Monitoring of the Cross-Linking Reaction and Determining the Mechanical Properties of Hydrogel Films
A rheometer-type Physica MCR-301 (Anton Paar, Sint-Martens-Latem, Belgium) with a parallel plate geometry (upper plate diameter of 25 mm) was applied. For monitoring the cross-linking reaction, 300 μL of each solution containing 2 mol % Irgacure 2959 (relative to the amount of cross-linkable functionalities) was placed between the plates using a gap setting of 0.35 mm. The edges were trimmed and sealed using silicone grease (Bayer, Sigma-Aldrich, Diegem, Belgium) to prevent sample drying. An oscillation frequency of 1 Hz and a strain of 0.1% were applied as these values are within the linear viscoelastic range as determined by isothermal measurements (37 °C) of the storage (G′) and loss moduli (G″) as a function of deformation at a constant frequency (1 Hz) and varying strain (0.01–10%). Next, the solutions were either cooled to 5 °C to induce physical gelation, which was monitored during 10 min prior to irradiation or immediately irradiated at 37 °C using UV-A light (10 min, EXFO Novacure 2000 UV light source at 365 nm using a fluence of 500 mW/cm2), followed by 2 min of postcuring monitoring. To assess the effect of the irradiation dose on the final mechanical properties, the same protocol was performed using 215, 360, or 500 mW/cm2 fluence, and the final storage modulus was plotted. The obtained fluence was obtained by entering a UV dose of 1500, 2500, or 3500 mW/cm2 on the light source followed by measuring the actual value at the site of cross-linking using a smart UV intensity meter (Accu-Cal-50, DYMAX).
Rheology on thin films was performed by cutting equilibrium swollen gelatin films (1 mm thick, 48 h in double distilled water at 37 °C) with a diameter of 14 mm and placing them between the spindle (d = 15 mm) and the bottom plate of the rheometer at 37 °C. Next, the spindle was lowered with increments of 25 μm until a normal force of around 0.6–1 N was observed to ensure proper contact between the thin film and both plates. Then, the storage modulus was monitored at 37 °C using an amplitude of 0.1% over a frequency range of 0.01–10 Hz.
Enzymatic Degradation Assay
The in vitro degradation of the hydrogels was studied by freeze-drying thin films (1 mm thick, 0.8 cm diameter) followed by determining their initial dry mass. Next, the samples were incubated in 0.5 mL of Tris–HCl buffer (0.1 M, pH 7.4) in the presence of 0.005% w/v NaN3 and 5 mM CaCl2 at 37 °C. After 1 h, 0.5 mL of collagenase (200 U/ml) dissolved in Tris–HCl buffer was added. At different time points, enzyme degradation was inhibited through the addition of 0.1 mL of EDTA solution (0.25 M) and subsequent cooling of the sample on ice. Next, the hydrogels were washed three times during 10 min with ice-cooled Tris–HCl buffer and three times with DDW; after freeze-drying, the gel fraction of the samples for each time point was determined.
Cell Lines
Mouse fibroblast cells (L929) obtained from Sigma and mouse calvaria-derived preosteoblast cells (MC3T3-E1 Subclone 4) from ATCC-LGC Standards were used for cell viability testing. The L929 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 4500 mg/L of glucose, l-glutamine, and sodium bicarbonate without sodium pyruvate (Sigma). The MC3T3-E1 cells were expanded in alpha minimum essential medium (aMEM) containing ribonucleases, deoxyribonucleases, and 2 mM l-glutamine in the absence of ascorbic acid (Gibco). Both media were supplemented with 10% fetal bovine serum (Sigma) and 1% of 10000 U/mL Penicillin/Streptomycin (Lonza). The cells were cultivated in an incubator in a humid atmosphere at 37 °C containing 5% carbon dioxide. The cell medium was refreshed every other day.
Metabolic Activity Assay
Ten w/v % solutions of the gelatin derivatives were prepared in PBS using 2 mol % of Irgacure 2959. For each sample, an aliquot of 15 μL was pipetted onto a Teflon plate, and a glass coverslip activated with 3-(trimethoxysilyl)propyl methacrylate was pressed on top to evenly cover the glass surface. Afterward, samples were stored at 4 °C for 1 h to induce physical cross-linking followed by 10 min UV-A-induced cross-linking (365 nm, 4 mW/cm2). Next, samples were removed from the Teflon surface, transferred to a 12-well plate, and soaked in medium. To sterilize the coated samples, UV–C irradiation (254 nm, 30 min) was applied prior to storage in the incubator overnight (5% carbon dioxide, 37 °C) in appropriate medium to remove any un-cross-linked material and induce equilibrium swelling in all samples. Next, all medium was aspirated from the samples, and 50 μL of medium containing either 20000 MC3T3-E1 or L929 cells was seeded per well. After 30 min of settling time, 1 mL of appropriate medium was added. During further culturing, the appropriate cell medium was replaced every other day. At specific time points (1, 2, 3, and 7 days), the metabolic activity was assessed using a Presto Blue Cell Viability test (Life technologies). For these tests, Presto Blue, a Resazurin-based reagent, was diluted 1:10 with appropriate medium, and 500 μL of solution was applied per well followed by incubation for 1 h. In the presence of viable cells, resazurin is reduced, thereby becoming highly fluorescent. From each well, 100 μL of solution was transferred to a 96-well plate for fluorescence measurements, and the remaining cell medium was aspirated and replaced by new appropriate medium followed by incubation. The fluorescence was measured with a plate reader (Synergy Bio-Tek, excitation 560 nm, emission 590 nm). After subtraction of sample blank (diluted PrestoBlue incubated for 1 h in appropriated medium), the different substrates were compared to each other and to the “dead cell” control (cells in 50% DMSO and 50% medium for 1 h). The fluorescence value obtained for the cells cultivated on tissue culture plastic (TCP) after 7 days of culture was considered as 100% viability. Next, all fluorescence values were normalized against this control and expressed relative to this 100% viability.
Two-Photon Polymerization of Gelatin Derivatives
Two-photon polymerization experiments were performed on a setup previously reported.23,38 A water immersion objective (C-Achroplan 32×, NA = 0.85, water immersion, Zeiss) was used in combination with a femtosecond pulsed NIR laser with 70 fs pulse duration. The scan speed was set at 100 mm/s for all samples. The CAD design was sliced with a layer spacing of 1 μm and hatched with 0.5 μm line spacing. In every layer, the focal spot was scanned in both the x and y directions for all samples. Average laser powers varying from 10 to 100 mW in 5, 10, and 15 w/v % hydrogel precursor solutions in DMEM medium containing 2 mol % P2CK as a two-photon initiator (relative to the amount of double bonds present) were applied. To prevent sample drying, approximately 50 μL of each solution was placed in a microwell (μ-Dish 35 mm, Ibidi) consisting of two glass plates separated by a silicon spacer with a diameter of 6 mm and a thickness of 1 mm. The bottom plate was silanized with 3-(trimethoxysilyl)propyl methacrylate to enable sufficient attachment.23 After structuring, 2 mL of PBS was added to each sample, and the samples were stored in the incubator at 37 °C for at least 24 h to wash away all un-cross-linked material and induce equilibrium swelling of the microstructures.
Swelling of Microstructures
For each hydrogel building block concentration, an array of ten cubes was structured (each 100 × 100 × 100 μm) at a scan speed of 100 mm/s using average laser powers in the range from 10 up to 100 mW in steps of 10 mW. Laser scanning microscopy (LSM 700, Carl Zeiss) images using the same objective as for structuring were obtained for structure analysis. The surface of the bottom part of the structures where swelling is constrained due to attachment to the glass slide was analyzed using ImageJ software and compared to the surface of the top of the structures, which is not constrained in swelling by the glass slide after 24 h incubation in PBS buffer.
Statistical Analysis
To evaluate the statistical significance of the obtained data, we first performed an F-test on two groups of variables to determine whether their variances are different. Next, a student t test was performed. Two values are considered significantly different when p < 0.05.
Results and Discussion
The aim of the present paper is the development of novel photo-cross-linkable gelatin derivatives resolving swelling-related deformations and compromised spatial resolution as the two most persistent issues in laser-based processing of hydrogel building blocks. The amount of photo-cross-linkable functionalities in gel-MOD is determined by the amount of primary amine functions and is thus limited. Therefore, we anticipated that an increase in photoreactive functionalities using the gelatin carboxylic acids would positively affect the cross-link density of the resulting hydrogels.
Methacrylation of the Gel-MOD Carboxylic Acids: Reaction Conditions Study
Starting from gel-MOD with a degree of substitution (DS) of 97% (0.37 mmol methacrylamides/g of gelatin), we targeted the partial modification of the carboxylic acid functionalities present in the glutamate and aspartate side chains. To this end, carboxylic acid activation was performed via conventional carbodiimide coupling chemistry using ethyl(dimethylaminopropyl) carbodiimide (EDC). N-Hydroxysuccinimide (NHS) was also added to stabilize the activated carboxylic acid groups. Next, a nucleophilic substitution was realized using the primary amine present in 2-aminoethyl methacrylate (AEMA). As a result, both methacrylate as methacrylamide functionalities are introduced to gelatin (Figure 1B). The DS of the obtained derivatives was determined via proton NMR spectroscopy by comparing the integration of the characteristic methacrylate signals at 6.20 and 5.80 ppm with the reference signal for valine, leucine and isoleucine at 1.01 ppm (Figure 1C). The integration of this reference signal corresponds to 18 protons and a total of 0.3836 mol/100 g of gelatin.18 Because the methacrylate signal corresponds to two protons, and a total of 0.1098 mol/100 g of Asp and Glu is present in gelatin type B, the DS can be calculated using the formula depicted in Figure 1B.
Variation of the selected reaction conditions led to gel-MOD-AEMA derivatives with a different carboxylic acid DS. Experiments indicated that rather than increasing the amount of added reagents (data not shown), the gelatin concentration of the reaction mixture provides control over the DS. Indeed, a decreasing amount of methacrylate functions of 56% (0.60 mmol methacrylates/g of gelatin) to 35% (0.38 mmol methacrylates/g of gelatin) was obtained when increasing the gel-MOD concentration from 2.5 to 10 w/v % in the reaction mixture (Figure 1D). This trend is a consequence of the superior gelatin chain mobility in DMSO at lower concentrations (compare the concentration-dependent viscosity).39 As a result, the accessibility of the carboxylic acids is increased, making them more prone to react.39
Comparing the amount of cross-linkable double bonds of gel-MOD to gel-MOD-AEMA, it can be concluded that the proposed gelatin functionalization scheme enables a tripling of the total amount of cross-linkable functionalities (0.99 mmol/g of gel-MOD-AEMA vs 0.37 mmol/g of gel-MOD).
The original aim of the newly developed gel-MOD-AEMA was to obtain higher cross-linking densities to reduce postproduction swelling and improve the mechanical properties of the resulting hydrogels in comparison to gel-MOD. Therefore, all further experiments throughout the paper will be performed using gel-MOD-AEMA with the highest carboxylic acid DS (i.e., 56%).
As the proposed functionalization scheme involves the reaction of gelatin with the primary amine functionalities of AEMA, gel permeation chromatography measurements were performed to reveal possible effects on the molecular weight. The results indicate that the influence of carboxylic acid modification on the molecular weight can be considered moderate in comparison to the hydrolysis occurring during the established primary amine modification procedure to obtain gel-MOD (Table 1).
Table 1. Effect of Functionalization on the Gelatin Molecular Weight As Determined by Gel Permeation Chromatography.
| gelatin type B | gel-MOD | gel-MOD-AEMA | |
|---|---|---|---|
| Mn (Da) | 47900 | 35400 | 32800 |
| Mw (Da) | 97900 | 90600 | 77500 |
| ĐM | 2.04 | 2.56 | 2.36 |
Influence of the Chemical Modification of Gelatin on the Physical and Covalent Cross-Linking Properties
Determination of the Physical Gelation Behavior of Functionalized Gelatins via Differential Scanning Calorimetry
Gelatin is a protein that exhibits upper critical solution temperature (UCST) behavior. The material forms collagen-like triple helices below the UCST resulting in the formation of a physical network. The UCST is influenced by several key factors including the polymer molecular weight, the hydrophilicity/hydrophobicity, and the DS (i.e., the number of incorporated functionalities).40 Visual observations of the herein-developed materials indicated that, in contrast to unmodified gelatin and gel-MOD, solutions of gel-MOD-AEMA at concentrations of 15 w/v % and below remain soluble at room temperature rather than forming a physical gel. The latter observation was further substantiated by differential scanning calorimetry (DSC) experiments following a protocol previously described in the literature.37,39
Similar melting temperatures (∼30 °C) were observed for both gelatin derivatives and pristine gelatin type B (Figure 2). The latter implies that triple helix formation occurs for all evaluated gelatin derivatives.37,41 However, large differences in denaturation enthalpy could be distinguished (Figure 2B). The denaturation enthalpy is proportional to the number of hydrogel bonds associated with triple helix formation.42 Indeed, introduction of methacrylamides into the side chains of gelatin thereby forming gel-MOD resulted in a 7% decrease in intramolecular interactions including hydrogen bonds. The introduction of both methacrylamides and methacrylates in gel-MOD-AEMA resulted in a drastic decrease (70%) in denaturation enthalpy (see Figure 2B). This drastic decrease can potentially be attributed to several factors. First, it is known that variation of the average molecular weight of gelatin alters the physical gelation properties. However, because GPC measurements indicated only moderate hydrolysis, the contribution of the molecular weight to the denaturation enthalpy can be anticipated to be limited. A more important effect can be attributed to the functionalization of the side chains thereby hampering efficient triple helix formation. The observed effect is more pronounced for gel-MOD-AEMA in comparison to that for gel-MOD because more carboxylic acids are present in gelatin in comparison to primary amines. As a consequence, the introduced functionalities will interfere more in triple helix formation resulting in the formation of less extended junction zones (shorter helices). The latter is in accordance with literature reports illustrating that the DS of gelatin can influence its physical gelation properties.32,33,40 For the herein developed gel-MOD-AEMA, the amount of physical cross-links, as revealed by DSC, apparently is insufficient to induce gel-like behavior as observed by the inverted tube method (data not shown). As a consequence, the derivative exhibits liquid-like behavior when solubilized in an aqueous environment at room temperature. To the best of our knowledge, this is the first report describing such behavior for a gelatin derivative without the introduction of additional compounds (e.g., citric acid, ascorbic acid).43,44 As a consequence, the material becomes more versatile for processing via additive manufacturing techniques that require room temperature solubility including digital light processing, widened objective working range (WOW) 2PP, as well as stereolithography.45
Figure 2.

(A) DSC thermograms of 10 w/v % (functionalized) gelatin solutions. (B) Physical gelation temperature and associated physical interactions of 10 w/v % gel-MOD and 10 w/v % gel-MOD-AEMA relative to the denaturation enthalpy of pristine gelatin type B.
Determination of the Mechanical Properties of Hydrogels Based on Functionalized Gelatins via Rheology
The observed differences in physical gelation properties of gel-MOD-AEMA were further studied through rheology experiments with the aim of revealing possible effects on the hydrogel’s mechanical properties. In a first assay, the cross-linking, which occurred via a photoinduced, chain-growth free radical polymerization mechanism, was monitored using rheology. In this respect, the storage modulus G′ was monitored as this provides an indication of the elastic behavior of a sample, which is related to the number of cross-links present in a material.26 During the experiment, a comparison based on the evolution of G′ was made between cross-linking in the presence (Figure 3A) or absence (Figure 3B) of physical interactions. To this end, 10 w/v % precursor solutions were either cooled down to 5 °C to induce physical gelation prior to UV exposure (Figure 3 A) or heated to 37 °C prior to UV-induced cross-linking to preclude the influence of physical interactions on the efficiency of chemical cross-linking (Figure 3B).
Figure 3.

Evolution of the storage modulus of 10 w/v % gel-MOD and 10 w/v % gel-MOD-AEMA during UV-A-induced cross-linking at 500 mW/cm2 with (A) and without (B) 10 min physical gelation at 5 °C as determined by rheology. (C) Influence of applied UV-A irradiance on the final mechanical properties of 10 w/v % gel-MOD-AEMA with prior cooling at 5 °C for 10 min and subsequent cross-linking for 10 min. (D) Effect of gelatin functionalization and concentration on the storage modulus of 1 mm thick equilibrium-swollen hydrogel films prepared via film casting at a UV-A irradiance of 2 × 4 mW/cm2 after 30 min cross-linking. (E, F) Mass swelling ratio and gel fraction of these thin films. All experiments were performed in the presence of 2 mol % Irgacure 2959.
A clear difference in mechanical properties due to physical gelation can be observed between 10 w/v % solutions of gel-MOD and gel-MOD-AEMA. Gel-MOD clearly outperforms gel-MOD-AEMA in terms of physical gelation (i.e., 2000 Pa for gel-MOD vs 1000 Pa for gel-MOD-AEMA) within the observed time frame, which is in good agreement with the DSC results discussed earlier. However, the presence of these physical interactions does significantly and positively affect the final stiffness after covalent cross-linking for both derivatives. When inducing triple helices by lowering the temperature below the UCST prior to UV irradiation, the gelatin chains will organize. On the one hand, this brings the cross-linkable functionalities in closer proximity to each other, leading to more efficient cross-linking.46,47 This hypothesis is substantiated by the literature as similar observations were reported by Houben et al. for synthetic cross-linkable hydrogel building blocks, where self-organization due to crystallization increased the observed cross-linking reactivity.48 On the other hand, the formed triple helices are partially locked by the covalent cross-links thereby further increasing the final mechanical properties46,47 (see Figure S3). As a consequence, UV-A irradiation of a physical network results in a more efficient cross-linking reaction, which is reflected by a substantially higher storage modulus obtained after cross-linking for both gel-MOD (i.e., 18 ± 1.3 vs 4.7 ± 0.3 kPa) and gel-MOD-AEMA (i.e., 60.6 ± 0.6 vs 14.9 ± 0.2 kPa) (comparison of Figure 3A and B).
Furthermore, the introduction of additional cross-linkable functionalities also positively affects the kinetics of the photoinduced cross-linking as indicated by the steeper slope of the G′ curve during cross-linking for gel-MOD-AEMA in Figure 3A and B. Additionally, the increased cross-link-density results in a higher stiffness of the cross-linked hydrogels as evidenced by a 3.0–3.6-fold increase in the final storage modulus for gel-MOD-AEMA vs gel-MOD both in the presence (i.e., from 18 ± 1.3 to 60.6 ± 0.6 kPa) and absence (i.e., from 4.7 ± 0.3 to 14.9 ± 0.2 kPa) of physical interactions prior to cross-linking (comparison of Figure 3A and B).
Furthermore, variation of the applied light intensity provides control over the final mechanical properties (see Figure 3C). Lower light intensities result in a less densely cross-linked network and concomitant lower storage modulus.21 This phenomenon can be considered very relevant when applying 2PP for material processing because the technique is characterized by high spatiotemporal control in terms of the locally applied irradiation dose. As a consequence, the material could be applied to obtain structures with locally tuned stiffness, thereby mimicking natural tissue to a greater extent.
Because these in situ cross-linking experiments only provide insight into the mechanical properties of the hydrogels in the relaxed state prior to equilibrium swelling, cross-linked hydrogel films were also monitored after equilibrium swelling. Therefore, hydrogel films were prepared from different concentrations of both gelatin derivatives. Next, these films were equilibrium swollen at physiological temperature (37 °C), and the storage modulus was determined under the same conditions over a frequency range of 0.01–10 Hz. The average storage moduli and associated standard deviations are presented in Figure 3D. First, the experiment indicated frequency-independent G′ and G″, which indicates the existence of a cross-linked rubbery network49 (data not shown). Second, next to variations in applied UV irradiation dose and the gelatin DS, the initial precursor concentration is another parameter that provides control over the final hydrogel mechanical properties.21,50 The results indicate that gel-MOD-AEMA outperforms gel-MOD in equilibrium swollen conditions over the entire concentration range in terms of stiffness (Figure 3D). As a consequence, a higher amount of cross-linkable functionalities in the hydrogel precursor allows gel-MOD-AEMA to reach the same mechanical properties as gel-MOD albeit at lower concentrations. This is very relevant as previous findings from our group indicated that high gelatin concentrations negatively affect the biocompatibility.21 It should be noted that similar gel fractions (close to 100%, no significant differences p > 0.05) were obtained for all studied hydrogel films. As a consequence, stable hydrogel films were formed for both derivatives, and observed differences in stiffness cannot be attributed to incomplete cross-linking (Figure 3F).
In conclusion, when looking into potential tissue engineering applications, literature reports state that the obtained mechanical properties match the mechanical properties of a series of tissues (see Figure S1 and Table S1). These tissues range from soft tissue including brain tissue (G′ = 3–12 kPa)51 to relatively hard tissues including the intervertebral discs (G′ = 8 – 93 kPa).52 Furthermore, a comparison between the obtained mechanical properties and those reported earlier for biomaterials indicates that gel-MOD-AEMA scores more toward the higher end of the mechanical spectrum. In this respect, it outperforms all reported gelatin derivatives to date that have been cross-linked in the absence of a second material (e.g., chondroitin sulfate, hyaluronic acid)42 (see Figure S1 and Table S1). Additionally, the obtained mechanical properties are comparable with those described earlier for cross-linked collagen despite the less pronounced physical interactions inherent to gelatin.53,54
The combination of the observed faster cross-linking kinetics with the superior mechanical properties of gel-MOD-AEMA are anticipated to be beneficial for lithography-based additive manufacturing purposes, as this can lead to shorter structuring times.
Effect of Gelatin Functionalization and Concentration on the Hydrogel Gel Fraction, Water Uptake Capacity, and Network Density
Hydrogel materials are generally excellent candidates for tissue
culture because they mimic the aqueous environment present in the
extracellular matrix while providing mechanical support to the cells.
Therefore, the equilibrium swelling degree of a hydrogel material
is an important characteristic for ECM mimics. To this end, swelling
at equilibrium was determined gravimetrically for both derivatives
at varying precursor concentrations (Figure 3E). The assay indicated that, while still
being able to absorb large quantities of water (≥350%), the
gel-MOD-AEMA derivative exhibits a significant reduction in equilibrium
swelling compared to that of gel-MOD, which can again be attributed
to a more densely cross-linked network. To further substantiate these
observations, a more thorough comparison of the obtained network density
can be calculated via the rubber elasticity theory using the average
molecular weight, the equilibrium swelling ratio, and the mechanical
properties.21,55,56 This theory allows for calculating an estimation of several important
parameters including the polymer volume fraction in the swollen state
(v2,s), the volumetric swelling ratio
(Q), the average molecular weight between cross-links
(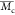 ), the network mesh size (ξ), and
the cross-link density (ρx) (detailed
calculations are available in the Supporting Information). A summary of the experimentally obtained results based on GPC,
rheology, and gravimetric swelling assays is presented in Table 2 along with the calculated
results obtained using the rubber elasticity theory.
), the network mesh size (ξ), and
the cross-link density (ρx) (detailed
calculations are available in the Supporting Information). A summary of the experimentally obtained results based on GPC,
rheology, and gravimetric swelling assays is presented in Table 2 along with the calculated
results obtained using the rubber elasticity theory.
Table 2. Influence of Concentration and Gelatin Derivative on Gel Fraction, Mass Swelling Ratio, Q, G′, Mc, ξ, and ρx.
| sample | # cross-linkable functionalities (mmol/ggelatin) | initial concn (% w/v) | gel fraction | mass swelling ratio | G′ at 37 °C (kPa) | Q | Mc (g/mol) | ξ (Å) | ρx (× 10–4) (mol/cm3) |
|---|---|---|---|---|---|---|---|---|---|
| gel-MOD | 0.37 | 5 | 83.0 ± 6.5 | 18.5 ± 1.0 | 5.7 ± 0.3 | 26.21 | 5326 | 160 | 2.55 |
| 10 | 94.5 ± 3.0 | 10.1 ± 0.2 | 22.1 ± 0.3 | 14.68 | 3752 | 110 | 3.63 | ||
| 15 | 97.7 ± 2.5 | 8.9 ± 0.1 | 47.5 ± 0.8 | 13.11 | 2892 | 93 | 4.70 | ||
| gel-MOD-AEMA | 0.99 | 5 | 91.0 ± 4.9 | 10.4 ± 2.2 | 7.7 ± 0.4 | 15.11 | 4783 | 126 | 2.84 |
| 10 | 95.1 ± 1.2 | 6.4 ± 0.2 | 56.2 ± 1.2 | 9.76 | 1899 | 69 | 7.16 | ||
| 15 | 93.7 ± 2.9 | 4.7 ± 0.1 | 147.1 ± 2.9 | 7.37 | 1248 | 51 | 10.90 |
The results clearly indicate a correlation between the initial gelatin concentration, the amount of cross-linkable functionalities, and the density of the obtained network. This is reflected by a decreased average molecular weight between cross-links and increased cross-link density both upon increasing the precursor concentration and increasing the amount of cross-linkable functionalities. As a result, the observations from swelling assays as well as rheological measurements can be justified to be attributed to network density. Furthermore, it should be noted that rheology and swelling experiments were performed above the UCST of the gelatin derivatives (27–30 °C as determined by DSC) (Figure 2). Consequently, the obtained cross-link densities can only be attributed to the presence of covalent cross-links without the influence of interfering physical interactions. However, as previously discussed, physical interactions prior to covalent cross-linking did result in an increased final gel strength and, therefore, a more densely cross-linked network.
Effect of Gelatin Functionalization and Concentration on Enzymatic Degradation
To assess to what extent the biodegradable properties of gelatin were preserved upon derivatization and subsequent cross-linking, in vitro degradation experiments have been performed in the presence of collagenase (100 CDU/ml). The results indicated that gel-MOD-AEMA remains fully enzymatically degradable as previously reported for other cross-linked gelatin derivatives.57 However, the presence of additional cross-links (i.e., 25.5 and 36.3 mmol/cm3 for 5 and 10 w/v % gel-MOD and 28.4 and 71.6 mmol/cm3 for gel-MOD-AEMA, respectively) in combination with decreased water uptake capacity results in a reduced intercross-link chain mobility. This effect combined with the fact that more bonds need to be cleaved for denser cross-linked networks results in a longer degradation time. Indeed, the degradation time for gel-MOD-AEMA is substantially larger (up to a factor of 7.5 for both 5 and 10 w/v % hydrogels) (Figure 4). It should be noted that for gel-MOD-AEMA only the extrapolated final degradation times are presented. However, the materials were fully degradable because after somewhat less than 30 h, no material was left after washing and freeze-drying for all studied samples.
Figure 4.

In vitro degradation behavior of gel-MOD (gray) and gel-MOD-AEMA in the presence of 100 CDU/ml collagenase starting from different polymer concentrations (5 versus 10 w/v %). The extrapolated final degradation times are given between brackets.
Influence of Gelatin Functionalization on Biocompatibility
Despite the favorable material properties, the developed derivative has to retain its favorable cell interactivity to remain suitable for tissue engineering purposes. Therefore, in vitro biological tests were performed on hydrogel-coated glass slides using both MC3T3-E1 preosteoblasts and L929 fibroblasts. To this end, the metabolic activity of the cells was monitored over the course of 7 days at regular time points using a PrestoBlue assay. The results of the assays are depicted in Figure 5. To ensure a more quantitative comparison, all samples were normalized against the TCP control after 7 days of culture (= 100%). Because a clear increase in metabolic activity is observed as a function of time, the cells can be considered healthy and proliferating on all substrates throughout the course of the experiment. It should be noted, however, that for the MC3T3 cells, the increase in metabolic activity between days 3 and 7 is less pronounced. This is a phenomenon also observed in the literature for this cell type as typically the metabolic activity reaches a plateau corresponding to the confluence state.58 In the performed assay, confluence was indeed reached between days 3 and 7 resulting in a plateau in metabolic activity. Furthermore, no significant difference in metabolic activity can be observed between gel-MOD-AEMA and gel-MOD, which is currently one of the gold standards in the field of biofabrication.34,59 In addition, for the MC3T3 cells, all substrates exhibited a metabolic activity of >70% after 7 days of culture, and the metabolic activity for the L929 cells was even above 90% for all substrates. As a consequence, both materials can be considered biocompatible and suitable for tissue culture of both cell types.
Figure 5.

Presto blue assay performed on hydrogel-coated glass slides expressing the metabolic activity of MC3T3-E1 preosteoblasts (left panel) and L929 fibroblasts (right panel) relative to a tissue culture plastic (TCP) reference and DMSO as a negative control.
Influence of Gelatin Functionalization and Concentration on Processing through Two Photon Polymerization
To prove the suitability of the material for laser-based additive manufacturing purposes, 2PP experiments were performed comparing gel-MOD and gel-MOD-AEMA solutions.
By scanning a tightly focused femtosecond pulsed near-infrared (800 nm) laser beam through the solutions in the presence of a suitable photoinitiator, local polymerization can occur in the focal spot (voxel) as a result of simultaneous absorption of two photons by the photoinitiator (Figure 6A). As a result, the photoinitiator will generate radicals thereby locally inducing a free radical-polymerization cross-linking reaction (see Figure 6A). Consequently, a complex 3D hydrogel construct can be fabricated by scanning the focal spot through the precursor solution according to a CAD model followed by dissolution and washing away of un-cross-linked material. To the best of our knowledge, to date, 2PP fabrication using gelatin-based solutions have only been reported for concentrations starting from 20 wt % functionalized gelatin or when using an additional cross-linker.5,6,23−25 Because of the higher reactivity and superior mechanical properties, the reported gel-MOD-AEMA precursors are anticipated to be a superior alternative for gel-MOD from a processing perspective. For proving this hypothesis, the swelling properties and the CAD mimicry of conventionally applied gel-MOD and the novel gel-MOD-AEMA derivative have been compared using different polymer concentrations (5–15 w/v %) and various average laser powers (10–100 mW) in solutions containing 2 mol % P2CK, a biocompatible and efficient 2PP photoinitiator23,31,38 (Figure 6 B and C).
Figure 6.

(A) Schematic representation of the two-photon polymerization (2PP) principle on gel-MOD-AEMA in the presence of P2CK photoinitiator including a Jablonski diagram demonstrating the theoretical background for which single photon excitation (blue) is compared to two photon excitation (red). (B) Applied CAD model and structured university logos expressing clear differences in swelling and swelling-related deformations between gel-MOD and gel-MOD-AEMA at different concentrations. (TU Wien logo printed with permission from the TU Wien; PBM logo printed with permission from the Polymer Chemistry and Biomaterials research group at Ghent University.) (C) Semiquantitative analysis of structuring range and related swelling (observed as a “halo” around the square) for both derivatives via 3D renderings of the generated cubes (100 μm × 100 μm × 100 μm) imaged through the glass slide after 24 h of incubation at 37 °C. (All experiments were performed in the presence of 2 mol % P2CK.)
The degree of volumetric swelling obtained after 2PP was assessed. This not only provides insight into the cross-link density of a material, it is also very relevant when targeting additive manufacturing and more specifically, 2PP. Postproduction swelling generally requires a correction in the CAD model to realize reproducible computer-aided manufacturing (CAM) based on the implemented design.60 Furthermore, swelling is also correlated with the applied irradiation dose (see Figure 6C). Unfortunately, swelling is often not uniform or even design dependent. As a result, it becomes challenging to anticipate and correct for postproduction swelling-related morphological changes when generating the CAD design. Inhomogeneous swelling can induce local stress areas and result in distortions of the construct architecture as depicted for gel-MOD in Figure 6B.25 Therefore, the correlation between swelling, irradiation dose, and precursor concentration was qualitatively assessed by imaging cubes polymerized on methacrylated glass via 2PP. The bottom (100 × 100 μm) of the hydrogel structures is covalently attached to the methacrylated glass, thereby preventing swelling and mimicking the dimensions of the CAD model. The top part of the structure, however, is free to swell during development of the sample. Consequently, a “halo”-like contour is present behind the bottom surface (Figure 6 C) when imaged through the glass slide. These qualitative observations were quantified by comparing the surface area of the bottom slice attached to the glass with the equilibrium swollen top section of the cube (Figure 7). Clearly, the concentration and applied average laser power affect the swelling of gel-MOD structures, especially at lower polymer concentrations. However, for gel-MOD-AEMA, the effect, although still present, is less pronounced because swelling is nearly absent for average laser powers of 80 mW onward both for 10 and 15 w/v % solutions. As a consequence, the “true“ CAD/CAM shape fidelity of gel-MOD-AEMA hydrogels is demonstrated (Figures 6C and 7). This significantly reduced swelling behavior is a consequence of a decrease in the average molecular weight between the cross-linking points. As a result, the mobility of the polymer chains within the network is lowered, and the water uptake capacity is decreased, as discussed earlier.21,61 A second observation was the fact that no reproducible structures could be obtained starting from 5 w/v % solutions of gel-MOD or gel-MOD-AEMA. To obtain more insight in this matter, the amount of cross-linkable functionalities present in one voxel was estimated for both derivatives at the applied precursor concentrations. An estimation of the two-photon excitation (TPE) volume of one voxel was calculated by approximating the illumination point spread function2 (IPSF2) (see Figure 7B) as a three-dimensional Gaussian analytical integration.62
| 4 |
The calculation of ωxy and ωz can be found in the Supporting Information. For the amount of double bonds present in the voxel to be calculated, the concentration of the applied gelatin solution was combined with the calculated amount of double bonds present in the material to obtain the number of double bonds/volume (see Figure 7C).
Figure 7.
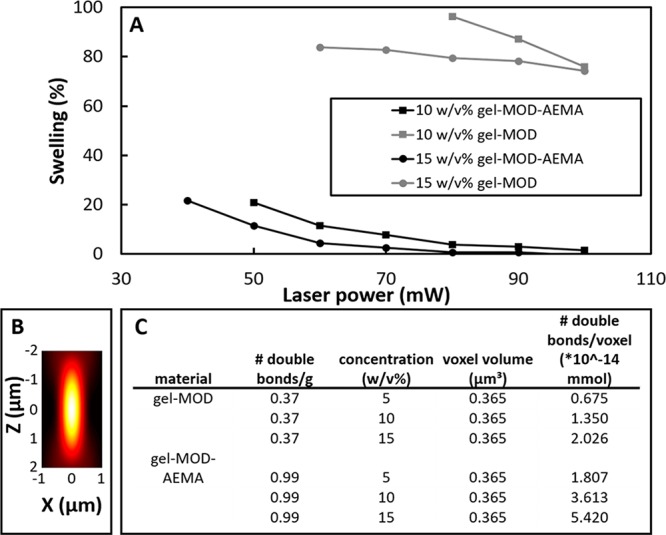
(A) Semiquantitative swelling analysis performed on printed cubes (100 × 100 × 100 μm) by comparison of the surface area of the bottom slice attached to the glass and the top slice of the printed cube after equilibrium swelling. The structuring range of both derivatives as well as the influence of the applied average laser power on the swelling is demonstrated. (B) Exemplary schematic representation of the point spread function as a consequence of tight focusing exhibiting an ellipsoidal morphology.63 (C) Table estimating number of double bonds within the same volume voxel of different materials.
It should be noted that, although more cross-linkable functionalities are present in 5 w/v % solutions of gel-MOD-AEMA as compared to 10 w/v % gel-MOD, no reproducible structures could be obtained starting from 5 w/v % gel-MOD-AEMA although the polymerization could be monitored during structuring. However, the poor mechanical properties of the material starting from a 5 w/v % concentration render it insufficiently strong to support its own weight during structuring and lead to a loss of part of the structure during the development. As a consequence, only parts of the structure remained after development (see Figure S4). Furthermore, the combination of more cross-linkable functionalities per voxel (see Figure 7B) combined with more favorable cross-linking kinetics for gel-MOD-AEMA results in the formation of denser networks even at low concentrations (as discussed earlier). Therefore, lower irradiation doses enable the generation of similar mechanical properties compared to those of gel-MOD even at higher concentrations and average laser powers (e.g., 10 w/v % gel-MOD-AEMA exhibits a similar stiffness compared to that of 15 w/v % gel-MOD, Figure 3D). Consequently, it is anticipated that higher writing speeds can be applied to gel-MOD-AEMA to obtain similar mechanical properties in combination with a lower swelling degree relative to that of gel-MOD.
To visualize the true CAD/CAM fidelity for the novel gel-MOD-AEMA precursors, more complex structures were also constructed starting from 10 and 15 w/v % concentrations (see Figure 6B). Furthermore, it is demonstrated that even fine features (down to 1 μm with a high aspect ratio of 1:40 as evidenced by the small features apparent in the PBM logo) were closely reproduced using the novel gelatin derivative (Figure 6 B). Consequently, subcellular dimensions (≤10 – 20 μm)64 could be realized with high aspect ratios using gel-MOD-AEMA as starting material. It is therefore anticipated that the combination of gel-MOD-AEMA and 2PP will become a powerful tool in the study of cellular responses toward ultrasmall biocompatible hydrogel structures, thereby influencing cellular behavior, and to guide cells into a desired morphology or pathway.65
Furthermore, because in contrast to gel-MOD gel-MOD-AEMA does not form a physical gel at room temperature (as discussed above), it becomes possible to dip the microscope objective used for 2PP inside the solution. As a consequence, dip-in laser lithography as well as WOW-2PP is in reach on gelatin derivatives at room temperature, thereby drastically increasing the maximum attainable construct size from the μm to the mm scale.45,66
Conclusions
The combination of primary amine functionalization with subsequent carboxylic acid modification to introduce cross-linkable moieties proves to be an elegant tool to increase the 2PP potential of gelatin-based hydrogel precursors. In addition to superior material stiffness, gel-MOD-AEMA also exhibits faster cross-linking kinetics compared to those of conventionally applied photo-cross-linkable gelatin derivatives. Furthermore, nearly no postprocessing swelling occurred upon applying gel-MOD-AEMA while the material biocompatibility with respect to tissue culture potential was sufficiently maintained. These factors render the material ideal for 2PP processing at high-resolution (feature sizes of around 1 μm) and increase the additive manufacturing potential of gelatin precursors in general for which faster cross-linking kinetics, lower swelling ratios, and superior mechanical integrity can increase the maximum attainable writing speeds in combination with a higher shape fidelity for the applied CAD without compromising biocompatibility. Furthermore, the absence of visually observable physical gelation at room temperature increases the potential of gel-MOD-AEMA for layer-by-layer and dip-in laser lithography techniques, which drastically increases the maximum attainable construct sizes. This aspect clears the road toward the production of patient-specific macrostructures up to several millimeters in size containing ultraprecise microfeatures to optimize the desired cellular behavior. Furthermore, the applied polymer functionalization can be translated toward other (bio)polymers containing free carboxylic acids including collagen, elastin, and glycosaminoglycans. In this respect, the ECM-mimicking toolbox can be further expanded toward a new platform consisting of highly specific processable material enabling close reproductions of living tissue.
Acknowledgments
The authors acknowledge Tim Courtin of the NMR department for helping with the NMR measurements. Furthermore, Veerle Boterberg of PBM is acknowledged for her continouous efforts in keeping the lab and all its devices up and running. The authors acknowledge the financial support of Ghent University, the UGent Multidisciplinary Research Partnership Nanoand Biophotonics, the Vrije Universiteit Brussel, Flanders Make, and Methusalem. S.V.V would like to acknowledge the Research Foundation Flanders (FWO, Belgium) for several research grants. (FWOKN273, G00516N, G0F0516N, FWOAL843). J.V.H holds an SB PhD grant from the research foundation flanders (FWO). A.O. acknowledges the financial support of the European Research Council (Starting Grant-307701, A.O.) Furthermore, the FWF-FWO grant (Austrian Science Fund project #I2444N28) is acknowledged for financial support.
Glossary
ABBREVIATIONS
- 2PP
two photon polymerization
- α′
expansion factor
- αMEM
alpha minimum essential medium
- ξ
average mesh size at equilibrium swelling
- ν̅
specific volume of gelatin
- ρx
cross-link density
- ρgelatin
gelatin density
- AEMA
2-aminoethyl methacrylate
- c
concentration
- CAD
computer-aided design
- CAM
computer-aided manufacturing
- CDU
collagenase digestion units
- ĐM
polydispersity
- DDW
double distilled water
- DS
degree of substitution
- DSC
differential scanning calorimetry
- DMSO
dimethyl sulfoxide
- DMEM
Dulbecco’s modified Eagle medium
- ECM
extracellular matrix
- EDC
1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide
- FDA
Food and Drug Administration
- G
shear modulus
- G′
storage modulus
- G″
loss modulus
- gel-MOD
methacrylamide modified gelatin
- gel-MOD-AEMA
methacrylamide/methacrylate modified gelatin
- GPC
gel permeation chromatography
average molecular weight between cross-links
- Mn
average numerical molecular weight
- Mr
average molecular weight of 1 repeating unit
- Mw
mass average molecular weight
- NHS
N-hydroxysuccinimide
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- q
mass swelling ratio
- Q
volumetric swelling ratio
- R
universal gas constant
- RGD
(Arg-Gly-Asp; arginine-glycine-aspartate)
- T
temperature
- TCP
tissue culture plastic
- UCST
upper critical solution temperature
- UV-A
ultraviolet (315 < λ < 400 nm)
- UV-C
ultraviolet (100 < λ < 280 nm)
- v2,s
polymer volume fraction in the swollen state
- Vg
hydrogel volume at equilibrium swelling
- Vp
polymer volume at equilibrium swelling
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biomac.7b00905.
GPC data, a detailed mechanical comparison of the reported derivatives to different types of tissue and previously reported biomaterials, detailed background and calculations concerning the rubber elasticity theory, detailed formulas applied for calculation of the voxel volume, and structures obtained via 2PP in low concentration gel-MOD-AEMA solutions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Vishnoi T.; Kumar A. J. Mater. Sci.: Mater. Med. 2013, 24 (2), 447–459. 10.1007/s10856-012-4795-z. [DOI] [PubMed] [Google Scholar]
- Chang K.-H.; Liao H.-T.; Chen J.-P. Acta Biomater. 2013, 9 (11), 9012–9026. 10.1016/j.actbio.2013.06.046. [DOI] [PubMed] [Google Scholar]
- Djagny V. B.; Wang Z.; Xu S. Crit. Rev. Food Sci. Nutr. 2001, 41 (6), 481–492. 10.1080/20014091091904. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Samal S. K.; Dubruel P. Macromol. Symp. 2011, 309–310 (1), 173–181. 10.1002/masy.201100030. [DOI] [Google Scholar]
- Ovsianikov A.; Deiwick A.; Van Vlierberghe S.; Dubruel P.; Moller L.; Dräger G.; Chichkov B. Biomacromolecules 2011, 12 (4), 851–858. 10.1021/bm1015305. [DOI] [PubMed] [Google Scholar]
- Ovsianikov A.; Deiwick A.; Van Vlierberghe S.; Pflaum M.; Wilhelmi M.; Dubruel P.; Chichkov B. Materials 2011, 4 (12), 288–299. 10.3390/ma4010288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues S. C.; Salgado C. L.; Sahu A.; Garcia M. P.; Fernandes M. H.; Monteiro F. J. J. Biomed. Mater. Res., Part A 2013, 101 (4), 1080–1094. 10.1002/jbm.a.34394. [DOI] [PubMed] [Google Scholar]
- Dainiak M. B.; Allan I. U.; Savina I. N.; Cornelio L.; James E. S.; James S. L.; Mikhalovsky S. V.; Jungvid H.; Galaev I. Y. Biomaterials 2010, 31 (1), 67–76. 10.1016/j.biomaterials.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Elvin C. M.; Vuocolo T.; Brownlee A. G.; Sando L.; Huson M. G.; Liyou N. E.; Stockwell P. R.; Lyons R. E.; Kim M.; Edwards G. a.; Johnson G.; McFarland G. a.; Ramshaw J. a M.; Werkmeister J. a. Biomaterials 2010, 31 (32), 8323–8331. 10.1016/j.biomaterials.2010.07.032. [DOI] [PubMed] [Google Scholar]
- Chen Y.-C.; Su W.-Y.; Yang S.-H.; Gefen A.; Lin F.-H. Acta Biomater. 2013, 9 (2), 5181–5193. 10.1016/j.actbio.2012.09.039. [DOI] [PubMed] [Google Scholar]
- Skoog S. a; Goering P. L.; Narayan R. J. J. Mater. Sci.: Mater. Med. 2014, 25 (3), 845–856. 10.1007/s10856-013-5107-y. [DOI] [PubMed] [Google Scholar]
- Hölzl K.; Lin S.; Tytgat L.; Van Vlierberghe S.; Gu L.; Ovsianikov A. Biofabrication 2016, 8 (3), 32002. 10.1088/1758-5090/8/3/032002. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Dubruel P.; Schacht E. H. Biomacromolecules 2011, 12 (5), 1387–1408. 10.1021/bm200083n. [DOI] [PubMed] [Google Scholar]
- Dubruel P.; Unger R.; Van Vlierberghe S.; Cnudde V.; Jacobs P. J. S.; Schacht E.; Kirkpatrick C. J. Biomacromolecules 2007, 8 (2), 338–344. 10.1021/bm0606869. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Cnudde V.; Dubruel P.; Masschaele B.; Cosijns A.; De Paepe I. De; Jacobs P. J. S.; Van Hoorebeke L. Van; Remon J. P.; Schacht E. Biomacromolecules 2007, 8 (2), 331–337. 10.1021/bm060684o. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Dubruel P.; Lippens E.; Cornelissen M.; Schacht E. J. Biomater. Sci., Polym. Ed. 2009, 20 (10), 1417–1438. 10.1163/092050609X12457418905508. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Dubruel P.; Lippens E.; Masschaele B.; Van Hoorebeke L.; Cornelissen M.; Unger R.; Kirkpatrick C. J.; Schacht E. J. Mater. Sci.: Mater. Med. 2008, 19 (4), 1459–1466. 10.1007/s10856-008-3375-8. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Fritzinger B.; Martins J. C.; Dubruel P. Appl. Spectrosc. 2010, 64 (10), 1176–1180. 10.1366/000370210792973550. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Sirova M.; Rossmann P.; Thielecke H.; Boterberg V.; Rihova B.; Schacht E.; Dubruel P. Biomacromolecules 2010, 11 (10), 2731–2739. 10.1021/bm100783h. [DOI] [PubMed] [Google Scholar]
- Hoch E.; Hirth T.; Tovar G. E. M.; Borchers K. J. Mater. Chem. B 2013, 1 (41), 5675–5685. 10.1039/c3tb20745e. [DOI] [PubMed] [Google Scholar]
- Billiet T.; Van Gasse B.; Gevaert E.; Cornelissen M.; Martins J. C.; Dubruel P. Macromol. Biosci. 2013, 13 (11), 1531–1545. 10.1002/mabi.201300143. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Dubruel P.; Schacht E. J. Bioact. Compat. Polym. 2010, 25 (5), 498–512. 10.1177/0883911510377254. [DOI] [Google Scholar]
- Ovsianikov A.; Mühleder S.; Torgersen J.; Li Z.; Qin X.-H.; Van Vlierberghe S.; Dubruel P.; Holnthoner W.; Redl H.; Liska R.; Stampfl J. Langmuir 2014, 30 (13), 3787–3794. 10.1021/la402346z. [DOI] [PubMed] [Google Scholar]
- Engelhardt S.; Hoch E.; Borchers K.; Meyer W.; Krüger H.; Tovar G. E. M.; Gillner A. Biofabrication 2011, 3 (2), 25003. 10.1088/1758-5082/3/2/025003. [DOI] [PubMed] [Google Scholar]
- Brigo L.; Urciuolo A.; Giulitti S.; Della Giustina G.; Tromayer M.; Liska R.; Elvassore N.; Brusatin G. Acta Biomater. 2017, 55, 373–384. 10.1016/j.actbio.2017.03.036. [DOI] [PubMed] [Google Scholar]
- Graulus G.-J.; Mignon A.; Van Vlierberghe S.; Declercq H.; Fehér K.; Cornelissen M.; Martins J. C.; Dubruel P. Eur. Polym. J. 2015, 72, 494. 10.1016/j.eurpolymj.2015.06.033. [DOI] [Google Scholar]
- Daniele M. A.; Adams A. A.; Naciri J.; North S. H.; Ligler F. S. Biomaterials 2014, 35 (6), 1845–1856. 10.1016/j.biomaterials.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Visser J.; Melchels F. P. W.; Jeon J. E.; van Bussel E. M.; Kimpton L. S.; Byrne H. M.; Dhert W. J. a; Dalton P. D.; Hutmacher D. W.; Malda J. Nat. Commun. 2015, 6, 6933. 10.1038/ncomms7933. [DOI] [PubMed] [Google Scholar]
- Markovic M.; Van Hoorick J.; Hölzl K.; Tromayer M.; Gruber P.; Nürnberger S.; Dubruel P.; Van Vlierberghe S.; Liska R.; Ovsianikov A. J. Nanotechnol. Eng. Med. 2015, 6 (2), 21004. 10.1115/1.4031466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rie J.; Declercq H.; Van Hoorick J.; Dierick M.; Van Hoorebeke L.; Cornelissen R.; Thienpont H.; Dubruel P.; Van Vlierberghe S. J. Mater. Sci.: Mater. Med. 2015, 26 (3), 5465. 10.1007/s10856-015-5465-8. [DOI] [PubMed] [Google Scholar]
- Ciuciu A. I.; Cywiński P. J. RSC Adv. 2014, 4 (85), 45504–45516. 10.1039/C4RA06892K. [DOI] [Google Scholar]
- Van Hoorick J.; Declercq H.; De Muynck A.; Houben A.; Van Hoorebeke L.; Cornelissen R.; Van Erps J.; Thienpont H.; Dubruel P.; Van Vlierberghe S. J. Mater. Sci.: Mater. Med. 2015, 26 (10), 247. 10.1007/s10856-015-5566-4. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Schacht E.; Dubruel P. Eur. Polym. J. 2011, 47 (5), 1039–1047. 10.1016/j.eurpolymj.2011.02.015. [DOI] [Google Scholar]
- Klotz B. J.; Gawlitta D.; Rosenberg A. J. W. P.; Malda J.; Melchels F. P. W. Trends Biotechnol. 2016, 34 (5), 394–407. 10.1016/j.tibtech.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue K.; Trujillo-de Santiago G.; Alvarez M. M.; Tamayol A.; Annabi N.; Khademhosseini A. Biomaterials 2015, 73, 254–271. 10.1016/j.biomaterials.2015.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Cnudde V.; Dubruel P.; Masschaele B.; Cosijns A.; De Paepe I.; Jacobs P. J. S.; Van Hoorebeke L.; Remon J. P.; Schacht E. Biomacromolecules 2007, 8, 331–337. 10.1021/bm060684o. [DOI] [PubMed] [Google Scholar]
- Prado J. R.; Vyazovkin S. Macromol. Chem. Phys. 2014, 215 (9), 867–872. 10.1002/macp.201400029. [DOI] [Google Scholar]
- Li Z.; Torgersen J.; Ajami A.; Mühleder S.; Qin X.; Husinsky W.; Holnthoner W.; Ovsianikov A.; Stampfl J.; Liska R. RSC Adv. 2013, 3 (36), 15939. 10.1039/c3ra42918k. [DOI] [Google Scholar]
- Van Nieuwenhove I.; Stubbe B.; Graulus G.-J.; Van Vlierberghe S.; Dubruel P. Macromol. Rapid Commun. 2014, 35 (15), 1351–1355. 10.1002/marc.201400103. [DOI] [PubMed] [Google Scholar]
- Van Den Bulcke A. I.; Bogdanov B.; De Rooze N.; Schacht E. H.; Cornelissen M.; Berghmans H. Biomacromolecules 2000, 1 (1), 31–38. 10.1021/bm990017d. [DOI] [PubMed] [Google Scholar]
- Dranca I.; Vyazovkin S. Polymer 2009, 50 (20), 4859–4867. 10.1016/j.polymer.2009.06.070. [DOI] [Google Scholar]
- Bigi a.; Cojazzi G.; Panzavolta S.; Rubini K.; Roveri N. Biomaterials 2001, 22 (8), 763–768. 10.1016/S0142-9612(00)00236-2. [DOI] [PubMed] [Google Scholar]
- Donnely T. H.; McGinnis R. S.. Preparation of Water Soluble Gelatin. US 3904771 A, 1975.
- Cahn F. J.; Hagerty E. B.. Cold water-soluble gelatin composition and method of preparing the same. US 2841498 A1958.
- Obata K.; El-Tamer A.; Koch L.; Hinze U.; Chichkov B. N. Light: Sci. Appl. 2013, 2 (12), e116. 10.1038/lsa.2013.72. [DOI] [Google Scholar]
- Ma S.; Natoli M.; Liu X.; Neubauer M. P.; Watt F. M.; Fery A.; Huck W. T. S. J. Mater. Chem. B 2013, 1 (38), 5128. 10.1039/c3tb20851f. [DOI] [PubMed] [Google Scholar]
- Giraudier S.; Hellio D.; Djabourov M.; Larreta-Garde V. Biomacromolecules 2004, 5 (5), 1662–1666. 10.1021/bm049670d. [DOI] [PubMed] [Google Scholar]
- Houben A.; Roose P.; Van den Bergen H.; Declercq H.; Van Hoorick J.; Gruber P.; Ovsianikov A.; Bontinck D.; Van Vlierberghe S.; Dubruel P. Mater. Today Chem. 2017, 4, 84–89. 10.1016/j.mtchem.2017.01.005. [DOI] [Google Scholar]
- Kuijpers A. J.; Engbers G. H. M.; Feijen J.; De Smedt S. C.; Meyvis T. K. L.; Demeester J.; Krijgsveld J.; Zaat S. A. J.; Dankert J. Macromolecules 1999, 32 (10), 3325–3333. 10.1021/ma981929v. [DOI] [Google Scholar]
- Billiet T.; Gevaert E.; De Schryver T.; Cornelissen M.; Dubruel P. Biomaterials 2014, 35 (1), 49–62. 10.1016/j.biomaterials.2013.09.078. [DOI] [PubMed] [Google Scholar]
- Cheng S.; Clarke E. C.; Bilston L. E. Med. Eng. Phys. 2008, 30, 1318–1337. 10.1016/j.medengphy.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Yao J.; Ko C. W.; Baranov P. Y.; Regatieri C. V.; Redenti S.; Tucker B. a.; Mighty J.; Tao S. L.; Young M. J. Tissue Eng., Part A 2015, 21 (7–8), 1247–1260. 10.1089/ten.tea.2013.0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes R.; Yang X. B.; Dunne A.; Florea L.; Wood D.; Tronci G. Polymers (Basel, Switz.) 2017, 9, 226. 10.3390/polym9060226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. Z.; Xia Z. D.; Han Z. W.; Hulley P. a; Triffitt J. T.; Czernuszka J. T. J. Biomed. Mater. Res., Part B 2008, 85 (2), 519–528. 10.1002/jbm.b.30975. [DOI] [PubMed] [Google Scholar]
- Martens P.; Anseth K. S. Polymer 2000, 41 (21), 7715–7722. 10.1016/S0032-3861(00)00123-3. [DOI] [Google Scholar]
- Lee K. Y.; Bouhadir K. H.; Mooney D. J. Biomaterials 2004, 25 (13), 2461–2466. 10.1016/j.biomaterials.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe S.; Dubruel P.; Lippens E.; Cornelissen M.; Schacht E. J. Biomater. Sci., Polym. Ed. 2009, 20 (10), 1417–1438. 10.1163/092050609X12457418905508. [DOI] [PubMed] [Google Scholar]
- Jackson R. A.; Murali S.; Van Wijnen A. J.; Stein G. S.; Nurcombe V.; Cool S. M. J. Cell. Physiol. 2007, 210 (1), 38–50. 10.1002/jcp.20813. [DOI] [PubMed] [Google Scholar]
- Billiet T.; Vandenhaute M.; Schelfhout J.; Van Vlierberghe S.; Dubruel P. Biomaterials 2012, 33 (26), 6020–6041. 10.1016/j.biomaterials.2012.04.050. [DOI] [PubMed] [Google Scholar]
- Torgersen J.; Ovsianikov A.; Mironov V.; Pucher N.; Qin X.; Li Z.; Cicha K.; Machacek T.; Liska R.; Jantsch V.; Stampfl J. J. Biomed. Opt. 2012, 17 (10), 105008. 10.1117/1.JBO.17.10.105008. [DOI] [PubMed] [Google Scholar]
- Hu X.; Ma L.; Wang C.; Gao C. Macromol. Biosci. 2009, 9 (12), 1194–1201. 10.1002/mabi.200900275. [DOI] [PubMed] [Google Scholar]
- Zipfel W. R.; Williams R. M.; Webb W. W. Nat. Biotechnol. 2003, 21 (11), 1369–1377. 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- Nasse M. J.; Woehl J. C. J. Opt. Soc. Am. A 2010, 27 (2), 295–302. 10.1364/JOSAA.27.000295. [DOI] [PubMed] [Google Scholar]
- Auran J. D.; Koester C. J.; Kleiman N. J.; Rapaport R.; Bomann J. S.; Wirotsko B. M.; Florakis G. J.; Koniarek J. P. Ophthalmology 1995, 102 (1), 33–41. 10.1016/S0161-6420(95)31057-3. [DOI] [PubMed] [Google Scholar]
- Culver J. C.; Hoffmann J. C.; Poché R. a; Slater J. H.; West J. L.; Dickinson M. E. Adv. Mater. 2012, 24 (17), 2344–2348. 10.1002/adma.201200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bückmann T.; Stenger N.; Kadic M.; Kaschke J.; Frölich A.; Kennerknecht T.; Eberl C.; Thiel M.; Wegener M. Adv. Mater. 2012, 24 (20), 2710–2714. 10.1002/adma.201200584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.