

Abstract
Purpose of the review
This review will provide a timely assessment of MAP kinase actions in bone development and homeostasis with particular emphasis on transcriptional control of the osteoblast lineage
Recent findings
ERK and p38 MAP kinases function as transducers of signals initiated by the extracellular matrix, mechanical loading, TGF-β, BMPs and FGF2. MAPK signals may also affect and/or interact with other important pathways such as WNT and HIPPO. ERK and p38 MAP kinase pathways phosphorylate specific osteogenic transcription factors including RUNX2, Osterix, ATF4 and DLX5. For RUNX2, phosphorylation at specific serine residues initiates epigenetic changes in chromatin necessary for decondensation and increased transcription. MAPK also suppresses marrow adipogenesis by phosphorylating and inhibiting PPARγ, which may explain the well-known relationship between reduced skeletal loading and marrow fat accumulation.
Summary
MAPKs transduce signals from the extracellular environment to the nucleus allowing bone cells to respond to changes in hormonal/growth factor signaling and mechanical loading thereby optimizing bone structure to meet physiological and mechanical needs of the body.
Keywords: MAP kinase, transcription, osteoblast, chromatin, RUNX2
Introduction
The mitogen-activated protein kinase (MAPK) pathways function as important regulators of cell growth, differentiation and morphogenesis in most tissues including bone. Extracellular signal-regulated kinases (ERK), p38 kinases and c-June N-terminal kinases (JNK) constitute the three main classes of MAP kinases to be discussed in this review. Canonical components of the ERK pathway are Ras, RAF, the MAP kinase kinases, MEK1 and MEK2, and the terminal MAP kinases, ERK1 and ERK2. Intermediates of the p38 pathway are MAP kinase kinase 3 and 6 (MKK3, MKK6) and p38 α, β, γ and δ. JNK pathway components are MAP kinase kinase 4 and 7 and JNK1 and 2. Because MAPKs are activated by a wide variety of factors including growth factors, morphogens, extracellular matrix (ECM) components and biomechanical signals, they have the potential to mediate the skeletal response to both internal and external environmental cues. This article will specifically focus on possible mechanisms used by MAPKs to mediate bone responses to ECM and other signals as well as delve into potential control mechanisms and the major intracellular targets, tissue-specific transcription factors. This work is not intended to be comprehensive and instead reflects the unique perspective of the authors. For a more comprehensive treatment of this subject in bone and other tissues, the reader is referred to other recent reviews(1, 2).
MAPK Signaling in Skeletal Development
The MAPKs are among the most fundamental signal transduction pathways in biology, being present in all eukaryotes including yeasts, plants, insects and vertebrates. They are required at the earliest stages of vertebrate development including initial animal cap differentiation to mesenchyme in Xenopus laevis embryos and formation of primary mesenchyme in mice(3, 4). In addition, JNK1 and JNK2 are required for development of the neural tube while ERK, p38 and JNK all participate in formation of various components of the immune system(5, 6). In skeletal development, clear in vivo roles have been established for both ERK and P38 MAPK pathways, both of which will be discussed in detail.
Transgenic overexpression of a constitutively-active form of the ERK/MAPK intermediate, Mek1, in osteoblasts using the osteoblast-specific Bglap2 promoter accelerates formation of the both cranial and appendicular skeletons while a dominant-negative Mek1 slows development(7). Epistasis was demonstrated between Mek1 transgene activity and the osteoblast-related transcription factor, RUNX2, thereby providing strong evidence that major aspects of the MAPK response are mediated by this factor (see below). Specifically, crossing mice expressing constitutively-active MEK1 with Runx2+/− mice partially rescued the hypoplastic clavicles and hypomineralized calvaria characteristic of Runx2 haploinsufficiency in mice and humans(8) while crossing dominant-negative Mek1 mice with Runx2+/− animals exacerbated clavicular hypertrophy and calvarial hypomineralization resulting in embryonic lethality(7). Consistent with these results, calvarial osteoblasts isolated from transgenic mice showed the expected changes in differentiation with cells from dominant-negative Mek1 mice differentiating less than cells from wild type littermates and cells from constitutively-active Mek1 mice showing enhanced differentiation. Similarly, in loss of function studies Prx1-cre-mediated inactivation of Erk2 in mesenchymal cells of the appendicular skeleton in Erk1-null mice blocked osteoblast differentiation leading to ectopic cartilage formation in regions of the perichondrium that normally form bone. Furthermore, increased ERK/MAPK signaling in the same cell population increased osteoblast differentiation and inhibited chrondrogenesis(9). In these mice, osteoclast numbers are also reduced, consistent with ERK also being important for RANKL induction in both hypertrophic chondrocytes and osteoblasts, possibly via a RUNX2-dependent mechanism. This may explain the inability of these mice to remove hypertrophic chondrocytes to make way for new bone formation.
A important role for ERK/MAPK signaling was also identified in FGF-mediated cranial suture fusion. FGF ligands, particularly FGF8, are necessary for normal growth and development of craniofacial structures(10). Gain of function mutations in FGFR2 cause certain forms of craniosynostosis (premature suture fusion) including Apert syndrome and Crouzon syndrome(11). Activated FGFRs signal through the ERK/MAPK pathway leading to elevated levels of P-ERK1/2(12). In mice harboring the Fgfr2S252W mutation, which is causal for Apert syndrome, inhibition of ERK phosphorylation with the specific inhibitor, U0126, or with an shRNA specific to mutant Fgfr2, can block pathological suture fusion(13). These studies strongly support a model wherein FGFR2 stimulates suture fusion via activation of the ERK/MAPK pathway.
The p38 MAPK pathway is also required for osteoblast differentiation where it functions as a down-stream signal activated by the TGF-β and BMP responsive kinase, TAK1(14, 15). Conditional deletion of Tak1 in preosteoblasts using an Osx-cre leads to reduced cortical and trabecular bone, clavicular hypoplasia and delayed fontanelle fusion. Effects of TAK1 deficiency were specifically attributed to reduced p38 signaling in that mice deficient in the p38 intermediates, Mkk3, Mkk6, p38α or p38β all have decreased bone mass(14). Interestingly, some selectivity was seen in the requirement for p38α or p38β in bone development with p38β-deficient mice showing defects in long bone formation without major effects on calvarial development. The phenotype of Tak1-deficient mice is similar to that observed with Runx2 haploinsufficiency, which suggest that p38 may regulate RUNX2 (see below).
Involvement of the JNK pathway in bone development has not been extensively examined although several studies suggest a role for this pathway in osteoblast differentiation. For example, accumulation of MEKK2 as a consequence of Smurf1 inactivation leads to JNK activation and increased bone mass while overexpression of a constitutively-active JNK1 increases in vitro osteoblast differentiation(16). Also, the craniosynostosis-associated factor, Nell-1, activates JNK1 and ERK1/2 in calvarial osteoblasts and is associated with enhanced differentiation(17). However, the interpretation of these studies is complicated by the known role of JNK signaling in cell survival and apoptosis(5).
MAPK Targets
All MAPKs are serine/threonine kinases with a broad range of substrates. Of specific relevance to bone is the observation that a number of osteoblast-related transcriptions factors can be phosphorylated by ERK and/or p38 MAPKs.
RUNX2
The first MAPK target to be identified was RUNX2, an essential transcription factor for osteoblast and hypertrophic chondrocyte development(18–20). This discovery arose from initial observations showing that the electrophoretic mobility of RUNX2 was slightly altered in differentiating osteoblast cultures. Changes in mobility were correlated with increased binding of RUNX2 to DNA and increase ability of RUNX2 to stimulate transcription in the absence of changes in RUNX2 protein levels(21, 22). Because RUNX2 activation was dependent on de novo collagen synthesis and binding of type I collagen to α2β1 integrins, the author’s laboratory postulated that RUNX2 is phosphorylated and activated by the ERK MAPK pathway, a known mediator of integrin signaling(21–25). Subsequent mass spectroscopy and related analysis established RUNX2 as a direct ERK1/2 target that is phosphorylated at Ser43, Ser301, Ser319 and Ser510 (murine Type II RUNX2 isoform). Of these, Ser301 and Ser319 were most important for MAPK-dependent transcriptional activation of osteoblast related genes such as Bglap2, Ibsp and Alpl(26). Significantly, Ser to Ala mutations of individual phosphorylation sites had little effect on transcriptional activity. Only when combined S301A/S319A mutations were examined was a major drop in MAPK-stimulated transcription observed. In addition, ERK was shown to directly bind RUNX2 using a consensus ERK docking “D” site between amino acid residues 201–215 of the runt DNA-binding domain(27). This site is similar to those for other MAPK-responsive transcription factors in that it is immediately N-terminal to a transcription activation domain, called the proline/serine/threonine-rich (PST) domain in RUNX2, which also contains the critical Ser301 and Ser319 phosphorylation sites(28). Although initial studies focused on ECM/integrin-mediated activation of ERK/MAPK and RUNX2 phosphorylation, other factors known to signal through MAPK such as FGF2 and BMPs were subsequently shown to also increase RUNX2 phosphorylation and activity(29–31).
Relationship between RUNX2 Phosphorylation and Chromatin Remodeling
Interestingly, binding of P-ERK to RUNX2 occurs on the chromatin of target genes rather than in the cytoplasm or other nuclear/perinuclear sites(32). In undifferentiated preosteoblasts, RUNX2 is already present in the nucleus bound to specific enhancer regions of Ibsp and Bglap2. Once cells are exposed to differentiating conditions, ERK translocates from perinuclear to nuclear sites where it is detected in complex with chromatin-associated RUNX2. The binding of P-ERK to chromatin requires RUNX2 and intact RUNX2-binding enhancer sequences in both genes. In subsequent work, nuclear translocation of P-ERK was shown to be tightly correlated with appearance of chromatin-associated Ser319 P-RUNX2, p300/CBP (CREB binding protein) and RNA polymerase II as well as specific changes in histone acetylation and methylation(33). The following differentiation-dependent histone changes were measured: increased H3K9 and H4K5 acetylation and H3K4 di-methylation, histone marks associated with transcriptional activation, and decreased H3K9 mono-, di- and tri-methylation, histone marks associated with transcriptional repression(34). Significantly, most of these chromatin changes as well as RUNX2-dependent transcription were blocked by MAPK inhibition or by mutation of Ser301 and Ser319 residues in RUNX2 to alanine. These results support a model wherein ERK-dependent RUNX2 phosphorylation plays a pivotal role in recruiting chromatin-modifying factors such as p300/CBP to gene enhancers thereby allowing the epigenetic changes necessary for transcription and osteoblast differentiation. Consistent with this model, genetic interactions have been demonstrated between RUNX2, p300/CBP and its binding partner, CREB in the maintenance of bone mass(35).
It should be noted that the full repertoire of factors recruited to chromatin after RUNX2 phosphorylation is still not known. The observed increase in chromatin-associated p300/CBP, which contains histone acetyltransferase (HAT) activity, may explain some of the observed increases in histone acetylation(36). Other changes in acetylation may be due to inhibition of histone deacetylases (HDACs) such as HDACs 3, 7 and 8, which are known to affect bone formation and osteoblast activity(37, 38). Similarly, the basis for changes in histone methylation remain to be explored. KDM4B, which demethylates trimethyl H3K9, and KDM6B, which demethylates H3K27, were shown to be required for osteoblast differentiation of mesenchymal cells(39), but their relationship to RUNX2 phosphorylation has not been examined.
Thus far, effects of MAPK-dependent RUNX2 phosphorylation on chromatin have only been examined in the proximal promoter regions of two osteoblast-related genes (Bglap2 and Ibsp). However, recent ChIP-Seq analysis suggests that the changes observed may be generally associated with RUNX2-dependent transcriptional activation(40–42). In all these studies, chromatin-associated RUNX2 was widely distributed throughout the genome; 30 percent of bound RUNX2 was in the promoter regions of putative target genes and 70 percent in nonpromoter regions including intron, exon and intergenic regions. Significantly, in differentiated osteoblasts where RUNX2 is likely in the phosphorylated state, bound RUNX2 was generally near chromatin regions enriched in Me2H3K4 and AcH3K9 and AcH4K5, the same histone marks shown to be dependent on MAPK activity and RUNX2 phosphorylation(33). Based on this work, it may be inferred that MAPK-dependent RUNX2 phosphorylation at Ser301 and Ser319 has broad effects on the genome leading to the accumulation of activating histone marks in genes necessary for osteoblast differentiation and function. Through this pathway, extracellular signals can be broadly delivered throughout the genome to alter global patterns of gene expression.
The association of ERK with RUNX2 on target gene chromatin is a specific example of a more general concept where tissue-specific transcription factors can serve as docking sites for nuclear kinases in progenitor cells of various lineages. Subsequent transcription factor phosphorylation can then direct a tissue-specific pattern of gene expression necessary for differentiation or response to specific stimuli. Examples of this include the association of p38α/β with MyoD and MEF2 on the chromatin of muscle-related genes, which are necessary for myoblast differentiation, the binding of P-ERK to Beta2, MafA and PDX-1 transcription factors in pancreatic β cells in response to elevated glucose and binding of yeast homologues of p38, ERK and PKA with the chromatin of multiple genes to globally alter patterns of gene expression in response to external stimuli such as osmotic stress, pheromones or glucose(43–46).
ERK1/2 and p38 Interactions in the Control of RUNX2 Activity
Consistent with the role of p38 in bone formation described above, RUNX2 is also a substrate for p38α and β, which phosphorylate at Ser31, Ser254 and Ser319 (14, 31). As was the case with ERK1/2 phosphorylation, separate mutation of each site had a minimal effect on transcriptional activity while combined mutation blocked most p38-dependent transcription. Also like ERK phosphorylation, p38 increased binding of RUNX2 to p300/CBP, which suggests that both MAPKs activate RUNX2 through a similar mechanism involving phosphorylation on shared as well as separate serine residues.
In spite of these similarities, more recent evidence suggests that ERK and p38 may be responsible for different osteoblast responses(31). Specifically, although ERK1, p38α and p38β all stimulate RUNX2 Ser319 phosphorylation and transcriptional activity, direct comparisons indicated that ERK1 was the most active of the three kinases. Also, although ERK and p38 bind RUNX2 through a common MAPK D site, the apparent affinity of this site for ERK is greater than for p38. Lastly, RUNX2 Ser319 phosphorylation and osteoblast differentiation whether measured in calvarial organ cultures, primary osteoblasts or osteoblast cell lines, is preferentially sensitive to ERK, but not p38 inhibition. In contrast, p38 inhibition does partially inhibit BMP2/7-dependent differentiation/gene expression, although this response is not accompanied by a reduction in RUNX2 Ser319 phosphorylation. These results suggests involvement either of other RUNX2 phosphorylation sites or p38 regulation of other nuclear factors and are consistent with p38 preferentially mediating TGF-β/BMP responses likely through the MAPK kinase kinase, TAK1(14).
Effects of MAPK Phosphorylation on RUNX2 Stability
Although the increased osteoblast gene expression associated with ERK or p38-dependent RUNX2 phosphorylation has been attributed to P-RUNX2 effects on nuclear factor recruitment and transcription, phosphorylation may also stabilize RUNX2 by facilitating acetylation and subsequent resistance to proteosomal degradation(47–49). Specifically, BMP2 or FGF2 can both increase p300-dependent RUNX2 acetylation which renders RUNX2 resistant to Smurf-1 mediated ubiquitination and proteosomal degradation. Both BMP2 and FGF2 increase ERK signaling, which is required for subsequent RUNX2 stabilization. In the case of FGF2, ERK activation requires the RUNX2 Ser319 phosphorylation site(49).
Other MAPK Targets
Both p38 and ERK have additional substrates that affect osteoblast differentiation using either direct or indirect mechanisms of action.
In the first category are the transcription factors, Osterix (OSX or SP7) and DLX5, which are substrates for p38. Osterix is a bone-specific transcription factor functioning downstream of RUNX2. It is necessary for overt osteoblast differentiation, but unlike RUNX2, it is not required for cartilage hypertrophy(50). In addition to being indirectly induced by BMP2 at the mRNA level, it is activated and phosphorylated by p38(51–53). Furthermore, in MKK6-transfected cells, OSX was phosphorylated at Ser73 and Ser77. Phosphorylation at these sites was necessary for recruitment of RNA polymerase II, p300 and Brg-1 to promoter regions of Ibsp and Fmod genes as well transcriptional activation(51). The second transcription factor, DLX5, is preferentially associated with craniofacial development; Dlx5-deficient mice have defects in cranial ossification with only minor changes in the axial and appendicular skeletons(54). DLX5 regulates expression of osteoblast-related genes like Ibsp and Sp7 by directly binding homeobox sequences in the promoter region(53, 55). DLX5 is induced by BMP2 with a time course that precedes induction of osteoblast markers including SP7 and IBSP. DLX5-dependent transcriptional activity is dependent on p38-dependent phosphorylation at Ser34 and Ser217(53). As suggested by Greenblatt and coworkers, it is possible that the preferential effects of p38α deficiency on craniofacial versus long bone mineralization may be explained by selective phosphorylation of DLX5 by this MAPK although the specific p38 (α or β) phosphorylating DLX5 has not been determined(1).
MAPK substrates that regulate osteoblast differentiation through indirect mechanisms include RSK2, ERF and GSK-3β. RSK2, a protein kinase A/protein kinase G/protein kinase C (AGC) family member, is activated by ERK phosphorylation(56). RSK2 then phosphorylates and activates ATF4, an osteoblast-enriched transcription factor and mediator of the unfolded protein response that is critical for proper collagen production in mature osteoblasts(57, 58). Activation of MAPK signaling by disruption of the Ras GTPase activating protein (RasGAP), neurofibromin (NF1), in mature osteoblasts increases ATF4 phosphorylation and transcriptional activity as well as bone mass in mice(59). Paradoxically, neurofibromatosis patients with germline loss-of-function mutations in NF1, have a low bone mass phenotype(60). As was subsequently shown, this discrepancy may be explained by the stage at which Nf1 is disrupted since crossing mice harboring a conditional Nf1 allele with a Col2α1-Cre expressed in cartilage and osteochondroprogenitor cells of bone marrow resulted in mice with reduced bone mass, osteoblast number and increased resorption(61). Thus, NF1 can function in a stage-specific manner to either suppress or stimulate bone mass and this response is mediated by ATF4. The ETS family transcription factor, ERF, is an additional ERK1/2 nuclear target that may function to suppress RUNX2 activity(62). Loss-of-function and hypomorphic mutations in Erf cause craniosynostosis in humans and mice due to accelerated cranial suture fusion. ChIP-seq analysis revealed frequent localization of RUNX2 binding motifs within ERF target genes, suggesting possible interactions between these two factors. It was further shown that ERF could inhibit RUNX2 transcriptional activity by interacting with a hybrid RUNX2-ETS binding site. Since nuclear export of ERF is stimulated by ERK phosphorylation(63), the MAPK pathway was proposed to stimulate osteogenesis by reversing ERF inhibition of RUNX2. This pathway may work in consort with direct ERK phosphorylation and activation of RUNX2 to stimulate osteogenesis. A final MAPK target that indirectly regulates osteoblast differentiation is GSK-3β. In the absence of WNT signaling, GSK-3β is a component of an inhibitory complex that phosphorylates β-catenin to target it for proteasome-mediated degradation. The ERK/MAPK pathway indirectly activates the WNT pathway by phosphorylating and inactivating GSK-3β (Serine9 phosphorylation) thereby allowing nuclear translocation of β-catenin and activation of osteoblast gene expression(64). This ERK-dependent activation of the WNT pathway is negatively regulated by the scaffolding protein, Schnurri-3 (SHN3), which selectively inhibits the phosphorylation and inactivation of GSK-3β by ERK, to inhibit WNT signaling(65). Consistent with this model, Shn3-deficient mice have a high bone mass sclerotic phenotype that is partially rescued by crossing with Lrp5−/− mice.
MAPKs and the Response of Skeletal Progenitor Cells to Mechanical Stimuli
Bone has the remarkable ability to alter its structure in response to changes in mechanical loading. Well-known examples of this are the increase in bone mass associated with weight-bearing exercise and, conversely, loss of bone after prolonged bed rest or exposure to microgravity during space flight(66–69). Loading stimulates a large number of systemic responses in bone including activation of WNT, BMP and nitric oxide signaling (for review, see Knapik et al.(70). However, this review will selectively focus on cellular responses to load with emphasis on the role of MAPK signaling.
Both dynamic and static loads can alter the differentiation of skeletal progenitor cells in bone marrow. For example, when mice are exposed to low-magnitude mechanical signals, marrow stromal cells (MSCs), which include skeletal progenitors, preferentially differentiate to osteoblasts while adipocyte differentiation is suppressed(71). In contrast, skeletal unloading of mice stimulates marrow adipogenesis and decreases bone formation(72). This is also seen in paraplegic patients whose disuse osteopenia is associated with increased marrow fat(73). Some of these responses to loading are also seen when MSCs are exposed to biaxial mechanical strain or fluid shear in vitro, leading to inhibition of adipocyte differentiation and stimulation of osteoblastogenesis(74–76). Variation of static loads (tension) on cells also profoundly affects MSC differentiation. This is accomplished by altering adhesive surface and cell spreading or through the use of synthetic scaffolds of varying stiffness(77–80). In all cases, the same general trends prevail; high static loads favor differentiation to osteoblasts while progressively decreasing loads favor cartilage, then muscle and, finally, adipocyte differentiation.
One of the primary mechanisms used by cells to detect mechanical forces is through the integrin-mediated activation of focal adhesion kinase (FAK), a component of focal adhesions. These multiprotein structures link the extracellular matrix, primarily composed of type I collagen in bone, with the actin cytoskeleton and act as force transducers to match intracellular contractility generated by the cytoskeleton with extracellular tension provided by the ECM. The main integrins responsible for the binding of bone cells to type I collagen are α1 β1, α2β1 and α11β1 although other integrins such as α5β1 may also be involved in mechanotransduction(81, 82). Exposure of integrins to mechanical force activates FAK and Src kinases and stimulates RhoA and Rho-associated coiled-coil containing protein kinase (ROCK). Rho/ROCK signaling then stimulates strengthening of the actin-myosin cytoskeleton resulting in cell stiffening (reviewed in (83)). FAK also activates ERK, p38 and JNK MAP kinases as well as phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathways followed by induction of mechanoresponsive genes and cell differentiation(84–86). Consistent with its role in mechanoresponsiveness, conditional deletion of FAK in osteoblasts using a Col1a1-Cre renders mice resistant to the local effects of mechanical loading(87).
FAK-mediated activation of ERK/MAPK signaling and subsequent transcription factor phosphorylation are likely responsible for at least some of the observed loading-dependent shift in MSCs differentiation from adipocytes to osteoblasts. The transcription factors, RUNX2 and PPARγ, are two critical regulators of this process. As recently shown, MAPK-dependent phosphorylation of RUNX2 and PPARγ in MSCs can simultaneously increase osteoblast differentiation and suppress adipogenesis(88). This is accomplished using the two activating phosphorylation sites in RUNX2 described above (i.e. Ser301 and Ser319) and a single inhibitory MAPK phosphorylation site in PPARγ (Ser112)(89, 90). Mechanical stimulation of osteoblasts by exposure to fluid flow shear stress rapidly (i.e. within minutes) increases P-ERK-dependent phosphorylation of RUNX2 at Ser301 and Ser319 on target gene chromatin and this phosphorylation is necessary for subsequent induction of histone acetylation and transcription(91). It is not currently known if P-ERK also phosphorylates PPARγ on the chromatin of adipocyte genes versus at other nuclear or cytoplasmic sites.
Interestingly, the phosphatase, PP5, was recently shown to reverse effects of P-ERK phosphorylation on RUNX2 and PPARγ(92). By preferentially dephosphorylating both transcription factors at their respective ERK phosphorylation sites, PP5 suppresses RUNX2 and stimulates PPARγ transcriptional activity. Consistent with these actions, mice deficient in Pp5 have increased osteoblast numbers, high rates of bone formation, increased bone mass and decrease marrow fat. In the presence of the PPARγ agonist, rosiglitazone, PP5 translocates to the nucleus where it binds RUNX2 and PPARγ and dephosphorylates both factors to promote marrow adipogenesis and inhibit bone formation. Interestingly, Pp5 deficiency blocks the negative effects of rosiglitazone on the skeleton.
YAP (Yes-associated protein) and TAZ (transcriptional co-activator with PDZ-binding motif) are two additional mediators of cellular responses to static and dynamic loads that function downstream of Rho/ROCK activation (for reviews, see(93, 94)). Both factors exist in a phosphorylated, inactive form in the cytoplasm following phosphorylation by the Hippo pathway kinases, LATS1/LATS1, and in an active dephosphorylated nuclear form. Through a mechanism that is not completely understood, the Rho/ROCK-mediated cytoskeletal stiffening prevents YAP/TAZ phosphorylation and promotes nuclear translocation. Once in the nucleus, YAP and TAZ interact with several transcription factors including TEA domain (TEAD) factors, T-box 5 (TBX5) and, interestingly, RUNX2 and PPARγ(95). On complexing with RUNX2, TAZ stimulates transcriptional activity; in contrast, TAZ inhibits PPARγ. While being highly dependent on Rho/ROCK activity and cytoskeletal reorganization, nuclear translocation of YAP/TAZ is surprisingly independent of FAK (96). Thus, FAK-mediated MAPK activation and subsequent regulation of RUNX2 and PPARγ activities and YAP/TAZ nuclear translocation can be viewed as separate arms of a coordinated cellular response to mechanic loads. Nevertheless, both pathways must be active for stimulation of osteoblast differentiation and suppression of adipogenesis since inhibition of either Rho/ROCK or ERK blocks osteoblast differentiation of MSCs grown of stiff hydrogels(79, 80, 97). Furthermore, there is apparent cross-talk between MAPK and Rho/ROCK signaling since ERK or JNK inhibitors can block TAZ nuclear localization(97).
Discoidin Receptors and Bone
Although integrins/focal adhesions are generally considered to be responsible for sensing mechanical signals in bone, this tissue contains other adhesion receptors including cadherins, syndecans, hyaluronan receptors, and discoidin receptors (for review, see(98)). With regard to MAPK signaling, the discoidin receptors are of particular interest. Unlike integrins, these molecules have intrinsic tyrosine kinase activity that is activated by binding to fibrillar collagens. Once activated, DDRs can stimulate several signal transduction pathways that, depending on the tissue, include ERK, JNK and p38 MAPKs, the PI-3 kinase/AKT and NFKβ pathways (for review, see(99)). The two DDRs in mammals, DDR1 and DDR2, have different tissue distributions and are preferentially activated by different collagens. DDR1 is mainly expressed by epithelium and has broad ligand specificity for most collagens while DDR2 is expressed by mesenchymal cells including osteoblasts and preferentially binds collagens I, II, III and X. Consistent with its distribution and collagen selectivity, DDR2 has important functions in bone. Loss-of-function mutations in DDR2 cause spondylo-meta-epiphyseal dysplasia (SMED) in humans, resulting in dwarfism, bone weakness, abnormal calcifications and craniofacial abnormalities(100). Furthermore, polymorphisms in the DDR2 locus are associated with increased fracture risk and low bone density (101). DDR2-deficient mice have SMED-like characteristics including dwarfism and reduced bone mineral density(102–104). Detailed analysis of the bone phenotype of mice globally deficient in DDR2 revealed dramatic reductions in mineral density in bones of the appendicular, axial and cranial skeletons in males and females that were explained by reduced osteoblast activity and bone formation in the absence of changes in resorption(104). Bone changes were accompanied by a large increase in marrow fat. Furthermore, MSCs from deficient mice showed reduced ability to differentiate into osteoblasts and increased adipogenesis. The defective osteoblast differentiation in DDR2-deficient cells was directly attributed to a reduction in ERK/MAPK signaling and RUNX2 Ser301 Ser319 phosphorylation and could be rescued with a phosphomimetic RUNX2 mutant (S301E/S319E mutant) that did not require phosphorylation for optimal activity. Consistent with these results, overexpression of a constitutively-active DDR2 stimulated ERK/MAPK signaling and phosphorylation of RUNX2 and PPARγ leading to increased RUNX2-dependent transcriptional activity and inhibition of PPARγ. Similar effects of DDR2 on the MAPK/RUNX2 axis were previously reported in studies with osteoblast and chondrocyte cell lines(105).
Summary and Conclusions
Many factors important in bone metabolism including growth factors, TGF-β/BMPs, extracellular matrix and mechanical loads stimulate MAP kinase signaling. In vivo gain and loss of function studies established clear roles for ERK and p38 MAP kinases in osteoblast differentiation. Both kinases largely act by directly phosphorylating and activating osteoblast related transcription factors including RUNX2, OSX and DLX5. In addition, they can function indirectly by activating secondary kinases such as RSK2 to stimulate osteoblast gene expression, or, alternatively, to inhibit the activity of factors like ERF and GSK-3β that themselves directly or indirectly suppress osteoblast activity. During MSC differentiation, ERK can simultaneously phosphorylate RUNX2 and PPARγ to stimulate osteoblast differentiation and suppress adipogenesis. Static and dynamic mechanical loads are detected in skeletal progenitor cells through focal adhesions and activation of focal adhesion kinase and subsequent activation of Src, Rho and ROCK activities. The Rho/ROCK-dependent stiffening of the actin-myosin cytoskeleton promotes dephosphorylation and nuclear translocation of YAP and TAZ while FAK activates ERK, p38, JNK and PI3 kinase. These two pathways may cooperate to stimulate osteoblast gene expression and suppress adipogenesis. DDR2 is a non-integrin receptor tyrosine kinase activated by fibrillar collagens including the type I collagen of bone that provides a second route for the ECM to activate MAPK signaling leading to increased osteoblast differentiation and suppression of adipogenesis. The nuclear functions of MAP kinases in bone are likely explained by their direct binding to and phosphorylation of tissue-specific transcription factors such as RUNX2 on the chromatin of target genes. This and related phosphorylation events stimulate recruitment of chromatin modifying factors leading to the epigenetic modification of histones necessary for osteoblast gene expression while also suppressing other lineages such as adipocytes. This provides a mechanism whereby an extracellular signal can be conveyed to the nucleus via translocation of MAPKs, which can then be targeted to all relevant genes necessary for specific control of mesenchymal stem cell lineage to osteoblasts (Figure 1 uses the specific example of RUNX2-dependent transcription to illustrate these control mechanisms).
Figure 1. Model for MAP Kinase Regulation of Osteoblast Gene Expression with emphasis on RUNX2-dependent transcription.
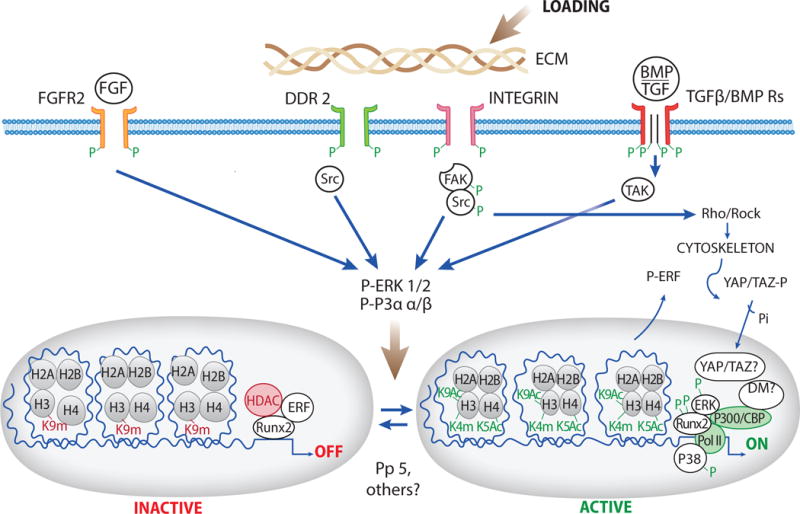
Major pathways in bone that signal at least in part through MAPKs are shown in top part of figure. Bottom of figure shows specific chromatin/histone modifications associated with ERK (and possibly p38)-dependent RUNX2 phosphorylation. Both stimulatory changes (H3K9Ac, H4K5Ac and H3K4m, YAP/TAZ dephosphorylation and nuclear translocation) as well as removal of inhibitory chromatin modifications/factors (decreased H3K9Me and increased DM activity, decreased HDAC activity, ERF-P/translocation to cytoplasm) are shown. The question mark next to nuclear YAP/TAZ and histone demethylase (DM) reflects uncertainty as to whether their association with chromatin is related to MAPK signaling.
Human and Animal Rights and Informed Consent.
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Footnotes
Compliance with Ethical Standards
Renny T. Franceschi and Chunxi Ge each declare no potential conflicts of interest.
Literature Cited
- 1*.Greenblatt MB, Shim JH, Glimcher LH. Mitogen-Activated Protein Kinase Pathways in Osteoblasts. Annu Rev Cell Dev Biol. 2013;29:2.1–2.17. doi: 10.1146/annurev-cellbio-101512-122347. A comprehensive review of MAPK functions in bone. [DOI] [PubMed] [Google Scholar]
- 2.Wellbrock C, Arozarena I. The Complexity of the ERK/MAP-Kinase Pathway and the Treatment of Melanoma Skin Cancer. Front Cell Dev Biol. 2016;4:33. doi: 10.3389/fcell.2016.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Umbhauer M, Marshall CJ, Mason CS, Old RW, Smith JC. Mesoderm induction in Xenopus caused by activation of MAP kinase. Nature. 1995;376(6535):58–62. doi: 10.1038/376058a0. [DOI] [PubMed] [Google Scholar]
- 4.Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, et al. Extracellular signalregulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci USA. 2003;100(22):12759–64. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22(4):667–76. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- 6.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 7.Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol. 2007;176(5):709–18. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia [see comments] Cell. 1997;89(5):773–9. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 9.Matsushita T, Chan YY, Kawanami A, Balmes G, Landreth GE, Murakami S. Extracellular signal-regulated kinase 1 (ERK1) and ERK2 play essential roles in osteoblast differentiation and in supporting osteoclastogenesis. Mol Cell Biol. 2009;29(21):5843–57. doi: 10.1128/MCB.01549-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Creuzet S, Schuler B, Couly G, Le Douarin NM. Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proc Natl Acad Sci U S A. 2004;101(14):4843–7. doi: 10.1073/pnas.0400869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Passos-Bueno MR, Serti Eacute AE, Jehee FS, Fanganiello R, Yeh E. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol. 2008;12:107–43. doi: 10.1159/000115035. [DOI] [PubMed] [Google Scholar]
- 12.Tsang M, Dawid IB. Promotion and attenuation of FGF signaling through the Ras-MAPK pathway. Sci STKE. 2004;2004(228):pe17. doi: 10.1126/stke.2282004pe17. [DOI] [PubMed] [Google Scholar]
- 13.Shukla V, Coumoul X, Wang RH, Kim HS, Deng CX. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39(9):1145–50. doi: 10.1038/ng2096. [DOI] [PubMed] [Google Scholar]
- 14.Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. 2010;120(7):2457–73. doi: 10.1172/JCI42285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shim JH, Greenblatt MB, Xie M, Schneider MD, Zou W, Zhai B, et al. TAK1 is an essential regulator of BMP signalling in cartilage. EMBO J. 2009;28(14):2028–41. doi: 10.1038/emboj.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita M, Ying SX, Zhang GM, Li C, Cheng SY, Deng CX, et al. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell. 2005;121(1):101–13. doi: 10.1016/j.cell.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang X, Ting K, Bessette CM, Culiat CT, Sung SJ, Lee H, et al. Nell-1, a key functional mediator of Runx2, partially rescues calvarial defects in Runx2(+/−) mice. J Bone Miner Res. 2011;26(4):777–91. doi: 10.1002/jbmr.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 19.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 20.Enomoto H, Enomoto-Iwamoto M, Iwamoto M, Nomura S, Himeno M, Kitamura Y, et al. Cbfa1 is a positive regulatory factor in chondrocyte maturation. J Biol Chem. 2000;275(12):8695–702. doi: 10.1074/jbc.275.12.8695. [DOI] [PubMed] [Google Scholar]
- 21.Xiao G, Wang D, Benson MD, Karsenty G, Franceschi RT. Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J Biol Chem. 1998;273(49):32988–94. doi: 10.1074/jbc.273.49.32988. [DOI] [PubMed] [Google Scholar]
- 22.Xiao G, Cui Y, Ducy P, Karsenty G, Franceschi RT. Ascorbic acid-dependent activation of the osteocalcin promoter in MC3T3-E1 preosteoblasts: requirement for collagen matrix synthesis and the presence of an intact OSE2 sequence. Mol Endocrinol. 1997;11(8):1103–13. doi: 10.1210/mend.11.8.9955. [DOI] [PubMed] [Google Scholar]
- 23.Xiao G, Jiang D, Thomas P, Benson MD, Guan K, Karsenty G, et al. MAPK pathways activate and phosphorylate the osteoblast-specific transcription factor, Cbfa1. J Biol Chem. 2000;275(6):4453–9. doi: 10.1074/jbc.275.6.4453. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi Y, Suzawa M, Kikuchi T, Nishida E, Fujita T, Matsumoto T. Differentiation and transforming growth factor-beta receptor down-regulation by collagen-alpha2beta1 integrin interaction is mediated by focal adhesion kinase and its downstream signals in murine osteoblastic cells. J Biol Chem. 1997;272(46):29309–16. doi: 10.1074/jbc.272.46.29309. [DOI] [PubMed] [Google Scholar]
- 25.Franceschi RT, Iyer BS. Relationship between collagen synthesis and expression of the osteoblast phenotype in MC3T3-E1 cells. J Bone Miner Res. 1992;7(2):235–46. doi: 10.1002/jbmr.5650070216. [DOI] [PubMed] [Google Scholar]
- 26.Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. 2009;284(47):32533–43. doi: 10.1074/jbc.M109.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akella R, Moon TM, Goldsmith EJ. Unique MAP Kinase binding sites. Biochim Biophys Acta. 2008;1784(1):48–55. doi: 10.1016/j.bbapap.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thirunavukkarasu K, Mahajan M, McLarren KW, Stifani S, Karsenty G. Two domains unique to osteoblast-specific transcription factor Osf2/Cbfa1 contribute to its transactivation function and its inability to heterodimerize with Cbfbeta. Mol Cell Biol. 1998;18(7):4197–208. doi: 10.1128/mcb.18.7.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao G, Gopalakrishnan R, Jiang D, Reith E, Benson MD, Franceschi RT. Bone morphogenetic proteins, extracellular matrix, and mitogen-activated protein kinase signaling pathways are required for osteoblast-specific gene expression and differentiation in MC3T3-E1 cells. J Bone Miner Res. 2002;17(1):101–10. doi: 10.1359/jbmr.2002.17.1.101. [DOI] [PubMed] [Google Scholar]
- 30.Xiao G, Jiang D, Gopalakrishnan R, Franceschi RT. Fibroblast growth factor 2 induction of the osteocalcin gene requires MAPK activity and phosphorylation of the osteoblast transcription factor, Cbfa1/Runx2. J Biol Chem. 2002;277(39):36181–7. doi: 10.1074/jbc.M206057200. [DOI] [PubMed] [Google Scholar]
- 31.Ge C, Yang Q, Zhao G, Yu H, Kirkwood KL, Franceschi RT. Interactions between extracellular signal-regulated kinase 1/2 and p38 MAP kinase pathways in the control of RUNX2 phosphorylation and transcriptional activity. J Bone Miner Res. 2012;27(3):538–51. doi: 10.1002/jbmr.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Ge C, Franceschi RT. Differentiation-dependent association of phosphorylated extracellular signal-regulated kinase with the chromatin of osteoblast-related genes. J Bone Miner Res. 2010;25(1):154–63. doi: 10.1359/jbmr.090705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Li Y, Ge C, Franceschi RT. MAP Kinase-Dependent RUNX2 Phosphorylation Is Necessary for Epigenetic Modification of Chromatin During Osteoblast Differentiation. J Cell Physiol. 2016 doi: 10.1002/jcp.25517. This study demonstrates that phosphorylation of RUNX2 in necessary for recruitment of chromatin modifying factors and epigenetic changes in histone necessary for induction of osteoblast-specific gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301(5634):798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- 35.Shim JH, Greenblatt MB, Singh A, Brady N, Hu D, Drapp R, et al. Administration of BMP2/7 in utero partially reverses Rubinstein-Taybi syndrome-like skeletal defects induced by Pdk1 or Cbp mutations in mice. J Clin Invest. 2012;122(1):91–106. doi: 10.1172/JCI59466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boumah CE, Lee M, Selvamurugan N, Shimizu E, Partridge NC. Runx2 recruits p300 to mediate parathyroid hormone’s effects on histone acetylation and transcriptional activation of the matrix metalloproteinase-13 gene. Mol Endocrinol. 2009;23(8):1255–63. doi: 10.1210/me.2008-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jensen ED, Schroeder TM, Bailey J, Gopalakrishnan R, Westendorf JJ. Histone deacetylase 7 associates with Runx2 and represses its activity during osteoblast maturation in a deacetylation-independent manner. J Bone Miner Res. 2008;23(3):361–72. doi: 10.1359/JBMR.071104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McGee-Lawrence ME, Westendorf JJ. Histone deacetylases in skeletal development and bone mass maintenance. Gene. 2011;474(1–2):1–11. doi: 10.1016/j.gene.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye L, Fan Z, Yu B, Chang J, Al Hezaimi K, Zhou X, et al. Histone demethylases KDM4B and KDM6B promotes osteogenic differentiation of human MSCs. Cell Stem Cell. 2012;11(1):50–61. doi: 10.1016/j.stem.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40*.Meyer MB, Benkusky NA, Lee CH, Pike JW. Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J Biol Chem. 2014;289(28):19539–54. doi: 10.1074/jbc.M114.578104. This paper describes ChIP-Seq analysis of chromatin binding sites for the vitamin D receptor and their relationship to RUNX2 and C/EBP binding sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41**.Meyer MB, Benkusky NA, Pike JW. The RUNX2 cistrome in osteoblasts: characterization, down-regulation following differentiation, and relationship to gene expression. J Biol Chem. 2014;289(23):16016–31. doi: 10.1074/jbc.M114.552216. This study provides a comprehensive ChIP-Seq analysis of RUNX2 binding to chromatin during osteoblast differentiation and association of RUNX2 binding with histone modifications associated with increased transcription. These sites were subsequently related to RUNX2 phosphorylation in ref. 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42**.Wu H, Whitfield TW, Gordon JA, Dobson JR, Tai PW, van Wijnen AJ, et al. Genomic occupancy of Runx2 with global expression profiling identifies a novel dimension to control of osteoblastogenesis. Genome Biology. 2014;15(3):R52. doi: 10.1186/gb-2014-15-3-r52. The first comprehensive ChIP-Seq analysis of Runx2 binding sites is described in this study of osteoblast differentiation. Both gain and loss of sites was observed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serra C, Palacios D, Mozzetta C, Forcales SV, Morantte I, Ripani M, et al. Functional interdependence at the chromatin level between the MKK6/p38 and IGF1/PI3K/AKT pathways during muscle differentiation. Mol Cell. 2007;28(2):200–13. doi: 10.1016/j.molcel.2007.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simone C, Forcales SV, Hill DA, Imbalzano AN, Latella L, Puri PL. p38 pathway targets SWI-SNF chromatin-remodeling complex to muscle-specific loci. Nature Genetics. 2004;36(7):738–43. doi: 10.1038/ng1378. [DOI] [PubMed] [Google Scholar]
- 45.Lawrence MC, McGlynn K, Shao C, Duan L, Naziruddin B, Levy MF, et al. Chromatin-bound mitogen-activated protein kinases transmit dynamic signals in transcription complexes in beta-cells. Proc Natl Acad Sci USA. 2008;105(36):13315–20. doi: 10.1073/pnas.0806465105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pokholok DK, Zeitlinger J, Hannett NM, Reynolds DB, Young RA. Activated signal transduction kinases frequently occupy target genes. Science. 2006;313(5786):533–6. doi: 10.1126/science.1127677. [DOI] [PubMed] [Google Scholar]
- 47.Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem. 2006;281(24):16502–11. doi: 10.1074/jbc.M512494200. [DOI] [PubMed] [Google Scholar]
- 48.Jun JH, Yoon WJ, Seo SB, Woo KM, Kim GS, Ryoo HM, et al. BMP2-activated Erk/MAP kinase stabilizes Runx2 by increasing p300 levels and histone acetyltransferase activity. J Biol Chem. 2010;285(47):36410–9. doi: 10.1074/jbc.M110.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park OJ, Kim HJ, Woo KM, Baek JH, Ryoo HM. FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J Biol Chem. 2010;285(6):3568–74. doi: 10.1074/jbc.M109.055053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 51.Ortuno MJ, Ruiz-Gaspa S, Rodriguez-Carballo E, Susperregui AR, Bartrons R, Rosa JL, et al. p38 regulates expression of osteoblast-specific genes by phosphorylation of osterix. J Biol Chem. 2010;285(42):31985–94. doi: 10.1074/jbc.M110.123612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Celil AB, Campbell PG. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem. 2005;280(36):31353–9. doi: 10.1074/jbc.M503845200. [DOI] [PubMed] [Google Scholar]
- 53.Ulsamer A, Ortuno MJ, Ruiz S, Susperregui AR, Osses N, Rosa JL, et al. BMP-2 induces Osterix expression through up-regulation of Dlx5 and its phosphorylation by p38. J Biol Chem. 2008;283(7):3816–26. doi: 10.1074/jbc.M704724200. [DOI] [PubMed] [Google Scholar]
- 54.Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, et al. Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999;126(17):3795–809. doi: 10.1242/dev.126.17.3795. [DOI] [PubMed] [Google Scholar]
- 55.Benson MD, Bargeon JL, Xiao G, Thomas PE, Kim A, Cui Y, et al. Identification of a homeodomain binding element in the bone sialoprotein gene promoter that is required for its osteoblast-selective expression. J Biol Chem. 2000;275(18):13907–17. doi: 10.1074/jbc.275.18.13907. [DOI] [PubMed] [Google Scholar]
- 56.Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. J Biol Chem. 1998;273(3):1496–505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
- 57.Yang X, Karsenty G. ATF4, the osteoblast accumulation of which is determined post-translationally, can induce osteoblast-specific gene expression in non-osteoblastic cells. J Biol Chem. 2004;279(45):47109–14. doi: 10.1074/jbc.M410010200. [DOI] [PubMed] [Google Scholar]
- 58.Yang X, Matsuda K, Bialek P, Jacquot S, Masuoka HC, Schinke T, et al. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117(3):387–98. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- 59.Elefteriou F, Benson MD, Sowa H, Starbuck M, Liu X, Ron D, et al. ATF4 mediation of NF1 functions in osteoblast reveals a nutritional basis for congenital skeletal dysplasiae. Cell Metabolism. 2006;4(6):441–51. doi: 10.1016/j.cmet.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elefteriou F, Kolanczyk M, Schindeler A, Viskochil DH, Hock JM, Schorry EK, et al. Skeletal abnormalities in neurofibromatosis type 1: approaches to therapeutic options. Am J Med Genet Part A. 2009;149A(10):2327–38. doi: 10.1002/ajmg.a.33045. [DOI] [PubMed] [Google Scholar]
- 61.Wang W, Nyman JS, Ono K, Stevenson DA, Yang X, Elefteriou F. Mice lacking Nf1 in osteochondroprogenitor cells display skeletal dysplasia similar to patients with neurofibromatosis type I. Hum Mol Genet. 2011;20(20):3910–24. doi: 10.1093/hmg/ddr310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62*.Twigg SR, Vorgia E, McGowan SJ, Peraki I, Fenwick AL, Sharma VP, et al. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet. 2013;45(3):308–13. doi: 10.1038/ng.2539. This paper describes an indirect mechanism for regulating RUNX2 activity during bone formation involving MAPK phosphorylation and nuclear export of the putative RUNX2 inhibitor, ERF. ERF mutations cause a characteristic craniosynostosis in humans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Le Gallic L, Virgilio L, Cohen P, Biteau B, Mavrothalassitis G. ERF nuclear shuttling, a continuous monitor of Erk activity that links it to cell cycle progression. Mol Cell Biol. 2004;24(3):1206–18. doi: 10.1128/MCB.24.3.1206-1218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149(6):1192–205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 65.Shim JH, Greenblatt MB, Zou W, Huang Z, Wein MN, Brady N, et al. Schnurri-3 regulates ERK downstream of WNT signaling in osteoblasts. J Clin Invest. 2013;123(9):4010–22. doi: 10.1172/JCI69443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gleeson PB, Protas EJ, LeBlanc AD, Schneider VS, Evans HJ. Effects of weight lifting on bone mineral density in premenopausal women. J Bone Miner Res. 1990;5(2):153–8. doi: 10.1002/jbmr.5650050208. [DOI] [PubMed] [Google Scholar]
- 67.Krahl H, Michaelis U, Pieper HG, Quack G, Montag M. Stimulation of bone growth through sports. A radiologic investigation of the upper extremities in professional tennis players. Am J Sports Med. 1994;22(6):751–7. doi: 10.1177/036354659402200605. [DOI] [PubMed] [Google Scholar]
- 68.Leblanc AD, Schneider VS, Evans HJ, Engelbretson DA, Krebs JM. Bone mineral loss and recovery after 17 weeks of bed rest. J Bone Miner Res. 1990;5(8):843–50. doi: 10.1002/jbmr.5650050807. [DOI] [PubMed] [Google Scholar]
- 69.Keyak JH, Koyama AK, LeBlanc A, Lu Y, Lang TF. Reduction in proximal femoral strength due to long-duration spaceflight. Bone. 2009;44(3):449–53. doi: 10.1016/j.bone.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 70.Knapik DM, Perera P, Nam J, Blazek AD, Rath B, Leblebicioglu B, et al. Mechanosignaling in bone health, trauma and inflammation. Antioxid Redox Signal. 2014;20(6):970–85. doi: 10.1089/ars.2013.5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luu YK, Capilla E, Rosen CJ, Gilsanz V, Pessin JE, Judex S, et al. Mechanical stimulation of mesenchymal stem cell proliferation and differentiation promotes osteogenesis while preventing dietary-induced obesity. J Bone Miner Res. 2009;24(1):50–61. doi: 10.1359/JBMR.080817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marie PJ, Kaabeche K. PPAR Gamma Activity and Control of Bone Mass in Skeletal Unloading. PPAR Res. 2006;2006:64807. doi: 10.1155/PPAR/2006/64807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Minaire P, Edouard C, Arlot M, Meunier PJ. Marrow changes in paraplegic patients. Calcif Tissue Int. 1984;36(3):338–40. doi: 10.1007/BF02405340. [DOI] [PubMed] [Google Scholar]
- 74.Tanabe Y, Koga M, Saito M, Matsunaga Y, Nakayama K. Inhibition of adipocyte differentiation by mechanical stretching through ERK-mediated downregulation of PPARgamma2. J Cell Sci. 2004;117(Pt 16):3605–14. doi: 10.1242/jcs.01207. [DOI] [PubMed] [Google Scholar]
- 75.David V, Martin A, Lafage-Proust MH, Malaval L, Peyroche S, Jones DB, et al. Mechanical loading down-regulates peroxisome proliferator-activated receptor gamma in bone marrow stromal cells and favors osteoblastogenesis at the expense of adipogenesis. Endocrinology. 2007;148(5):2553–62. doi: 10.1210/en.2006-1704. [DOI] [PubMed] [Google Scholar]
- 76.Sen B, Xie Z, Case N, Ma M, Rubin C, Rubin J. Mechanical strain inhibits adipogenesis in mesenchymal stem cells by stimulating a durable beta-catenin signal. Endocrinology. 2008;149(12):6065–75. doi: 10.1210/en.2008-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McBeath R, Pirone DM, Nelson CM, Bhadriraju K, Chen CS. Cell shape, cytoskeletal tension, and RhoA regulate stem cell lineage commitment. Dev Cell. 2004;6(4):483–95. doi: 10.1016/s1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- 78.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 79.Khatiwala CB, Kim PD, Peyton SR, Putnam AJ. ECM compliance regulates osteogenesis by influencing MAPK signaling downstream of RhoA and ROCK. J Bone Miner Res. 2009;24(5):886–98. doi: 10.1359/JBMR.081240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shih YR, Tseng KF, Lai HY, Lin CH, Lee OK. Matrix stiffness regulation of integrin-mediated mechanotransduction during osteogenic differentiation of human mesenchymal stem cells. J Bone Miner Res. 2011;26(4):730–8. doi: 10.1002/jbmr.278. [DOI] [PubMed] [Google Scholar]
- 81.Blumbach K, Niehoff A, Belgardt BF, Ehlen HW, Schmitz M, Hallinger R, et al. Dwarfism in mice lacking collagen-binding integrins alpha2beta1 and alpha11beta1 is caused by severely diminished IGF-1 levels. J Biol Chem. 2012;287(9):6431–40. doi: 10.1074/jbc.M111.283119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ekholm E, Hankenson KD, Uusitalo H, Hiltunen A, Gardner H, Heino J, et al. Diminished callus size and cartilage synthesis in alpha 1 beta 1 integrin-deficient mice during bone fracture healing. Am J Pathol. 2002;160(5):1779–85. doi: 10.1016/s0002-9440(10)61124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215(4):445–56. doi: 10.1083/jcb.201609037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Young SR, Gerard-O’Riley R, Kim JB, Pavalko FM. Focal adhesion kinase is important for fluid shear stress-induced mechanotransduction in osteoblasts. J Bone Miner Res. 2009;24(3):411–24. doi: 10.1359/JBMR.081102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boutahar N, Guignandon A, Vico L, Lafage-Proust MH. Mechanical strain on osteoblasts activates autophosphorylation of focal adhesion kinase and proline-rich tyrosine kinase 2 tyrosine sites involved in ERK activation. J Biol Chem. 2004;279(29):30588–99. doi: 10.1074/jbc.M313244200. [DOI] [PubMed] [Google Scholar]
- 86.Sen B, Styner M, Xie Z, Case N, Rubin CT, Rubin J. Mechanical loading regulates NFATc1 and beta-catenin signaling through a GSK3beta control node. J Biol Chem. 2009;284(50):34607–17. doi: 10.1074/jbc.M109.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leucht P, Kim JB, Currey JA, Brunski J, Helms JA. FAK-Mediated mechanotransduction in skeletal regeneration. PLoS ONE. 2007;2(4):e390. doi: 10.1371/journal.pone.0000390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88**.Ge C, Cawthorn WP, Li Y, Zhao G, Macdougald OA, Franceschi RT. Reciprocal Control of Osteogenic and Adipogenic Differentiation by ERK/MAP Kinase Phosphorylation of Runx2 and PPARgamma Transcription Factors. J Cell Physiol. 2016;231(3):587–96. doi: 10.1002/jcp.25102. A lineage switching mechanism is described in this study wherein MAPK phosphorylation of RUNX2 and PPARγ transcription factors preferentially stimulates osteoblast differentiation from mesenchymal progenitors while simultaneously suppressing adipocyte formation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hu E, Kim JB, Sarraf P, Spiegelman BM. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARgamma. Science. 1996;274(5295):2100–3. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 90.van Beekum O, Fleskens V, Kalkhoven E. Posttranslational modifications of PPARgamma: fine-tuning the metabolic master regulator. Obesity (Silver Spring) 2009;17(2):213–9. doi: 10.1038/oby.2008.473. [DOI] [PubMed] [Google Scholar]
- 91.Li Y, Ge C, Long JP, Begun DL, Rodriguez JA, Goldstein SA, et al. Biomechanical stimulation of osteoblast gene expression requires phosphorylation of the RUNX2 transcription factor. J Bone Miner Res. 2012;27:1263–74. doi: 10.1002/jbmr.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92**.Stechschulte LA, Ge C, Hinds TD, Jr, Sanchez ER, Franceschi RT, Lecka-Czernik B. Protein Phosphatase PP5 Controls Bone Mass and the Negative Effects of Rosiglitazone on Bone through Reciprocal Regulation of PPARgamma (Peroxisome Proliferator-activated Receptor gamma) and RUNX2 (Runt-related Transcription Factor 2) J Biol Chem. 2016;291(47):24475–86. doi: 10.1074/jbc.M116.752493. This paper describes a phosphatase capable of dephosphorylating both RUNX2 and PPARγ that is required for accumulation of marrow fat. Knockout of Pp5 greatly stimulates RUNX2 and PPARγ phosphorylation leading to increased bone formation and almost complete blockage of marrow fat accumulation. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 93.Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nat Rev Mol Cell Biol. 2012;13(9):591–600. doi: 10.1038/nrm3416. [DOI] [PubMed] [Google Scholar]
- 94*.Hao J, Zhang Y, Wang Y, Ye R, Qiu J, Zhao Z, et al. Role of extracellular matrix and YAP/TAZ in cell fate determination. Cell Signal. 2014;26(2):186–91. doi: 10.1016/j.cellsig.2013.11.006. Good general review of cellular effects of ECM stiffness on cell signaling through the cytoskeleton and YAP/TAZ factors. [DOI] [PubMed] [Google Scholar]
- 95.Hong JH, Hwang ES, McManus MT, Amsterdam A, Tian Y, Kalmukova R, et al. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science. 2005;309(5737):1074–8. doi: 10.1126/science.1110955. [DOI] [PubMed] [Google Scholar]
- 96.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012;26(1):54–68. doi: 10.1101/gad.173435.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97*.Hwang JH, Byun MR, Kim AR, Kim KM, Cho HJ, Lee YH, et al. Extracellular Matrix Stiffness Regulates Osteogenic Differentiation through MAPK Activation. PLoS One. 2015;10(8):e0135519. doi: 10.1371/journal.pone.0135519. This study establishes a connection between ECM stiffness and activation of both MAPK and YAP/TAZ factors to stimulate osteoblast differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brunner M, Jurdic P, Tuckerman JP, Block MR, Bouvard D. New insights into adhesion signaling in bone formation. Int Rev Cell Mol Biol. 2013;305:1–68. doi: 10.1016/B978-0-12-407695-2.00001-9. [DOI] [PubMed] [Google Scholar]
- 99*.Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. 2014;310:39–87. doi: 10.1016/B978-0-12-800180-6.00002-5. Good general review of discoidin domain receptor functions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bargal R, Cormier-Daire V, Ben-Neriah Z, Le Merrer M, Sosna J, Melki J, et al. Mutations in DDR2 gene cause SMED with short limbs and abnormal calcifications. Am J Hum Genet. 2009;84(1):80–4. doi: 10.1016/j.ajhg.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guo Y, Yang TL, Dong SS, Yan H, Hao RH, Chen XF, et al. Genetic analysis identifies DDR2 as a novel gene affecting bone mineral density and osteoporotic fractures in Chinese population. PLoS One. 2015;10(2):e0117102. doi: 10.1371/journal.pone.0117102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Manes S, et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. 2001;2(5):446–52. doi: 10.1093/embo-reports/kve094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kano K, Marin de Evsikova C, Young J, Wnek C, Maddatu TP, Nishina PM, et al. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol Endocrinol. 2008;22(8):1866–80. doi: 10.1210/me.2007-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104**.Ge C, Wang Z, Zhao G, Li B, Liao J, Sun H, et al. Discoidin Receptor 2 Controls Bone Formation and Marrow Adipogenesis. J Bone Miner Res. 2016;31(12):2193–203. doi: 10.1002/jbmr.2893. This paper describes a detailed in vivo analysis of the requirement for Ddr2 in bone formation and osteoblast differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105*.Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, et al. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. 2011;26(3):604–17. doi: 10.1002/jbmr.225. This is the first study to show that DDR2 can regulate RUNX2 activity via MAPK-dependent phosphorylation. [DOI] [PubMed] [Google Scholar]