

Abstract
Next-generation sequencing technologies have provided insights into the biology and mutational landscape of cancer. Here we evaluate the relevance of cancer neoantigens in human breast cancers. Using patient-derived xenografts from three patients with advanced breast cancer (xenografts were designated as WHIM30, WHIM35, and WHIM37), we sequenced exomes of tumor and patient-matched normal cells. We identified 2091 (WHIM30), 354 (WHIM35), and 235 (WHIM37) nonsynonymous somatic mutations. A computational analysis identified and prioritized HLA class I–restricted candidate neoantigens expressed in the dominant tumor clone. Each candidate neoantigen was evaluated using peptide-binding assays, T-cell cultures that measure the ability of CD8+ T cells to recognize candidate neoantigens, and preclinical models in which we measured antitumor immunity. Our results demonstrate that breast cancer neoantigens can be recognized by the immune system, and that human CD8+ T cells enriched for prioritized breast cancer neoantigens were able to protect mice from tumor challenge with autologous patient-derived xenografts. We conclude that next-generation sequencing and epitope-prediction strategies can identify and prioritize candidate neoantigens for immune targeting in breast cancer.
Keywords: breast cancer, neoantigens, T cells, immunotherapy
INTRODUCTION
Next-generation sequencing technologies have transformed our understanding of how somatic mutations contribute to cancer initiation and progression. Improvements in instrument performance together with cost reductions have enabled a systematic analysis of the mutational landscape in a variety of cancer types. The results provide opportunities to personalize therapy. In preclinical models, cancer neoantigens can be identified by next-generation sequencing, and neoantigens can be prioritized by epitope prediction algorithms. Some neoantigens are targets for checkpoint blockade therapy and personalized vaccine therapy (1–3). Ongoing clinical trials confirm the importance of cancer neoantigens in the response to therapies based on immune checkpoint inhibitors or personalized vaccines in non-small cell lung cancer, melanoma, and colorectal cancers with DNA mismatch repair deficiency (4–9). Here we explored neoantigens in breast cancer, which generally does not carry a high mutational load.
MATERIALS AND METHODS
Human subjects
All human subjects’ research was reviewed and approved by the Human Subjects Committee at Washington University School of Medicine. Three advanced stage breast cancer patients participated in these studies. Each subject consented to tissue banking and establishment of patient-derived xenografts (PDXs). PDXs were established in NSG mice (10) and early generation tumors were flash frozen. Each subject also consented to leukapheresis. The subjects were designated WHIM30, WHIM35 and WHIM37 based on the designation of their PDX. Leukapheresis was performed at Barnes Jewish Hospital. Peripheral blood mononuclear cells (PBMC) were isolated through density centrifugation using Ficoll-Paque PLUS, and cryopreserved as cell pellets. Each subject consented to genome sequencing. Aliquots of PBMC were frozen as cell pellets. DNA from PDX tumors and PBMC was extracted using the QIAamp DNA Mini Kit (Qiagen Sciences, Baltimore, MD) and RNA from PDX tumors was extracted using the High Pure RNA Paraffin kit (Roche, Indianapolis, IN). DNA and RNA quality was determined using a Nanodrop 2000 and quantitated using a Qubit Fluorometer (Life Technologies, Carlsbad, CA).
Exome sequencing
For each subject, tumor and normal matched DNA samples were processed for whole exome sequencing using standard protocols for Kapa Biosystems NGS libraries with corresponding barcoded adapters. The libraries were quantitated and combined at equimolar ratios into an exome capture using the Roche Nimblegen EZ Exome version 3.0 reagent. Exome sequence data were generated as 2 × 100 bp read pairs on an Illumina HiSeq2000 instrument. Our Genome Modeling System (GMS) (11) was used to align exome reads and identify somatic variants. The analysis pipeline uses BWA (version 0.5.9) (12) for alignment with default parameters except for the following: ‘-t 4 -q 5’. All alignments were against GRCh37-lite-build37 of the human reference genome and were merged and subsequently de-duplicated with Picard (version 1.46). Detection of somatic mutations was performed using a combination of different variant callers, including SAMtools (13,14), Somatic Sniper (15), VarScan Somatic (16,17) and Strelka (18).
cDNA-capture sequencing
All RNA samples were DNase-treated with a TURBO DNA-free kit (Invitrogen) as per the manufacturer’s instructions. RNA integrity and concentration were assessed using an Agilent Eukaryotic Total RNA 6000 assay (Agilent Technologies) and a Quant-iTTM RNA assay kit on a QubitTM Fluorometer (Life Technologies Corporation). The MicroPoly(A)PuristTM Kit (Ambion) was used to enrich for poly(A) RNA from three WHIM patients DNAse-treated total RNA, and the resulting RNA was converted to cDNA using the Ovation® RNA-Seq System V2 (NuGen, 20 ng of either total or polyA RNA). All NuGen cDNA sequencing libraries were generated using NEBNext® UltraTM DNA Library Prep Kit for Illumina® as described previously (19). Each library ligation reaction was PCR-optimized using the Eppendorf Epigradient S qPCR instrument, and PCR-amplified for limited cycle numbers based on the Ct value identified in the optimization step. Libraries were quantitated using the Quant-iTTM dsDNA HS Assay (Life Technologies) and for size using the BioAnalyzer 2100 (Agilent Technologies). The Illumina-ready libraries were enriched using the Nimblegen SeqCap EZ Human Exome Library v3.0 reagent. Each hybridization reaction was incubated at 47° C for 72 hours, and single-stranded capture libraries were recovered and PCR-amplified per the manufacturer’s protocol. Post-capture library pools were sized and purified with AmpureXP magnetic beads to remove residual primer-dimers and to enrich for a library fragment distribution between 300 and 500bp, then diluted to 2nM prior to Illumina paired-end sequencing. Paired end reads were trimmed with flexbar v 2.21 (params: –adapter CTTTGTGTTTGA - -adapter-trim-end LEFT –nono-length-dist –threads 4 –adapter-min-overlap 7 –max- uncalled 150 –min-readlength 25) to remove single primer isothermal amplification adapter sequences The resulting reads were analyzed with a pipeline that included Tophat v2.0.8 (params: –bowtie-version = 2.1.0 for Ovation; –library-type fr-firststrand – bowtie-version = 2.1.0 for Truseq) (2). Expression levels (FPKM) were calculated with Cufflinks v2.0.2 (params–max-bundle-length=10000000–num-threads 4). cDNA capture data was reviewed visually to evaluate the expression of mutations identified by exome data and neoantigen prediction pipeline from pVacSeq. Both cDNA-capture (Alt-read number) and FPKM values were considered for candidate neoantigen prioritization.
HLA Typing
The subject’s HLA type was determined by PCR-SSOP (ProImmune).
Neoantigen Identification
We developed a pipeline for the identification and prioritization of potential neoantigens resulting from the somatic missense mutations detected from exome sequencing analysis. Briefly, thresholds for filtering (binding- and coverage-based) using both the exome and cDNA-cap sequencing data detailed above were used to compile a list of expressed somatic missense mutations. Next, amino-acid substitutions corresponding to each of the coding missense mutations were translated into a 21-mer amino acid FASTA sequence, with 10 amino acids flanking the substituted amino acid on each side. For each patient, these 21-mer amino-acid sequences were evaluated through the HLA class I peptide-binding algorithm NetMHC v3.2 (20,21) to identify high affinity neoantigens predicted to bind with high affinity to the patient’s HLA alleles. We similarly evaluated the corresponding wildtype sequences to compare differences in predicted binding affinities, wherein any candidate neoantigens with a predicted binding affinity IC50 value < 500nm were considered for further evaluation. This pipeline evolved into the pVAC-Seq pipeline (2) pipeline for identification of candidate neoantigens.
Sanger Sequencing of DNA samples
DNA of patient PBMCs: xenograft and parental tumor tissue was extracted with Qiagen QIAamp DNA Mini Kit and AllPrep DNA/RNA FFPE Kit, respectively. Primers were designed with Primer-3. PCR reactions were carried out in 20 μl with 40 ng of DNA according to the manufactural recommendation of Phusion High-Fidelity DNA Polymerase (ThermoFisher Scientific). PCR products were purified with QIAquick gel extraction (QIAGEN) followed by sequencing (GeneWiz).
Peptides
Peptides were obtained lyophilized from Peptide 2.0 Inc. (> 95% purity), and were dissolved in sterile water or in 10% DMSO dependent on the amino acid sequence.
Mice
NOD SCID gamma (NSG) mice were purchased from The Jackson Laboratory and housed in a specific pathogen-free animal facility. All in vivo experiments used 8- to 12-week old female NSG mice. All studies were performed in accordance with procedures approved by the AAALAC accredited Animal Studies Committee of Washington University in St. Louis.
Flow cytometry
The following anti-human monoclonal antibodies (mAb) were used for cell surface staining: CD3-APC-Cy7 (clone: OKT3), CD4-FITC (clone: OKT4), CD8-AF700 (clone: SK1), CD45-BV785 (clone: HI30). All antibodies were obtained from BioLegend (San Diego, CA). Samples were analyzed on an LSR Fortessa flow cytometer (BD Biosciences), and data were analyzed using FlowJo software.
Peptide binding assay
Binding of synthetic peptides was assessed by measuring induction of surface expression of HLA class I molecules: Peptide binding to several commonly expressed human class I alleles was determined using T2 cells and genetically-modified T2 cells, specifically T2-A3 (kindly provided by Dr. Storkus from UPMC/UPC) and T2-B7 cells (kindly provided by Dr. Lutz, University of Kentucky). Following an established protocol (22) with a few modifications, peptides (100 μM) were incubated with T2, T2-A3 or T2-B7 cells in serum-free RPMI at room temperature for one hour, then transferred to 28°C in a CO2 incubator. The following day, cells were stained with HLA allele-specific mAb (anti-HLA-A2 (BD Pharmingen); anti-HLA-A3 (BD Pharmingen); and anti-HLA-B7 (provided by Dr. Ted Hansen), and analyzed by flow cytometry.
In vitro T cell analysis
In vitro studies to evaluate the immunogenicity of candidate neoantigens was performed using patients’ autologous PBMC. Briefly, PBMC were cultured with individual peptides corresponding to candidate neoantigens at 50 μg/mL in RPMI with 5% human serum, 10 units/mL Penicillin-Streptomycin, 10 mM HEPES buffer, 2mM L-glutamine, 1 × non-essential amino acid. IL2 (50 U/mL) was added every 2 days. Control PBMC were stimulated with peptides corresponding to known viral antigens. On day 12, the peptide and tumor reactivity of the T cells was determined by IFNγ ELISPOT assay. Cultured T cells were stimulated with peptide-pulsed or autologous tumor-pulsed, irradiated autologous PBMC in the ELISPOT plate followed by 20 hours incubation at 37°C. Developed spots were counted in an ELISPOT reader (C.T.L., Shaker Heights, OH). In a different set of experiments, patients’ PBMC were cultured with irradiated autologous tumor cells instead of neoantigens for 12 days, as above. Tumor-primed T cells were tested for recognition of neoantigens by IFNγ ELISPOT assay.
In vivo T-cell analysis
PDX tumor cells were subcutaneously injected into immunodeficient NSG mice (1 × 106 cells per mouse). When tumors became palpable (around 3–4 mm in dimension), 5 × 106 PBMC stimulated with prioritized neoantigens or control viral antigens were transferred weekly into tumor-bearing NSG mice through tail vein injection (23–25). Mice were observed daily and tumor size was measured every two days. In selected cases, PDX tumor-bearing NSG mice were sacrificed at 30 days, and peripheral blood and tumor were harvested from each mouse. Tumor tissue was processed into a single cell suspension through mechanical (Miltenyi gentle MACS) and enzymatic dissociation followed by filtration through a 70 μM cell strainer. The single cell suspension and PBMC were stained for immune subsets followed by flow cytometry. Functional analysis was performed by IFNγ ELISPOT assay using neoantigen peptides identified from each patient.
Statistical analyses
Samples were compared using an unpaired, two-tailed Student’s t-test, unless specified.
RESULTS
To determine whether breast cancer neoantigens can be targeted with immunotherapy, we established patient-derived xenografts from three patients with advanced breast cancer, designated WHIM30, WHIM35, and WHIM37 (Supplementary Table S1 and Supplementary Fig S1). We sequenced tumor and normal whole exomes and identified 2091 (WHIM30), 354 (WHIM35), and 235 (WHIM37) nonsynonymous single-nucleotide variant (SNV) mutations using a published somatic variant pipeline (Table 1; Supplementary Tables S2–S4) (11). We filtered the results of this pipeline analysis using stringent criteria for tumor and normal read coverage, applying a minimal variant allele frequency (VAF) of 40%. This filter prioritized 74 (WHIM30), 33 (WHIM37), and 55 (WHIM37) candidate neoantigens from the dominant tumor clone of the patient-derived xenografts. We next examined cDNA-capture sequencing from the tumor RNA (3), to confirm that each mutant allele that was identified by our DNA-based analysis was expressed in RNA, and to exclude any gene represented at fewer than 1 fragment per kilobase of transcripts per million mapped reads (FPKM) (Supplementary Table S5). We also generated HLA typing data for each patient using PCR-SSOP (Supplementary Table S6). We selected nonsynonymous SNV mutations with a predicted binding affinity to the restricting HLA alleles of < 500 nM. The final prioritized candidate neoantigens and their corresponding peptide sequences (Supplementary Tables S7–S9) were pursued for further study.
Table 1.
Overview of identified neoantigens
| Patient | Number of Tier 1 mutations† | Number of mutations expressed‡ | Number predicted to bind§ | Number actually binding/tested* | Number immunogenic in vitro** | Number conferring tumor recognition*** |
|---|---|---|---|---|---|---|
| WHIM30 | 2091 | 15 | 9 | 7/8 | 2/9 | 2/2 |
| WHIM35 | 354 | 8 | 8 | 3/3 | 1/7 | 1/1 |
| WHIM37 | 235 | 11 | 8 | 6/8 | 1/8 | 1/1 |
As determined by whole-exome sequencing and (see Supplementary Methods) from each patient xenograft.
As determined by cDNA Cap-Seq and other filters described in Supplementary Methods.
As determined by NetMHC 3.2 (affinity of mutant peptides<500 nM).
Binding assay evaluated using the T2 assay (see Supplementary Methods).
Reactivity as determined by IFN-γ ELISPOT assay in vitro as described in Supplementary Methods.
Reactivity as determined by tumor growth inhibition in NSG mouse model in vivo as described in Supplementary Methods.
Peptides corresponding to the candidate neoantigens were synthesized and assessed for binding to the corresponding HLA class I allele using T2 cells (HLA-A02:01), and genetically modified T2 cells expressing HLA-A03:01 or HLA-B07:02 (26). Of the 18 peptides tested, 15 (83%) bound to the predicted HLA allele (Table 1; stabilization of MHC expression was measured by flow cytometry, Supplementary Fig. S2). To evaluate the ability of autologous T cells to recognize the candidate neoantigens in vitro, we cocultured peptides corresponding to the candidate neoantigens with corresponding autologous PBMC for 12 days in the presence of IL2, after which the T-cell response to each peptide was assessed in an IFNγ ELISPOT assay. Two of nine (22%) WHIM30 candidate neoantigens (PALB2 and ROBO3) induced significant peptide-specific T-cell responses (*P < 0.05, ** P < 0.01, respectively) (Table 1; Fig. 1A). One of eight (12.5%) WHIM35 candidate neoantigens (PTPRS), and one of eight (12.5%) WHIM 37 candidate neoantigens (ZDHHC16) also induced significant peptide-specific CD8+ T-cell responses (*P < 0.05, ** P < 0.01, respectively) (Table 1; Fig. 1B and C). The same candidate neoantigens induced 1.6–3 times increased CD8+ T-cell responses compared with control peptides after autologous mixed lymphocyte-tumor cultures using PBMC and irradiated PDX-tumor cells (Supplementary Fig. S3).
Fig. 1. Identification and validation of candidate neoantigens.
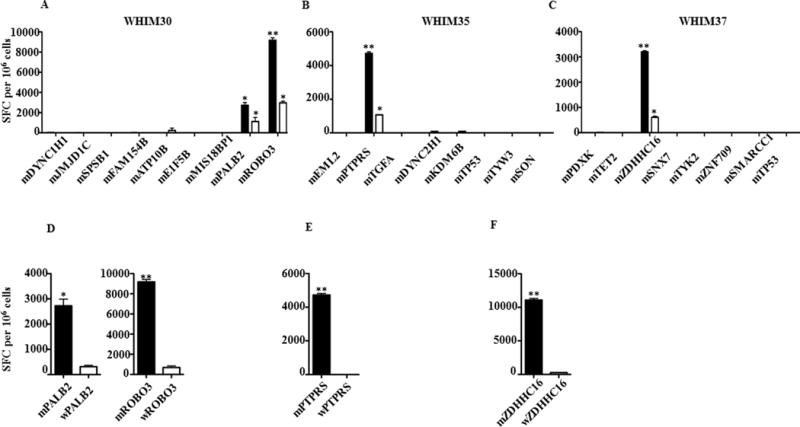
Autologous PBMCs were stimulated with candidate breast cancer neoantigens for 12 days. CD8+ T-cell IFNγ ELISPOT assays were performed on day 12 by co-culturing stimulated PBMC overnight with autologous, irradiated PBMCs pulsed with the candidate neoantigens (black) or irradiated PDX tumor cells (white). The immune response induced by candidate neoantigens and PDX tumor cells is shown in (A) WHIM30, (B) WHIM35 and (C) WHIM37. Negative controls in the ELISPOT assays included responder T cells cultured with no peptide (number of spot forming cells per 106 cells was 50–120) or irrelevant peptide (number of spot forming cells per 106 cells was 250–400). The background with irrelevant peptide was subtracted from the experimental condition in each case. To confirm the specificity of the immune response induced by candidate neoantigens, CD8+ T-cell IFNγ ELISPOT assays were performed against mutant (black) and wildtype peptides (white) after 12 day stimulation with mutant peptides. The results are shown in (D) WHIM30, (E) WHIM35 and (F) WHIM37. Data are presented as means ± s.e.m (n = 3 wells per peptide in ELISpot assay) and are representative of three independent experiments. Samples were compared using unpaired, two tailed Student’s test (*P < 0.05, ** P < 0.01), SFC is spot forming cells.
To further characterize the specificity of the T-cell response to the prioritized breast cancer neoantigens, we stimulated each patient’s PBMC with peptides corresponding to the mutant antigens for 12 days, then measured response to the antigens by IFNγ ELISPOT assays. Mutant PALB2-stimulated T cells did not cross-react with wild-type PALB2 (Fig. 1D). A similar pattern of reactivity was observed with the other breast cancer neoantigens: mutant, but not wild-type, ROBO3 (Fig. 1D), PTPRS (Fig. 1E) and ZDHHC16 (Fig. 1F) induced T-cell responses. These T-cell responses were not cross-reactive with the corresponding wild-type antigens. The recognition of mutant antigens was HLA restricted, as both allele-specific and HLA class I framework antibodies significantly reduced IFNγ secretion (*P < 0.05, **P < 0.01, respectively) (Supplementary Fig. S4).
Two breast cancer neoantigens, PTPRS and ZDHHC16, are HLA-A02:01 restricted. We tested the immunogenicity of these neoantigens in HHD mice. HHD mice are transgenic for a mutant form of HLA-A2:01 that can interact with murine CD8 molecules, but they lack murine MHC class I alleles. HHD mice were immunized with mutant PTPRS or ZDHHC16 peptide plus poly I:C. The neoantigen-specific T-cell response was then measured by IFNγ ELISPOT. The mutant PTPRS and ZDHHC16 peptides, but not other HLA-A02:01-specific breast cancer neoantigens, induced a 3–6 times increasedneoantigen-specific immune response (Supplementary Fig. S5).
To assess whether neoantigen-specific CD8+ T cells can recognize tumors, PBMC cultured in vitro in the presence of mutant peptides were tested for reactivity against autologous tumor cells in IFNγ ELISPOT assays. Both PALB2- and ROBO3-specific T cells (WHIM30) recognized WHIM30 tumor cells (Fig. 1A). Similarly, PTPRS-specific T cells (WHIM35) and ZDHHC16-specific T cells (WHIM37) recognized WHIM35 and WHIM37 tumor cells, respectively (Fig. 1B and C). In contrast, PBMC cultured with control peptides, including an immunogenic peptide derived from mammaglobin-A (27), an immunodominant viral peptide, or EML2, a HLA-A02:01–binding but nonimmunogenic mutant peptide (WHIM30), did not recognize tumor cells. PBMCs stimulated with control peptides were responsive to the control peptides, except peptide EML2, which is a nonimmunogenic neoantigen. Thus, neoantigen-specific CD8+ T cells do recognize their cognate tumors.
Breast cancer neoantigens might be useful as targets for personalized cancer vaccines or other immunotherapeutic strategies. Here, we assessed whether neoantigen-specific CD8+ T cells are associated with antitumor immunity in vivo. We began by implanting WHIM30 tumor sections subcutaneously in immune-compromised NOD SCID gamma (NSG) mice. After tumors became palpable, 5 × 106–10 × 106 autologous PBMC stimulated in vitro with PALB2, ROBO3 or control CMV peptides were adoptively transferred into tumor-bearing mice. Adoptive transfer was repeated every seven days. Tumor growth was measured every two days. Adoptive transfer of autologous PBMC stimulated with PALB2 and ROBO3 was associated with decreased tumor growth, whereas adoptive transfer of PBMC stimulated with CMV had no impact on tumor growth (Fig. 2A and C). Adoptive transfer of autologous PBMC from patient WHIM35 stimulated in vitro with PTPRS but not FluM1 peptide decreased WHIM 35 tumor growth (P<0.05) (Fig. 2B and D).
Fig. 2. Adoptive transfer of neoantigen-stimulated T cells inhibits growth of PDXs in vivo.
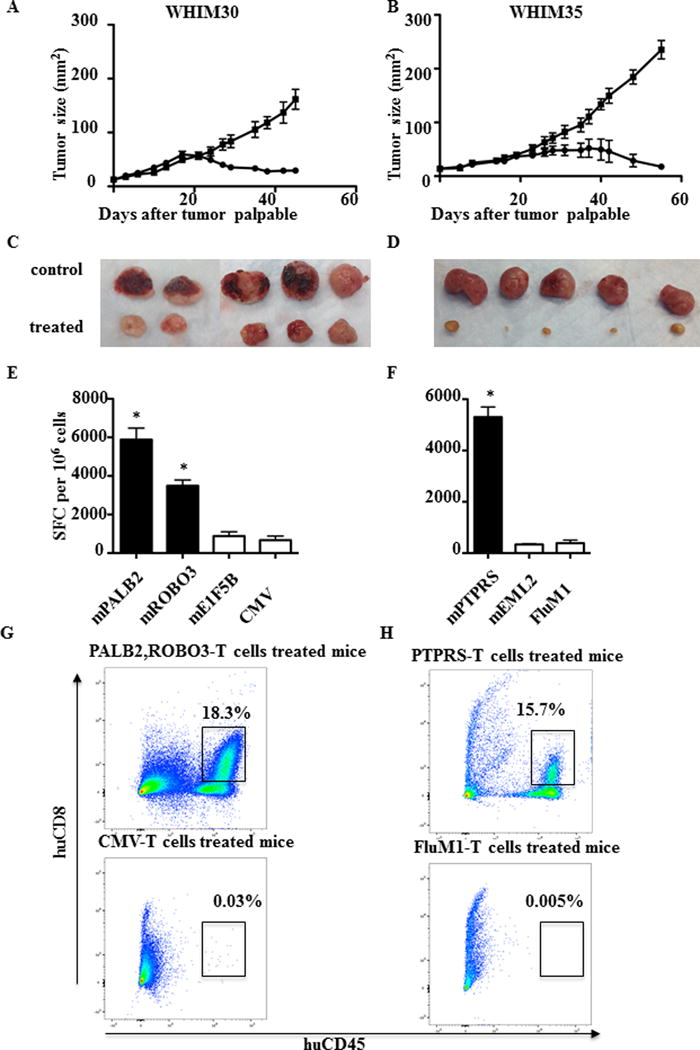
PDXs were established by injection of 1 × 106 tumor cells subcutaneously into NSG mice. 5–10 × 106 neoantigen-stimulated T cells and control viral-antigen stimulated T cells were adoptively transferred into tumor-bearing mice after the tumor became palpable. T cells were transferred at days 0, 7, and 14. Tumor size was measured every two days. (A) WHIM30 tumor growth following treatment with PBMC stimulated with mutant PALB2/ROBO3 (black solid circle) or CMV peptides (black solid square). (B) WHIM35 tumor growth following treatment with PBMC stimulated with mutant PTPRS (black solid circle) or FluM1 peptides (black solid square). (A) and (B) Data are presented as tumor size (mm2) ± s.e.m. of 5 mice per group (*P < 0.05). (C) WHIM30 tumor size (52 days after tumor challenge) following treatment with PBMC stimulated with mutant PALB2/ROBO3 or CMV peptides. (D) WHIM35 tumor size (62 days after tumor challenge) following treatment with PBMC stimulated with mutant PTPRS or CMV peptides. (E, G) TILs were isolated from WHIM30 tumors following treatment with PBMC stimulated with mutant PALB2/ROBO3 (black) or negative control mutant E1F5B and CMV (white) peptides. CD8+ T-cell IFNγ ELISPOT assay and flow cytometry were performed with experimental and control peptides as indicated. (F, H) TILs were isolated from WHIM30 tumors following treatment with PBMC stimulated with mutant PTPRS (black) or negative control mutant EML2 and FluM1 peptides (white). CD8+ T-cell mIFNγ ELISPOT assay and flow cytometry were performed with experimental and control peptides as indicated. (E) and (F) Data shown are mean ± s.e.m (n = 3 wells per peptide in ELISpot assay). Data shown are representative of three independent experiments. Samples were compared using an unpaired, two-tailed Student t test (P < 0.05).
To understand the mechanism of action of tumor rejection following adoptive transfer, we isolated tumor-infiltrating lymphocytes (TILs) from rejecting tumors and assayed them ex vivo by IFNγ ELISPOT. TILs from rejecting WHIM30 tumors generated IFNγ in response to both mPALB2 and mROBO3 (Fig. 2E). TILs from rejecting WHIM 35 tumors generated IFNγ in response to mPTPRS (Fig. 2F). In both cases, no response to control peptides was observed. Phenotypic analysis of TIL confirmed the presence of neoantigen-stimulated T cells in rejecting tumors. In contrast, no T cells were detectable in growing tumors following adoptive transfer of control, viral antigen-stimulated T cells (Fig. 2G, 2H).
Sanger sequencing results showed that the PTPRS mutation (G to A) was present in the WHIM35 parental tumor as well as in the WHIM35 PDX tumor, but not in the patient’s PBMC. DNA degradation in parental WHIM 30 and WHIM37 tumors prevented detection of neoantigens.
DISCUSSION
We have used next-generation sequencing and computational analysis to identify and prioritize candidate breast cancer neoantigens. Some of these candidate neoantigens were recognized by the immune system, thus presenting potential targets for cancer immunotherapy. Successful design of precision vaccines for treatment of human cancer will depend on rapid identification and prioritization of candidate neoantigens (1–4, 28–30), Based on the data presented here, we have initiated two phase 1 clinical trials testing neoantigen DNA and synthetic long-peptide vaccines in patients with triple-negative breast cancer (TNBC) (NCT02348320 and NCT02427581, respectively).
TNBC lacks expression of estrogen receptor, progesterone receptor, and HER2, and follows an aggressive clinical course. Clonal and mutational analysis of primary TNBC suggests that TNBC is characterized by a higher mutational frequency than other breast cancer subtypes (31–33). The relative abundance of somatic mutations in TNBC suggests that neoantigens are more likely to be present in this breast cancer subtype. Here, we observed that WHIM30, derived from a patient with TNBC, had over 2000 somatic mutations. We identified and credentialed two candidate neoantigens using algorithms designed to prioritize neoantigens from the dominant tumor clone, followed by in vitro and in vivo immune assays. WHIM35 and WHIM37 are luminal subtype breast cancers, with correspondingly lower mutational loads than WHIM30. Nonetheless, we identified and credentialed neoantigens in both these tumors, consistent with recent reports that human tumors containing limited mutations harbor neoantigens that can be targeted by immune therapies (34).
The limited number of neoantigens identified in TNBC may be a shortcoming of our identification process or may be related to the underlying biology of TNBC. Our sequencing and epitope prediction algorithms are similar to those of other investigators: we identified somatic missense mutations through exome sequencing of tumor and normal tissue, followed by expression analysis by RNA sequencing, as many mutations are not expressed. Our final step towards identification of candidate neoantigens involved the use of computer algorithms that predict which mutations can give rise to peptides that can be presented by the patient’s HLA alleles (30,35,36). Preclinical studies performed to date suggest that the majority of candidate neoantigens, i.e. those that are expressed and predicted to bind with high affinity, do not trigger detectable T-cell responses. For example, of the top 62 predicted binding epitopes, only 2 conferred tumor rejection in a mouse sarcoma model (1). Likewise, Verdegaal et al. (37) identified 501 and 226 non-synonymous mutations in two patients with advanced melanoma, respectively, but only 2/501 and 3/226 triggered antigen-specific CD8+ T cells. Relevant data from human breast cancers are not readily available. In the studies presented here we validated one or two neoantigens per patient out of eight to nine candidate neoantigens in the dominant tumor clone. Although the number of validated neoantigens is similar to that in other studies, the number of candidate neoantigens is substantially lower. We used a stringent variant allele frequency of 40%; lowering this number increases the number of candidate neoantigens.
It is too early to assess the clinical importance of targeting neoantigens. Immune therapy has heretofore emphasized induction or enhancement of immune responses to tumor-associated antigens such as HER2, MUC1, mammaglobin-A, and others (38,39). Although clinical responses have been observed with vaccines targeting tumor-associated breast cancer antigens (27,38,40), there is also evidence for a positive correlation between the number of candidate neoantigens and patient survival (41). Perhaps targeting a combination of both types of antigens will confer the greatest clinical benefit.
Our results support targeting neoantigens using vaccine-based immunotherapy in breast cancer. Although cancer neoantigens are targets for immune checkpoint-inhibitor therapies (1,42), we here show their concurrent value for personalized vaccine therapy, extending the results obtained in a murine sarcoma model (1). Personalized vaccine therapy may provide many of the benefits of immune checkpoint inhibitor therapy with decreased risk of autoimmunity and other severe adverse events. Finally, our studies suggest that a combination of genomic, computational, and in vitro functional assays can be used to identify, prioritize, and validate candidate breast cancer neoantigens, thereby facilitating clinical translation of this approach.
Supplementary Material
Acknowledgments
We thank Dr. Robert Schreiber for his comments, suggestions, and support. We thank Dr. Matthew Gubin for technical advice. We thank the staff at the McDonnell Genome Institute for their expertise and support. We thank Dr. Seth D. Crosby and Chris Sawyer from the Genome Technology Access Center for technical consultant and expertise input.
Grant support: This work is supported by grants to W.E.G. from Susan G. Komen for the Cure (KG111025), NCI T32 CA 009621, NIH P30CA091842, the Siteman Cancer Center/Barnes-Jewish Hospital Foundation Cancer Frontier Fund (BJHF CFF 7538-55), and the Siteman Cancer Center Siteman Investment Program (SIP Pre-SPORE 3781).
Footnotes
Disclosure of Potential Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hundal J, Carreno BM, Petti AA, Linette GP, Griffith OL, Mardis ER, et al. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016;8(1):11. doi: 10.1186/s13073-016-0264-5. 10.1186/s13073-016-0264-5 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–4. doi: 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015 doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. doi: 10.1038/nature12912. doi nature12912 [pii] 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013;31(32):e439–42. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013;19(6):747–52. doi: 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science. 1995;269(5228):1281–4. doi: 10.1126/science.7652577. [DOI] [PubMed] [Google Scholar]
- 10.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4(6):1116–30. doi: 10.1016/j.celrep.2013.08.022. doi S2211-1247(13)00463-4 [pii] 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffith M, Griffith OL, Smith SM, Ramu A, Callaway MB, Brummett AM, et al. Genome Modeling System: A Knowledge Management Platform for Genomics. PLoS Comput Biol. 2015;11(7):e1004274. doi: 10.1371/journal.pcbi.1004274. doi 10.1371/journal.pcbi.1004274 PCOMPBIOL-D-14-02043 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. doi btp324 [pii] 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. doi btp352 [pii] 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–93. doi: 10.1093/bioinformatics/btr509. doi btr509 [pii] 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28(3):311–7. doi: 10.1093/bioinformatics/btr665. doi btr665 [pii] 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–5. doi: 10.1093/bioinformatics/btp373. doi btp373 [pii] 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22(3):568–76. doi: 10.1101/gr.129684.111. doi gr.129684.111 [pii] 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28(14):1811–7. doi: 10.1093/bioinformatics/bts271. doi bts271 [pii] 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 19.Cabanski CR, Magrini V, Griffith M, Griffith OL, McGrath S, Zhang J, et al. cDNA hybrid capture improves transcriptome analysis on low-input and archived samples. J Mol Diagn. 2014;16(4):440–51. doi: 10.1016/j.jmoldx.2014.03.004. doi S1525-1578(14)00072-5 [pii] 10.1016/j.jmoldx.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007–17. doi: 10.1110/ps.0239403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008;36:W509–12. doi: 10.1093/nar/gkn202. Web Server issue. doi gkn202 [pii] 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen T, Myers N. Peptide induction of surface expression of class I MHC. Curr Protoc Immunol. 2003 doi: 10.1002/0471142735.im1811s57. Chapter 18: Unit 18 1. [DOI] [PubMed] [Google Scholar]
- 23.Stromnes IM, Schmitt TM, Chapuis AG, Hingorani SR, Greenberg PD. Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol Rev. 2014;257(1):145–64. doi: 10.1111/imr.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eberlein TJ, Rosenstein M, Rosenberg SA. Regression of a disseminated syngeneic solid tumor by systemic transfer of lymphoid cells expanded in interleukin 2. J Exp Med. 1982;156(2):385–97. doi: 10.1084/jem.156.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. doi nrc2355 [pii] 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elvin J, Potter C, Elliott T, Cerundolo V, Townsend A. A method to quantify binding of unlabeled peptides to class I MHC molecules and detect their allele specificity. J Immunol Methods. 1993;158(2):161–71. doi: 10.1016/0022-1759(93)90210-x. [DOI] [PubMed] [Google Scholar]
- 27.Tiriveedhi V, Tucker N, Herndon J, Li L, Sturmoski M, Ellis M, et al. Safety and preliminary evidence of biologic efficacy of a mammaglobin-a DNA vaccine in patients with stable metastatic breast cancer. Clin Cancer Res. 2014;20(23):5964–75. doi: 10.1158/1078-0432.CCR-14-0059. doi 20/23/5964 [pii] 10.1158/1078-0432.CCR-14-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72(5):1081–91. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 29.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Sharma PK, Peter Goedegebuure S, Gillanders WE. Personalized cancer vaccines: Targeting the cancer mutanome. Vaccine. 2016 doi: 10.1016/j.vaccine.2016.05.073. doi S0264-410X(16)30399-1 [pii] 10.1016/j.vaccine.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liedtke C, Bernemann C, Kiesel L, Rody A. Genomic profiling in triple-negative breast cancer. Breast Care (Basel) 2013;8(6):408–13. doi: 10.1159/000357534. doi 10.1159/000357534 brc-0008-0408 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–9. doi: 10.1038/nature10933. doi nature10933 [pii] 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464(7291):999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014;344(6184):641–5. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kakimi K, Karasaki T, Matsushita H, Sugie T. Advances in personalized cancer immunotherapy. Breast Cancer. 2017;24(1):16–24. doi: 10.1007/s12282-016-0688-1. [DOI] [PubMed] [Google Scholar]
- 36.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 37.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536(7614):91–5. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- 38.Sanchez K, Page D, McArthur HL. Immunotherapy in breast cancer: An overview of modern checkpoint blockade strategies and vaccines. Curr Probl Cancer. 2016;40(2–4):151–62. doi: 10.1016/j.currproblcancer.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Li L, Goedegebuure P, Mardis ER, Ellis MJ, Zhang X, Herndon JM, et al. Cancer genome sequencing and its implications for personalized cancer vaccines. Cancers (Basel) 2011;3(4):4191–211. doi: 10.3390/cancers3044191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clifton GT, Mittendorf EA, Peoples GE. Adjuvant HER2/neu peptide cancer vaccines in breast cancer. Immunotherapy. 2015;7(11):1159–68. doi: 10.2217/imt.15.81. [DOI] [PubMed] [Google Scholar]
- 41.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743–50. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015 doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.