

Abstract
Dopaminergic modulation of prefrontal cortex (PFC) is thought to play key roles in many cognitive functions and to be disrupted in pathological conditions, such as schizophrenia. We have previously described a phenomenon whereby dopamine D2 receptor (D2R) activation elicits afterdepolarizations (ADPs) in subcortically projecting (SC) pyramidal neurons within L5 of the PFC. These D2R-induced ADPs only occur following synaptic input, which activates NMDARs, even when the delay between the synaptic input and ADPs is relatively long (e.g., several hundred milliseconds). Here, we use a combination of electrophysiological, optogenetic, pharmacological, transgenic, and chemogenetic approaches to elucidate cellular mechanisms underlying this phenomenon in male and female mice. We find that knocking out D2Rs eliminates the ADP in a cell-autonomous fashion, confirming that this ADP depends on D2Rs. Hyperpolarizing current injection, but not AMPA receptor blockade, prevents synaptic stimulation from facilitating D2R-induced ADPs, suggesting that this phenomenon depends on the recruitment of voltage-dependent currents (e.g., NMDAR-mediated Ca2+ influx) by synaptic input. Finally, the D2R-induced ADP is blocked by inhibitors of cAMP/PKA signaling, insensitive to pertussis toxin or β-arrestin knock-out, and mimicked by Gs-DREADD stimulation, suggesting that D2R activation elicits the ADP by stimulating cAMP/PKA signaling. These results show that this unusual physiological phenomenon, in which D2Rs enhance cellular excitability in a manner that depends on synaptic input, is mediated at the cellular level through the recruitment of signaling pathways associated with Gs, rather than the Gi/o-associated mechanisms that have classically been ascribed to D2Rs.
SIGNIFICANCE STATEMENT Dopamine D2 receptors (D2Rs) in the prefrontal cortex (PFC) are thought to play important roles in behaviors, including working memory and cognitive flexibility. Variation in D2Rs has also been implicated in schizophrenia, Tourette syndrome, and bipolar disorder. Recently, we described a new mechanism through which D2R activation can enhance the excitability of pyramidal neurons in the PFC. Here, we explore the underlying cellular mechanisms. Surprisingly, although D2Rs are classically assumed to signal through Gi/o-coupled G-proteins and/or scaffolding proteins, such as β-arrestin, we find that the effects of D2Rs on prefrontal pyramidal neurons are actually mediated by pathways associated with Gs-mediated signaling. Furthermore, we show how, via this D2R-dependent phenomenon, synaptic input can enhance the excitability of prefrontal neurons over timescales on the order of seconds. These results elucidate cellular mechanisms underlying a novel signaling pathway downstream of D2Rs that may contribute to prefrontal function under normal and pathological conditions.
Keywords: D2 receptor, dopamine, PFC, pyramidal cells
Introduction
Dopaminergic modulation plays a key role in the prefrontal cortex (PFC). Prefrontal neurons receive dopaminergic input from a specific subpopulation of ventral tegmental area neurons, which are strongly activated by aversive stimuli and have unique physiological properties (Lammel et al., 2011, 2014). The infusion of dopamine D2 receptor (D2R) agonists and antagonists into the PFC modulates working memory and set-shifting in rodents (Druzin et al., 2000; Floresco et al., 2006; St Onge et al., 2011). In nonhuman primates, prefrontal D2Rs are specifically necessary for neural activity associated with memory-guided saccades (Wang et al., 2004). Consistent with these animal studies, genetic variation in D2Rs modulates prefrontal activity and working memory in humans (Zhang et al., 2007). Prefrontal D2Rs have also been hypothesized to contribute to schizophrenia (Winterer and Weinberger, 2004; Durstewitz and Seamans, 2008), Tourette syndrome (Simonic et al., 1998; Minzer et al., 2004; Yoon et al., 2007; Steeves et al., 2010), and bipolar disorder (Minton et al., 2009). Thus, D2Rs play a major role in both normal and pathological prefrontal function.
Recently, our laboratory and others have shown that, in the mouse mPFC, three major subtypes of dopamine receptors (D1Rs, D2Rs, and D3Rs) are differentially expressed by distinct subtypes of layer 5 (L5) pyramidal neurons (Gee et al., 2012; Seong and Carter, 2012; Clarkson et al., 2017). Specifically, we found that D2Rs are selectively expressed within thick-tufted, subcortically projecting (SC) L5 pyramidal neurons, which exhibit prominent hyperpolarization-activated cationic current (Ih) (Gee et al., 2012). We found that, in this population, pharmacological activation of D2Rs by the agonist quinpirole elicits a profound afterdepolarization (ADP) following depolarizing current injection. This ADP was also associated with a progressive membrane depolarization and reduction in action potential amplitude during the depolarizing current pulse. The ADP was not observed under baseline conditions, and only occurred when NMDA receptors were activated, via either optogenetic stimulation of excitatory synapses or bath application of low-dose (4 μm) NMDA. Similar to plateau potentials previously described in L5 pyramidal neurons (Milojkovic et al., 2005; Major et al., 2008), this ADP seems to rely on the intracellular accumulation of Ca2+ following influx through a combination of NMDA receptors (NMDARs) and L-type Ca2+ channels. In particular, it could be blocked by the NMDAR antagonist AP5, the selective L-type Ca2+ channel antagonist nimodipine, and the intracellular Ca2+ chelator BAPTA; however, additional mechanistic details of intracellular signaling pathways related to this phenomenon are not known.
D2Rs are classically assumed to couple to inhibitory G-proteins, reducing neuronal excitability by activating Gαi/o and inhibiting adenylate cyclase (Bonci and Hopf, 2005). D2Rs are also known to signal through at least two other pathways: via the Gβγ subunits and β arrestin. Here we explore two aspects of the intracellular signaling pathways through which the activation of D2Rs elicits ADPs in prefrontal SC-projecting neurons using a combination of electrophysiological, transgenic, pharmacological, and chemogenetic approaches. We find that the ability of synaptic stimulation to facilitate D2R-dependent ADPs several hundred milliseconds later depends on voltage-dependent (likely Ca2+) currents. Furthermore, we show that the D2R-dependent ADP persists following disruptions to Gi or β-arrestin signaling, and can be mimicked by chemogenetic activation of Gs signaling, suggesting that this phenomenon reflects novel intracellular signaling downstream of D2Rs.
Materials and Methods
Electrophysiology.
Coronal brain slices (250 μm), including mPFC, were made from adult mice of either sex that were at least 8 weeks old. We used the following transgenic mouse lines: wild-type C57BL/6J mice (www.jax.org/strain/000664), Drd1-Cre+/− (line EY262; www.gensat.org), Drd2-Cre+/− (line ER44; www.gensat.org), Drd2fl/fl (https://www.jax.org/strain/020631), Rosa26PTX (from Shaun Coughlin, University of California, San Francisco), and βarr2-KO (https://www.jax.org/strain/011130). Slicing solution was chilled to 4°C and contained the following (in mm): 234 sucrose, 26 NaHCO3, 11 glucose, 10 MgSO4, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, bubbled with 5% CO2/95% O2. Slices were incubated in ACSF at 32°C for 30 min and then at room temperature until recording. ACSF contained (in mm) the following: 123 NaCl, 26 NaHCO3, 11 glucose, 3 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgCl2, also bubbled with 5% CO2/95% O2.
Neurons were visualized using differential interference contrast or DODT contrast microscopy on an upright microscope (Olympus). Recordings were made using a Multiclamp 700B (Molecular Devices) amplifier and acquired with pClamp. Patch pipettes (2–5 mΩ tip resistance) were filled with the following (in mm): 130 K-gluconate, 10 KCl, 10 HEPES, 10 EGTA, 2 MgCl2, 2 MgATP, and 0.3 Na3GTP. In some experiments, the internal solution contained 1 μm GDP-βS or 100 μm Rp-cAMPs. All recordings were made at 32°C–34°C. Series resistance was compensated in all current-clamp experiments and monitored throughout recordings. Recordings were discarded if Rs changed by >25%.
D2R-expressing, subcortically projecting neurons were often identified by fluorescent visualization of retrograde tracer AlexaFluor-tagged cholera toxin subunit B (CTb) injected into the mediodorsal (MD) thalamus. In some experiments, pyramidal neuron subtypes were identified based on characteristic firing patterns, specifically h-current-induced “sag” >3mV in response to hyperpolarizing current pulses.
All bath-applied drugs were dissolved in water (4 μm NMDA, 10 μm (−)quinpirole, 10 μm CNQX, 10 μm H89, 1 μm clozapine-N-oxide [CNO]), DMSO (5 μm sulpiride, 5 aripiprazole [ARP]), or 1 m HCl (200 μm baclofen) before being diluted in ACSF.
Viral injection for expression of ChR2, fluorescent reporter, or DREADDs.
Viral injections were performed using standard mouse stereotactical methods. Mice were anesthetized for the duration of the surgery using isoflurane gas. After cleaning, an incision was made in the scalp, the skull was leveled, and small burr holes were drilled over the brain region of interest using a dental drill. Virus was injected through the burr holes using a microinjector at a speed of 150 nl/min, and the scalp was closed using tissue adhesive (Vetbond).
For expression of ChR2 in CC neurons, we injected a Cre-dependent ChR2 virus (AAV5-Ef1-DIO-ChR2-eYFP, 0.5 μl) into the mPFC of Drd1-Cre+/− mice (>p28) and waited 3–4 weeks for trafficking of ChR2 to the axon terminals in mPFC.
For fluorescent reporters, we injected a Cre-dependent eYFP virus (AAV5-Ef1-DIO-eYFP, 1 μl) into mPFC of Drd2-Cre+/−, Drd2fl/fl, or Rosa26PTX mice (>p28) and waited 3–4 weeks. For retrogradely labeling SC-projecting neurons, AlexaFluor-tagged CTb (ThermoFisher Scientific) was first dissolved in PBS to make a 1.0 mg/ml solution; 350 nl of CTb was then injected into the MD thalamus at a rate of 100 nl/min and waited 3–5 d before electrophysiological experiments.
To selectively knock out D2Rs in SC-projecting cells, we injected canine adenovirus-2 encoding Cre virus (Cav2-Cre, 0.5 μl; Institut de Génétique Moléculaire de Montpellier) into the MD thalamus as well as 1.5 μl AAV5-Ef1-DIO-eYFP into mPFC of Drd2fl/fl mice (>p28) and patched from both fluorescent and nonfluorescent cells after waited 4–5 weeks.
For DREADD experiments in D2R-expressing pyramidal neurons, we injected a Cre-dependent virus expressing the Gq-DREADD (AAV5-hSyn-DIO-hM3D(Gq)-mCherry, 750 nl), Gs-DREADD (AAV5-hSyn-DIO-rM3D(Gs)-mCherry, 750 nl), or Gi-DREADD (AAV5-hSyn-DIO-hM4D(Gi)-mCherry, 750 nl) into the mPFC of Drd2-Cre+/− mice and patched from fluorescent cells after waiting 5 weeks for expression.
MD thalamus injection coordinates were anteroposterior = −1.7, mediolateral = ±0.3, dorsoventral = −3.45. mPFC injection coordinates were anteroposterior = 1.7, mediolateral = ±0.3, dorsoventral = −2.75.
ChR2 stimulation.
We stimulated ChR2 in projections from the contralateral PFC using 5 ms flashes of light generated by a Lambda DG-4 (Sutter Instruments) high-speed optical switch with a 300 W Xenon lamp delivered through a 470 nm excitation filter. For stimulation of fibers, 5 different trains of random light flashes (intensity: ∼2 mW, total train duration: 2.5 s, ∼60 flashes/train, flash duration: 2 ms) were delivered at through a 40× objective every 13 s.
Experimental design and statistical analysis.
All data are shown as mean ± SEM. Statistical significance was accepted at the level p < 0.05. All statistical computations were performed using GraphPad Prism 7.0 software. We used Student's t test to compare pairs of groups if data were normally distributed (verified using Lillie test). If more than two groups were compared, we used ANOVA with post hoc tests between groups corrected for multiple comparisons (Holm–Sidak or Bonferroni). The specific post hoc test as well as exact F and corrected p values can be found in the text.
Results
The quinpirole-induced ADP depends on D2Rs
First, to confirm the basic properties of the D2R-mediated ADP we had described previously (Gee et al., 2012), we labeled SC-projecting neurons by injecting a retrogradely transported fluorescent tracer (AlexaFluor-tagged CTb) into the MD thalamus (Fig. 1A). Then, we patched from CTb+ L5 pyramidal neurons in mPFC and measured their voltage responses to depolarizing current pulses (250 ms duration) (Fig. 1B). We did not observe an ADP following depolarizing current pulses in the presence of just NMDA (4 μm); however, following the addition of the D2R agonist (−)quinpirole (10 μm) to the bath, we observed a robust ADP for ∼100–300 ms after the end of each depolarizing current pulse (Fig. 1C; n = 11/9/11/4, baseline/NMDA/QPL/SUL; one-way ANOVA, F(3,31) = 16.36, p < 0.0001; t test, Bonferroni correction, baseline vs NMDA: t(18) = 0.1408, baseline vs QPL: t(20) = 6.178, p < 0.0001, NMDA vs QPL: t(18) = 5.721, p < 0.0001). Subsequent wash-in of the selective D2/D3 antagonist sulpiride (5 μm) eliminated this quinpirole-induced ADP (t test, Bonferroni correction, QPL vs SUL: t(13) = 3.723, p = 0.0031).
Figure 1.

The quinpirole-induced ADP depends on D2Rs. A, Schematic of coronal brain slice. Top, We recorded from SC L5 pyramidal neurons labeled with CTB488 in the mouse mPFC. Bottom, Images of a recorded neuron in DIC and exhibiting 488 nm fluorescence. B, Example pyramidal cell responses to hyperpolarizing and depolarizing current steps at baseline (black) and after application of drug (purple represents 4 μm NMDA; blue represents 4 μm NMDA + 10 μm quinpirole; green represents 4 μm NMDA + 10 μm quinpirole + 5 μm sulpiride). Arrow indicates the quinpirole-induced ADP. C, Changes in the ADP, quantified by the amount of time required for the membrane potential to return to baseline following the depolarizing current pulse (more precisely, the time for 90% decay), after pharmacological manipulations listed above. n = 11/9/11/4, baseline/NMDA/QPL/SUL. ***p < 0.0001. D, Experimental design. Drd2fl/fl mice were injected with Cav2-cre virus in the MD thalamus and DIO-EYFP virus in the mPFC 4–5 weeks before slice experiments. E, Top, Example pyramidal cell responses to hyperpolarizing and depolarizing current steps in 4 μm NMDA + 10 μm quinpirole for EYFP-negative (control) and EFYP-positive (Drd2 knock-out) cells. Bottom, Images of a recorded neuron in DIC showing EYFP fluorescence. F, The quinpirole-induced ADP (arrows) is absent in cells lacking Drd2. n = 8 cell pairs. ***p < 0.0001.
To further confirm that this ADP was mediated by D2Rs, we injected canine adenovirus-2 encoding Cre (Cav2-Cre) (Hnasko et al., 2006) into MD thalamus to drive Cre expression within a subset of SC-projecting neurons in D2R conditional KO mice (Drd2fl/fl, The Jackson Laboratory; Fig. 1D) (Bello et al., 2011). We then injected AAV to drive Cre-dependent expression of EYFP into the mPFC to identify SC-projecting neurons, which expressed Cre, and from which D2Rs should have been knocked out. Finally, we made simultaneous recordings from pairs of EYFP+ and EYFP− neurons in L5 of the mPFC. We identified EYFP− SC-projecting neurons based on the presence of prominent voltage sag and rebound during responses to hyperpolarizing current pulses (Gee et al., 2012). We confirmed that the quinpirole-induced ADP still occurred in EYFP-negative SC-projecting neurons but was completely absent from simultaneously recorded EYFP+ SC-projecting neurons (Fig. 1E,F; n = 8 cell pairs; mixed-model two-way ANOVA, F(1,14) = 20.15, p = 0.0005; t test, Holm–Sidak correction, eYFP+ baseline vs QPL: t(7) = 6.461, p < 0.0001, eYFP− baseline vs QPL: t(7) = 0.1137). This confirms that the quinpirole-induced ADP can be eliminated in a cell autonomous fashion by knocking out D2Rs.
Hyperpolarizing current suppresses the ability of synaptic input to facilitate the ADP
Next, we investigated mechanisms through which optogenetic stimulation of callosal synapses can unmask the quinpirole-induced ADP (Gee et al., 2012). Again, bath application of quinpirole alone does not elicit an ADP. The ADP is only observed when quinpirole application is combined with the optogenetic stimulation of synaptic inputs (e.g., from the contralateral mPFC) or by bath application of low-dose NMDA. Synaptic stimulation can facilitate a subsequent ADP even when this stimulation is relatively weak (i.e., insufficient to elicit spiking in the postsynaptic neuron) and occurs several hundred milliseconds before the current pulse. Synaptic stimulation appears to act by recruiting NMDARs because the ADP is blocked in the presence of AP5. We hypothesized that there could be at least two distinct mechanisms through which synaptic stimulation activates NMDARs and facilitates the ADP. First, synaptic stimulation may recruit significant NMDAR-mediated currents; in particular, even though the resulting EPSPs may be relatively modest when measured at the soma (e.g., ∼1 mV in amplitude), they may be substantially larger in the dendrites leading to significant NMDAR-mediated Ca2+ influx, which persists for several hundred milliseconds contributing to the subsequent ADP. Alternatively, synaptic stimulation may trigger ongoing network activity that continues for several hundred milliseconds.
To distinguish between these two possibilities, we compared the magnitude of the quinpirole-induced ADP under various conditions (Fig. 2B). In each case, we elicited the ADP as follows: we injected AAV5-Ef1α-DIO-ChR2-EYFP into the mPFC in one hemisphere of Drd1-Cre (D1-Cre) mice (to label callosally projecting D1+ neurons), then after waiting 3–4 weeks for ChR2 expression, we made recordings from L5 SC-projecting neurons in the contralateral hemisphere (Fig. 2A). After delivering a train of irregularly occurring light flashes (total train duration: 2.5 s, ∼60 flashes/train, flash duration: 2 ms) to stimulate callosal terminals, we waited for 500 ms, then injected a depolarizing current pulse (400 pA, 500 ms duration) to elicit the ADP. Each experiment consisted of four phases. First, we simply recorded the ADP following synaptic stimulation (control; Fig. 2C). Second, we injected hyperpolarizing current (−200 pA) while delivering the train of light flashes to suppress voltage-dependent currents (e.g., NMDAR-mediated Ca2+ influx) associated with synaptic stimulation (“Exp 1”; Fig. 2C). Third, we washed on CNQX (10 μm) to block AMPA receptors and eliminate recurrent network activity elicited by synaptic stimulation (“Exp 2”; Fig. 2D). Fourth, we delivered hyperpolarizing current during the light trains while CNQX remained in the bath (“Exp 1 + 2”; Fig. 2D). To quantify the ADP, we measured the amount of time required the membrane potential to return to baseline following the depolarizing current pulse (more precisely, the time for 90% decay).
Figure 2.
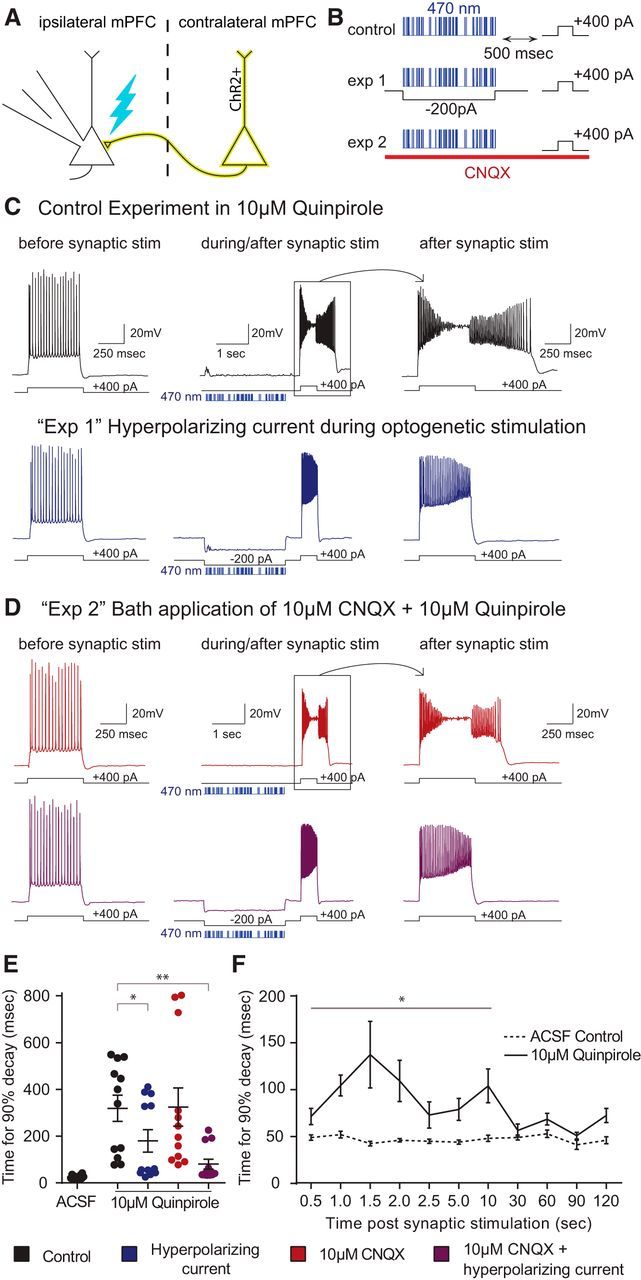
Hyperpolarizing current suppresses the ability of synaptic input to facilitate the quinpirole-induced ADP. A, Experimental design. We recorded from ChR2-negative layer 5 neurons while stimulating ChR2-expressing axon terminals from the contralateral mPFC with trains of light flashes (470 nm, 2.5 ms, ∼2 mW). B, For each cell with bath application of 10 μm quinpirole, we recorded the neuronal response under four different stimulation paradigms: synaptic stimulation (control, black); hyperpolarizing current (−200 pA) during the train of light flashes (“exp 1,” blue); synaptic stimulation in 10 μm CNQX (“exp 2,” red); hyperpolarizing current during light flashes while in 10 μm CNQX (purple). C, D, Responses of a layer V SC-projecting pyramidal neuron to current injection before (left) and immediately following (middle and right) optogenetic stimulation of synaptic inputs. Blue bars represent the times of light flashes. E, Quinpirole-induced ADP is reduced by injection of hyperpolarizing current. n = 12. *p = 0.0318. **p = 0.0037. F, The quinpirole-induced ADP persists up to 10 s after the synaptic stimulation. n = 5. *p < 0.0001–0.0338.
The duration of the ADP was significantly reduced when we injected hyperpolarizing current during the delivery of light flashes (Fig. 2E; n = 12; 46 ± 8% reduction in ADP duration; repeated-measures ANOVA, F = 8.659, p = 0.0024; t test, Holm–Sidak correction, for ACSF vs QPL without hyperpolarizing current: t(22) = 5.067, p = 0.0036; for QPL with vs without hyperpolarizing current: t(22) = 3.544, p = 0.0318). By contrast, there was relatively little effect of CNQX application alone (t test, QPL with vs without CNQX, no hyperpolarizing current: t(22) = 0.051). Notably, the combined effect of CNQX application and hyperpolarizing current injection was significantly larger than that of hyperpolarizing current injection alone; indeed, the ADP was almost eliminated in this case (70 ± 5% reduction in ADP duration; t test, QPL with vs without CNQX and hyperpolarizing current: t(22) = 4.988, p = 0.0037), suggesting that dendritic space clamp may be significantly improved in the presence of CNQX. Hyperpolarizing current delivered during the period of synaptic stimulation also seemed to attenuate other phenomena elicited by combined D2R activation + synaptic stimulation (e.g., the progressive membrane depolarization and reduction in spike amplitude during a current pulse).
Finally, we explored the timescales over which synaptic input could contribute to the quinpirole-induced ADP. For these experiments, we simply varied the delay between the trains of light flashes and the subsequent depolarizing current pulses. Remarkably, we could still observe an ADP lasting ∼100 ms even when current injection occurred up to 10 s following synaptic stimulation (Fig. 2F; n = 5; repeated measures two-way ANOVA: F(10,40) = 4.8, p = 0.0002; t test, Holm–Sidak correction), for individual time delays were as follows: p = 0.0071 (0.5 s), p = 0.0007 (1 s), p < 0.0001 (1.5 s), p < 0.0001 (2.0 s), p = 0.1190 (2.5 s), p = 0.0338 (5.0 s), p = 0.0003 (10 s), p = 0.6671 (30 s), p = 0.4942 (60 s), p = 0.6671 (90 s), p = 0.1248 (120 s).
Inhibiting adenylate cyclase/PKA-dependent signaling inhibits the quinpirole-induced ADP
Next, we turned our attention to the intracellular signaling pathways through which D2R activation recruits the ADP. First, to verify that the quinpirole-induced ADP depends on G-protein signaling, we performed experiments with 1 mm GDP-βS in our intracellular pipette. GDP-βS is a nonhydrolyzable form of GDP that binds to G-proteins and prevents the conformational changes, which normally follow receptor ligand binding, depend on the hydrolysis of GTP to GDP, and trigger downstream signaling (Seong and Carter, 2012). Indeed, whereas in control experiments (without GDP-βS in the intracellular pipette), we observed robust ADPs following application of (−)quinpirole (10 μm) + NMDA (4 μm), we observed no ADPs when 1 mm GDP-βS was included in the intracellular pipette (Fig. 3A,B; n = 7/6, K-gluconate/GDP-βs; two-way ANOVA: F(2,27) = 6.406, p = 0.0053; t test, Holm–Sidak correction, quinpirole + NMDA condition for K-gluconate vs GDP-βs: t(11) = 4.241, p = 0.0007).
Figure 3.
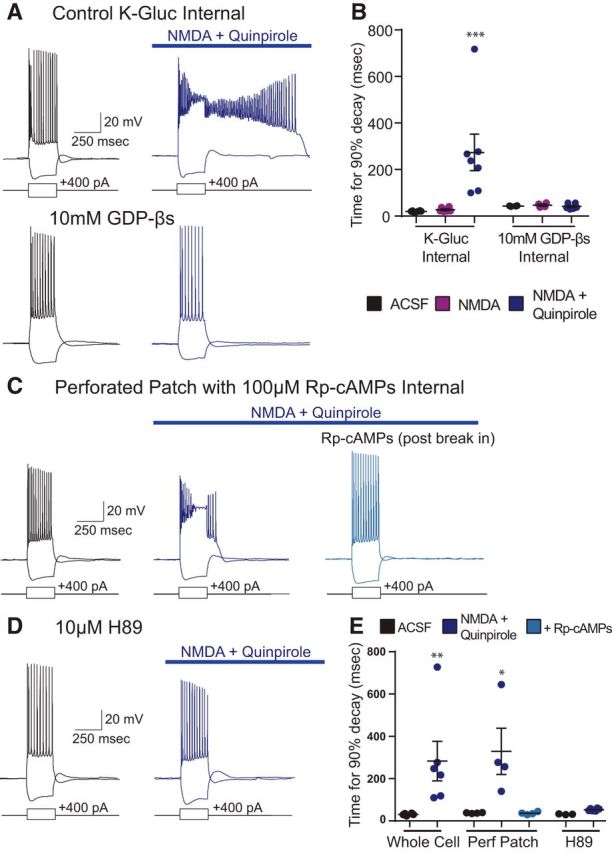
Inhibiting adenylate cyclase/PKA-dependent signaling inhibits the quinpirole-induced ADP. A, Sample traces in ACSF (black) and 4 μm NMDA + 10 μm quinpirole (blue) in control K-gluconate internal as well as internal, including 10 mm GDP-βS, a G-protein inhibitor. B, GDP-βS blocks the quinpirole-induced ADP in a cell-autonomous fashion. n = 7/6, K-gluconate/GDP-βs. ***p = 0.0007. C, Sample traces in ACSF (black) and 4 μm NMDA + 10 μm quinpirole (blue, left) in perforated patch and in whole-cell patch clamp dialyzed with internal solution containing 100 μm Rp-cAMPs (blue, right). D, Sample traces with bath application of 10 μm H89 in ACSF (black) and 4 μm NMDA + 10 μm quinpirole (blue). E, Inhibition of PKA suppresses the quinpirole induced ADP. n = 4 for perforated patch and 6 for H89. *p = 0.0285, **p = 0.0052.
Next, because D2Rs canonically signal through inhibitory G-proteins, we decided to test how inhibiting adenylate cyclase/PKA signaling would affect the ADP. Because inhibitory G-proteins normally inhibit these pathways, we expected PKA inhibitors, such as Rp-cAMPs or H89, to mimic or enhance the ADP. Instead, we found that, when we included Rp-cAMPs (100 μm) in the intracellular pipette, we could initially elicit a quinpirole-induced ADP in a perforated patch recording configuration, but that when we subsequently broke in, shifted to a whole-cell recording, and dialyzed the cell with our intracellular solution, the quinpirole-induced ADP was abolished (Fig. 3C,E; n = 4; ANOVA, F(2,9) = 7.154, p = 0.0138; t test, Bonferroni correction; ACSF vs QPL: t(6) = 3.271, p = 0.029, QPL vs Rp-cAMPs: t(6) = 3.281, p = 0.0285). This demonstrates that Rp-cAMPs is actually sufficient to abolish (not mimic) the quinpirole-induced ADP. Similarly, the presence of H89 (10 μm) in the bath prevented us from inducing an ADP with bath application of quinpirole and NMDA (Fig. 3D,E; n = 6; ANOVA, F(1,17) = 5.841, p = 0.0272, t test, Holm–Sidak correction, Control ACSF vs QPL: t(10) = 3.524, p = 0.0052, H89 control vs H89 + QPL: t(6) = 0.2426). These results suggest that inhibition of adenylate cyclase/PKA signaling is sufficient to block the quinpirole-induced ADP in a cell-autonomous fashion.
The quinpirole-induced ADP is independent of β-arrestin
Activation of D2Rs with relatively high doses of quinpirole (10 mm) and for somewhat prolonged periods of time (e.g., 15–20 min) may induce β-arrestin-mediated desensitization and internalization. Thus, the preceding observations could be explained by a model in which at baseline, D2Rs inhibit adenylate cyclase/PKA signaling, suppressing the ADP, but that quinpirole induces receptor internalization, terminating this suppression, and leading to the appearance of the ADP. To explore whether the ADP might depend on β-arrestin signaling via such a mechanism, we measured the ADP after disrupting β-arrestin signaling in two ways. First, we studied β-arrestin2 KO mice (βarr2-KO) (Bohn et al., 1999) and found that quinpirole and NMDA continued to elicit ADPs (Fig. 4A,B; n = 18/11, β-arrestin2 KO/C57BL/6; two-way ANOVA, Fgenotype(1,75) = 0.517, Fdrug(3,75) = 16.56, p < 0.0001; t test, Holm–Sidak correction, β-arrestin2 KO ACSF vs QPL: t(34) = 5.759, p < 0.0001). Moreover, in βarr2 KO mice, this quinpirole-induced ADP was still eliminated by the subsequent wash-in of the selective D2R antagonist sulpiride (5 μm), confirming that it still depends on D2Rs (Fig. 4A,B; t test, Holm–Sidak correction, β-arrestin2 KO QPL vs SUL: t(20) = 2.595, p = 0.0448). Second, we found that quinpirole and NMDA could still elicit an ADP, even in the presence of aripiprazole (ARP; 5 μm; Fig. 4C,D; n = 6; ANOVA, F(1,52) = 0.005, Fdrug(2,52) = 16.34, p < 0.0001, t test, Holm–Sidak correction, ARP ACSF vs ARP QPL: t(10) = 3.329, p < 0.0416). ARP is a functionally selective D2R ligand that is a complete antagonist with respect to β-arrestin signaling, but a partial agonist with respect to canonical G-protein signaling (Burris et al., 2002; Madhavan et al., 2013). Together, these two experiments show that D2Rs can elicit the ADP in the absence of β-arrestin.
Figure 4.
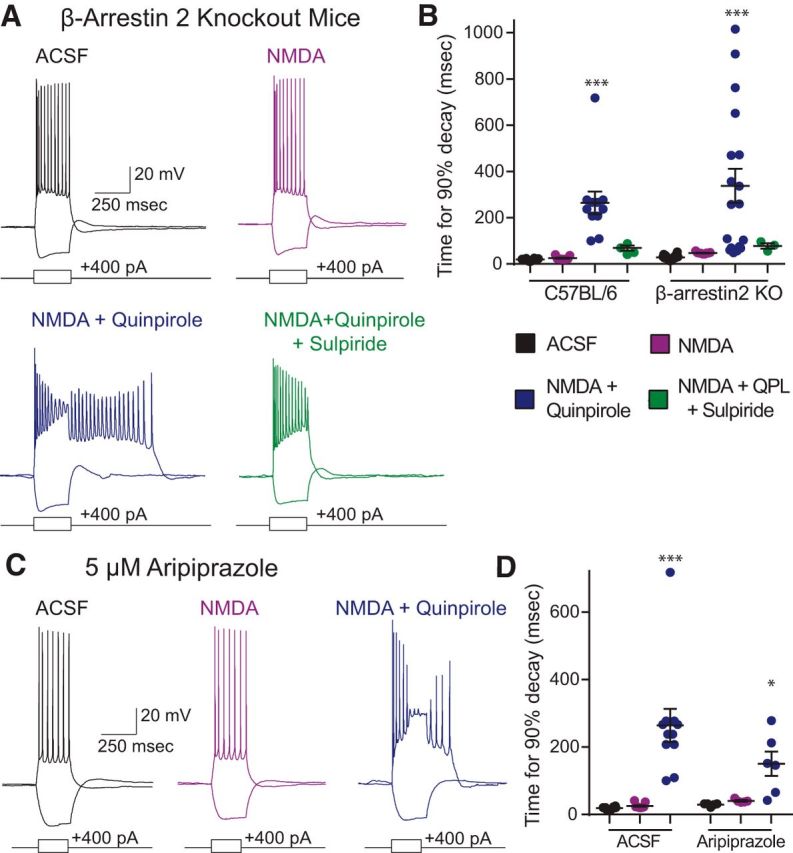
The quinpirole-induced ADP is independent of β-arrestin. A, Example recordings from β-arrestin2 KO mice in ACSF (black) and after application of drug (purple represents 4 μm NMDA; blue represents 4 μm NMDA + 10 μm quinpirole; green represents 4 μm NMDA + 10 μm quinpirole + 5 μm sulpiride). B, The quinpirole-induced ADP persists in β-arrestin2 KO mice. n = 18/11, β-arrestin2 KO/C57BL/6. ***p < 0.0001. C, Example traces in 5 m ARP at baseline (black) and after application of additional drugs (purple represents 4 μm NMDA; blue represents 4 μm NMDA + 10 μm quinpirole). D, The quinpirole-induced ADP persists in the presence of aripiprazole, a biased D2R ligand that selectively antagonizes β-arrestin signaling. n = 6. *p = 0.0416, ***p < 0.0001.
Pertussis toxin does not block the quinpirole-induced ADP
Next, we wanted to examine whether the quinpirole-induced ADP depends on Gi signaling at all. For this, we expressed pertussis toxin, which selectively blocks all Gi-mediated signaling. We crossed Drd2-Cre (D2-Cre) mice to a transgenic line expressing the Cre-dependent S1 catalytic subunit of pertussis toxin (PTX S1) inserted into the Rosa26 (Rosa26-PTX) (Regard et al., 2007) such that PTX S1 would be expressed in D2R+ cells, which normally exhibit the D2R-mediated ADP (Fig. 5A). We verified that pertussis toxin blocked Gi-mediated signaling in D2R+ neurons by comparing baclofen-induced currents in D2R+ (SC-projecting) neurons and D2R− (IT-projecting) neurons (distinguished based on the Cre-dependent expression of EYFP) (Fig. 5B). Baclofen is a GABAB receptor agonist, which normally recruits GIRK currents via Gi signaling. We found that, in voltage-clamp recordings in wild-type mice, bath application of baclofen (200 μm), elicits large outward currents in both D2R+ and D2R− neurons (n = 4 of each; Fig. 5C). In D2-Cre/Rosa26-PTX mice, baclofen continued to elicit GIRK currents normally in D2R− neurons; however, baclofen-induced currents were completely abolished in D2R+ neurons (Fig. 5C; n = 4/4, D2R+ and D2R− from each genotype; two-way ANOVA, F(1,12) = 620, p < 0.001, t test, Holm–Sidak correction, t(6) = 31.48, p < 0.0001). We found that the quinpirole-induced ADP was intact in D2R+ neurons from D2-Cre/Rosa26-PTX mice (Fig. 5D,E; n = 6/5, Drd2Cre::Rosa26-PTX/C57BL6; repeated-measures two-way ANOVA, Fgenotype(1,9) = 0.0002, Fdrug (2,18) = 45.4, p < 0.0001; t test, Holm–Sidak correction, C57BL/6 ACSF vs QPL: t(8) = 5.581, p < 0.0001, Drd2Cre::Rosa26-PTX ACSF vs QPL: t(10) = 6.047, p < 0.0001). These experiments show that D2Rs elicit the ADP even in the absence of Gi signaling.
Figure 5.

Pertussis toxin blocks Gi signaling but does not block the quinpirole-induced ADP. A, Experimental design. We crossed D2-Cre mice to Rosa26-PTX mice and injected the offspring in the mPFC with a Cre-dependent virus to drive EYFP expression. B, We then recorded from D2+ pyramidal neurons (i.e., EYFP+) and D2− pyramidal neurons (i.e., EYFP−) in L5 of mPFC. C, Pertussis toxin selectively suppresses baclofen-induced GIRK currents in D2+ neurons of Drd2Cre::Rosa26-PTX mice. n = 4. ***p < 0.0001. D, Sample traces from Drd2Cre::Rosa26-PTX mice in ACSF (black) and after application of drug (purple represents 4 μm NMDA; blue represents 4 μm NMDA + 10 μm quinpirole). E, The quinpirole-induced ADP is not blocked by pertussis toxin. n = 6/5, Drd2Cre::Rosa26-PTX/C57BL/6. ***p < 0.0001.
Gs-, but not Gi- or Gq-coupled, DREADDs elicits a quinpirole-like ADP
The preceding results strongly suggest that D2Rs do not act through Gi or β-arrestin to elicit the ADP. Furthermore, the fact that the PKA inhibitors Rp-cAMPs and H89 can inhibit the quinpirole-induced ADP raises the possibility that signaling through Gs or a Gs-like pathway may actually elicit this phenomenon. To test this directly, we injected virus to drive Cre-dependent expression of Gs-, Gi-, or Gq-coupled DREADDs into the mPFC of D2-Cre mice. We found that the presence of low dose (4 μm) NMDA together with CNO (1 μm), which activates each DREADD, could elicit an ADP identical to the sort normally induced by quinpirole in SC-projecting cells expressing Gs DREADDs (Fig. 6A,B; n = 8 for each DREADD; mixed-model two-way ANOVA, F(2,21) = 15.94, p < 0.0001; t test, Holm–Sidak correction, Gs-DREADD ACSF vs CNO + NMDA: t(14) = 7.01, p < 0.0001). Activation of the Gs-DREADD increases the number of action potential during and immediately following the current pulse (i.e., including the ADP). By contrast, we observed no effect of CNO in mice expressing the Gq DREADDs. Furthermore, in mice expressing the Gi DREADDs, bath application of 1 μm CNO decreased action potential firing in response to a depolarizing current step (Fig. 6C; n = 8 for each DREADD; mixed-model two-way ANOVA, F(2,21) = 20.79, p < 0.0001; t test, Holm–Sidak correction, Gi-DREADD ACSF vs CNO: t(14) = 3.319, p = 0.0065, Gs-DREADD ACSF vs CNO: t(14) = 5.621, p < 0.0001), confirming functional Gi DREADD expression in these neurons, and consistent with a Gi-induced reduction of neuronal excitability (Bonci and Hopf, 2005).
Figure 6.

Signaling through Gs-, but not Gi- or Gq-coupled, DREADDs elicits a quinpirole-like ADP. A, Example traces from L5 mPFC pyramidal neurons from D2-Cre mice injected with virus to derive Cre-dependent expression of Gi (left), Gs (middle), or Gq (right) coupled DREADDs, at baseline (top, black) or in 4 μm NMDA + 1 μm CNO (bottom, blue). B, Activation of the Gs-DREADD with 1 μm CNO together with low-dose NMDA elicits an ADP. n = 8. ***p < 0.0001. C, Activation of the Gi-DREADD with 1 μm CNO reduces the number of action potentials fired when 400 pA depolarizing current is injected for 250 ms. By contrast, activation of the Gs-DREADD increases the number of action potential during and immediately following the current pulse. n = 8. **p = 0.0065, ***p < 0.0001.
Discussion
Here we follow-up on our earlier work (Gee et al., 2012), which described a new mechanism through which D2R activation could elicit an increase in excitability, specifically an ADP, within subcortically projecting L5 pyramidal neurons in the mPFC. Several aspects of this phenomenon were unusual. First, we observed the ADP using relatively high doses of quinpirole (5–20 μm). This was in contrast to other effects of quinpirole can sometimes be observed using doses ∼1 μm (Tseng et al., 2007). Second, the ADP represents an increase in excitability, whereas D2R activation has classically (in the striatum) been assumed to inhibit neuronal excitability. Third, this phenomenon was not readily observed under baseline conditions in brain slices but required synaptic stimulation. All of these relatively unique features raised several questions: for example, is this phenomenon, which is elicited by quinpirole and blocked by sulpiride, really mediated by D2Rs? If so, is this phenomenon mediated by canonical D2R signaling, the desensitization or internalization of D2Rs, or another pathway (e.g., β-arrestin-mediated signaling) altogether? And finally, what is the mechanism through which synaptic input regulates the occurrence of an ADP several hundred milliseconds later?
This study answers those questions, at least in part. Knocking out D2Rs abolishes the quinpirole-induced ADP in a cell-autonomous manner, confirming that it is D2R-mediated. Hyperpolarizing current injection, but not CNQX, prevents synaptic stimulation from facilitating the quinpirole-induced ADP, showing that this effect depends on the ability of synaptic stimulation to recruit voltage-dependent currents, likely NMDAR-mediated Ca2+ influx. Finally, the quinpirole-induced depolarization is blocked by inhibitors of PKA signaling, insensitive to pertussis toxin, and mimicked by Gs-DREADD stimulation. Together, our observations, following numerous manipulations targeting multiple aspects of G-protein coupled signaling, suggest that D2R activation elicits the ADP by stimulating cAMP/PKA signaling that is downstream of Gs (Fig. 7).
Figure 7.
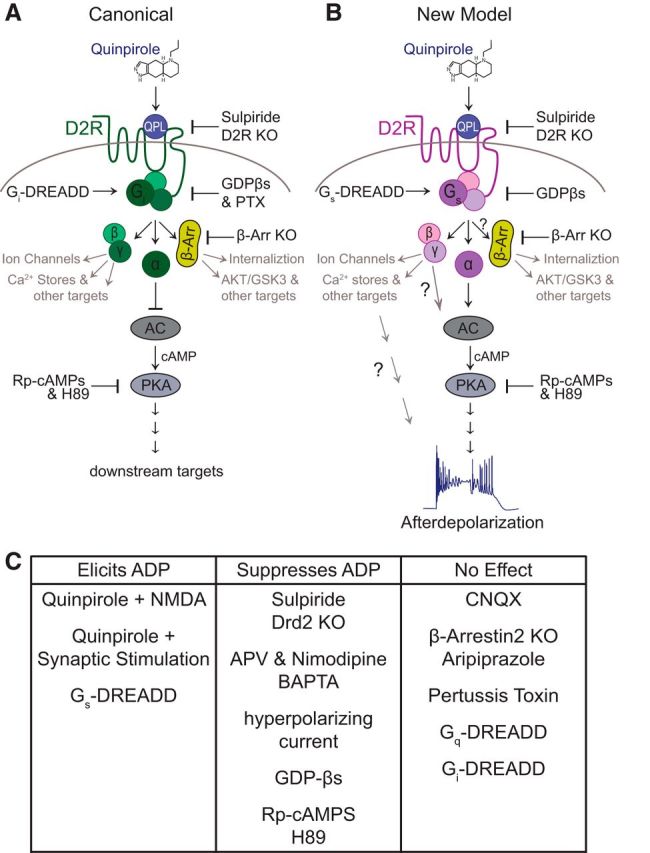
Schematic models for D2R signaling in mPFC SC-projecting projecting L5 pyramidal neurons. A, Schematic of the canonical D2R signaling cascade acting via Gi to inhibit cAMP/PKA activity. B, Schematic of the new model showing how D2R signaling might act via Gs- or a Gs-like pathway to stimulate cAMP/PKA and thereby elicit an ADP. C, Summary of how various experimental manipulations we tested affected the quinpirole-induced ADP.
Importantly, neither quinpirole nor sulpiride distinguishes between D2Rs and D3Rs. Thus, pharmacology alone could not rule out the possibility that D3Rs, along with D2Rs, contribute to the quinpirole-induced ADP. However, another recent study shows that D3Rs are expressed by a distinct population of prefrontal neurons, and not by the prefrontal SC-projecting neurons which are the subject of this study (Clarkson et al., 2017).
Limitations
Of course, our experiments have some important limitations. For example, H89 inhibits other kinases in addition to PKA, although Rp-cAMPs (which we delivered intracellularly) is more selective. Our experiments have not directly tested whether Gi-associated βγ subunits might contribute to the effects we have observed by stimulating PKA signaling, but we consider this possibility unlikely because the D2R-induced ADP is not blocked by pertussis toxin. By preventing dissociation of the heterotrimeric G-protein complex, pertussis toxin should block signaling by both α and βγ subunits associated with Gi. Similarly, although our results do suggest that D2Rs elicit an ADP via signaling pathways that are classically downstream of Gs, we have not determined to what extent Gs-associated βγ subunits might contribute to or modulate these effects (Fig. 7). Finally, although our experiments with pertussis toxin strongly argue against a role for Gi-mediated signaling in the D2R-induced ADP, and our DREADD experiments show that Gs-mediated signaling is sufficient to elicit this phenomenon, we cannot completely rule out the possibility that native Gq signaling might contribute to this phenomenon in ways that are not captured by simply stimulating Gq DREADDs.
Relationship to other modes of D2R signaling
D2R-mediated signaling is diverse. D2Rs have classically been assumed to inhibit neuronal excitability via Gi signaling (e.g., in indirect pathway medium spiny neurons and dopaminergic terminals) (Bonci and Hopf, 2005). However, other studies have described novel mechanisms whereby D2Rs can signal through Gβγ subunits or interactions with β-arrestin (Beaulieu et al., 2005), including in prefrontal interneurons (Urs et al., 2016). These results enlarge the family of possible signaling pathways downstream of D2R activation. At the same time, this study, which has focused on cellular physiology, does not elucidate the molecular or biochemical details of this pathway.
Relationship to D2R heterogeneity and heteroreceptors
D2Rs comprise 6 introns and are known to undergo alternative splicing giving rise to at least 2 major isoforms (Usiello et al., 2000; De Mei et al., 2009). There has also been controversy about whether D2Rs might form heteroreceptors with D1Rs in the cortex (Beaulieu and Gainetdinov, 2011; Zhang et al., 2011). Findings like these suggest that D2Rs exist in heterogeneous forms, which may explain how D2Rs can couple to various intracellular signaling pathways, giving rise to distinct effects on neuronal excitability, in different neuronal populations. It will be interesting for future studies to characterize the specific forms of D2Rs that are expressed in prefrontal SC-projecting neurons. In particular, there may be specific domains present in D2R isoforms expressed in prefrontal SC-projecting neurons, which enable coupling to the Gs pathways, D1Rs, or other effectors.
Functional implications
The phenomenon studied here is one whereby synaptic activity, even when subthreshold with respect to action potential generation in the postsynaptic neuron, can enhance neuronal excitability several hundreds of milliseconds or even up to 10 s later. This could enable postsynaptic neurons to integrate their inputs over relatively long durations. An alternative interpretation of this mechanism is that one set of inputs could switch neurons into a “high gain” mode whereby responses to subsequent inputs are potentiated, relative to the neuron's baseline state. These effects could help to generate patterns of prefrontal activity, e.g., the sequential patterns observed during the delay period of working memory or cued rule switching tasks (Bolkan et al., 2017; Schmitt et al., 2017). In these tasks, inhibiting prefrontal activity during the early portion of a delay disrupts subsequent sequential activity; early activity, if paired with D2R activation, may serve to “prime” the responses of neurons that fire later during the delay via this mechanism.
In this and previous studies (Gee et al., 2012), we have shown that optogenetically stimulating callosal inputs is sufficient to facilitate the D2R-dependent ADP. It will be interesting to explore whether other sources of inputs to mPFC can generate similar effects. We also have used trains of light flashes to stimulate synapses over periods of 2.5 s; it will be interesting to determine how this phenomenon depends on the duration of synaptic input. Finally, we have generally used patterns of synaptic stimulation that elicit EPSPs that are subthreshold for action potential generation in the postsynaptic neuron. An outstanding question is whether stronger patterns of stimulation are more or less effective for eliciting this phenomenon.
Clinical implications
Dopaminergic innervation of the PFC is disrupted in schizophrenia (Slifstein et al., 2015), mutations in D2Rs are associated with schizophrenia (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014), and all known clinically effective antipsychotics block D2Rs (Seeman, 2002). Together, these observations raise the possibility that prefrontal D2R function may be abnormal in schizophrenia, although the precise nature of such dysfunction and its relationship to symptoms remain largely unknown. As better models for schizophrenia are developed (e.g., based on neurons derived from patient iPS cells or mice with mutations in genetic loci strongly implicated in schizophrenia), it will be interesting to study whether this unusual form of prefrontal D2R signaling is altered.
In conclusion, D2Rs can enhance the excitability of subcortically projecting pyramidal neurons in layer 5 of mPFC through a pathway that depends on increased cAMP/PKA signaling. This increase in neuronal excitability also appears to require the activation of synaptic NMDARs and voltage-dependent Ca2+ influx during the preceding several seconds. Important next steps will be understanding how this phenomenon contributes to normal brain function and is disrupted in the setting of diseases, such as schizophrenia.
Footnotes
This work was supported by National Science Foundation Grant GRFP 1144247 to S.E.R., and National Institutes of Health Grant R01 MH100292 to V.S.S.
The authors declare no competing financial interests.
References
- Beaulieu JM, Gainetdinov RR (2011) The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 63:182–217. 10.1124/pr.110.002642 [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG (2005) An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell 122:261–273. 10.1016/j.cell.2005.05.012 [DOI] [PubMed] [Google Scholar]
- Bello EP, Mateo Y, Gelman DM, Noaín D, Shin JH, Low MJ, Alvarez VA, Lovinger DM, Rubinstein M (2011) Cocaine supersensitivity and enhanced motivation for reward in mice lacking dopamine D2 autoreceptors. Nat Neurosci 14:1033–1038. 10.1038/nn.2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT (1999) Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286:2495–2498. 10.1126/science.286.5449.2495 [DOI] [PubMed] [Google Scholar]
- Bolkan SS, Stujenske JM, Parnaudeau S, Spellman TJ, Rauffenbart C, Abbas AI, Harris AZ, Gordon JA, Kellendonk C (2017) Thalamic projections sustain prefrontal activity during working memory maintenance. Nat Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Hopf FW (2005) The dopamine D2 receptor: new surprises from an old friend. Neuron 47:335–338. 10.1016/j.neuron.2005.07.015 [DOI] [PubMed] [Google Scholar]
- Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T, Yocca FD, Molinoff PB (2002) Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J Pharmacol Exp Ther 302:381–389. 10.1124/jpet.102.033175 [DOI] [PubMed] [Google Scholar]
- Clarkson RL, Liptak AT, Gee SM, Sohal VS, Bender KJ (2017) D3 receptors regulate excitability in a unique class of prefrontal pyramidal cells. J Neurosci 37:5846–5860. 10.1523/JNEUROSCI.0310-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mei C, Ramos M, Iitaka C, Borrelli E (2009) Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr Opin Pharmacol 9:53–58. 10.1016/j.coph.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druzin MY, Kurzina NP, Malinina EP, Kozlov AP (2000) The effects of local application of D2 selective dopaminergic drugs into the medial prefrontal cortex of rats in a delayed spatial choice task. Behav Brain Res 109:99–111. 10.1016/S0166-4328(99)00166-7 [DOI] [PubMed] [Google Scholar]
- Durstewitz D, Seamans JK (2008) The dual-state theory of prefrontal cortex dopamine function with relevance to catechol-o-methyltransferase genotypes and schizophrenia. Biol Psychiatry 64:739–749. 10.1016/j.biopsych.2008.05.015 [DOI] [PubMed] [Google Scholar]
- Floresco SB, Magyar O, Ghods-Sharifi S, Vexelman C, Tse MT (2006) Multiple dopamine receptor subtypes in the medial prefrontal cortex of the rat regulate set-shifting. Neuropsychopharmacology 31:297–309. 10.1038/sj.npp.1300825 [DOI] [PubMed] [Google Scholar]
- Gee S, Ellwood I, Patel T, Luongo F, Deisseroth K, Sohal VS (2012) Synaptic activity unmasks dopamine D2 receptor modulation of a specific class of layer V pyramidal neurons in prefrontal cortex. J Neurosci 32:4959–4971. 10.1523/JNEUROSCI.5835-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnasko TS, Perez FA, Scouras AD, Stoll EA, Gale SD, Luquet S, Phillips PE, Kremer EJ, Palmiter RD (2006) Cre recombinase-mediated restoration of nigrostriatal dopamine in dopamine-deficient mice reverses hypophagia and bradykinesia. Proc Natl Acad Sci U S A 103:8858–8863. 10.1073/pnas.0603081103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, Malenka RC (2011) Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70:855–862. 10.1016/j.neuron.2011.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Malenka RC (2014) Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology 76:351–359. 10.1016/j.neuropharm.2013.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan A, Argilli E, Bonci A, Whistler JL (2013) Loss of D2 dopamine receptor function modulates cocaine-induced glutamatergic synaptic potentiation in the ventral tegmental area. J Neurosci 33:12329–12336. 10.1523/JNEUROSCI.0809-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major G, Polsky A, Denk W, Schiller J, Tank DW (2008) Spatiotemporally graded NMDA spike/plateau potentials in basal dendrites of neocortical pyramidal neurons. J Neurophysiol 99:2584–2601. 10.1152/jn.00011.2008 [DOI] [PubMed] [Google Scholar]
- Milojkovic BA, Radojicic MS, Antic SD (2005) A strict correlation between dendritic and somatic plateau depolarizations in the rat prefrontal cortex pyramidal neurons. J Neurosci 25:3940–3951. 10.1523/JNEUROSCI.5314-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minton GO, Young AH, McQuade R, Fairchild G, Ingram CD, Gartside SE (2009) Profound changes in dopaminergic neurotransmission in the prefrontal cortex in response to flattening of the diurnal glucocorticoid rhythm: implications for bipolar disorder. Neuropsychopharmacology 34:2265–2274. 10.1038/npp.2009.53 [DOI] [PubMed] [Google Scholar]
- Minzer K, Lee O, Hong JJ, Singer HS (2004) Increased prefrontal D2 protein in Tourette syndrome: a postmortem analysis of frontal cortex and striatum. J Neurol Sci 219:55–61. 10.1172/JCI32994 [DOI] [PubMed] [Google Scholar]
- Regard JB, Kataoka H, Cano DA, Camerer E, Yin L, Zheng YW, Scanlan TS, Hebrok M, Coughlin SR (2007) Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J Clin Invest 117:4034–4043. 10.1016/j.jns.2003.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt LI, Wimmer RD, Nakajima M, Happ M, Mofakham S, Halassa MM (2017) Thalamic amplification of cortical connectivity sustains attentional control. Nature 545:219–223. 10.1038/nature22073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P. (2002) Atypical antipsychotics: mechanism of action. Can J Psychiatry 47:27–38. [PubMed] [Google Scholar]
- Seong HJ, Carter AG (2012) D1 receptor modulation of action potential firing in a subpopulation of layer 5 pyramidal neurons in the prefrontal cortex. J Neurosci 32:10516–10521. 10.1523/JNEUROSCI.1367-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonic I, Gericke GS, Ott J, Weber JL (1998) Identification of genetic markers associated with Gilles de la Tourette syndrome in an Afrikaner population. Am J Hum Genet 63:839–846. 10.1086/302002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slifstein M, van de Giessen E, Van Snellenberg J, Thompson JL, Narendran R, Gil R, Hackett E, Girgis R, Ojeil N, Moore H, D'Souza D, Malison RT, Huang Y, Lim K, Nabulsi N, Carson RE, Lieberman JA, Abi-Dargham A (2015) Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 72:316–324. 10.1001/jamapsychiatry.2014.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steeves TD, Ko JH, Kideckel DM, Rusjan P, Houle S, Sandor P, Lang AE, Strafella AP (2010) Extrastriatal dopaminergic dysfunction in Tourette syndrome. Ann Neurol 67:170–181. 10.1002/ana.21809 [DOI] [PubMed] [Google Scholar]
- St Onge JR, Abhari H, Floresco SB (2011) Dissociable contributions by prefrontal D1 and D2 receptors to risk-based decision making. J Neurosci 31:8625–8633. 10.1523/JNEUROSCI.1020-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Lipska BK, O'Donnell P (2007) Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol Psychiatry 62:730–738. 10.1016/j.biopsych.2006.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, Jin J, Roth BL, O'Donnell P, Caron MG (2016) Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc Natl Acad Sci U S A 113:E8178–E8186. 10.1073/pnas.1614347113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usiello A, Baik JH, Rougé-Pont F, Picetti R, Dierich A, LeMeur M, Piazza PV, Borrelli E (2000) Distinct functions of the two isoforms of dopamine D2 receptors. Nature 408:199–203. 10.1038/35041572 [DOI] [PubMed] [Google Scholar]
- Wang M, Vijayraghavan S, Goldman-Rakic PS (2004) Selective D2 receptor actions on the functional circuitry of working memory. Science 303:853–856. 10.1126/science.1091162 [DOI] [PubMed] [Google Scholar]
- Winterer G, Weinberger DR (2004) Genes, dopamine and cortical signal-to-noise ratio in schizophrenia. Trends Neurosci 27:683–690. 10.1016/j.tins.2004.08.002 [DOI] [PubMed] [Google Scholar]
- Yoon DY, Gause CD, Leckman JF, Singer HS (2007) Frontal dopaminergic abnormality in Tourette syndrome: a postmortem analysis. J Neurol Sci 255:50–56. 10.1016/j.jns.2007.01.069 [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang H, Zhao H, Zhao C (2011) Calcium-related signaling pathways contributed to dopamine-induced cortical neuron apoptosis. Neurochem Int 58:281–294. 10.1016/j.neuint.2010.11.021 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Bertolino A, Fazio L, Blasi G, Rampino A, Romano R, Lee ML, Xiao T, Papp A, Wang D, Sadée W (2007) Polymorphisms in human dopamine D2 receptor gene affect gene expression, splicing, and neuronal activity during working memory. Proc Natl Acad Sci U S A 104:20552–20557. 10.1073/pnas.0707106104 [DOI] [PMC free article] [PubMed] [Google Scholar]