

Abstract
The α-1 adrenoreceptor antagonist prazosin has shown promise in the treatment of post-traumatic stress disorder (PTSD) symptoms, but its mechanisms are not well understood. Here we administered prazosin or placebo prior to threat conditioning (day 1) and tested subsequent extinction (day 2) and reextinction (day 3) in healthy human participants. Prazosin did not affect threat conditioning but augmented stimulus discrimination during extinction and reextinction, via lower responding to the safe stimulus. These results suggest that prazosin during threat acquisition may have influenced encoding or consolidation of safety processing in particular, subsequently leading to enhanced discrimination between the safe and threatening stimuli.
Pavlovian threat conditioning is a prominent model for understanding the significance of threat discrimination (i.e., the distinction of threatening and safe stimuli) learning and memory in post-traumatic stress disorder (PTSD). This model assumes that a conditioned stimulus (CS+, a formerly neutral stimulus such as a shape or a sound) is associated with an unconditioned stimulus (US, the aversive event) during threat learning, enabling the CS+ itself to become an unsafe cue and trigger a defensive response, while the control stimulus (CS−, a safe cue) may acquire inhibitory properties as it had never been paired with the US. Threat conditioning studies in PTSD generally found increased responses to the CS− but no consistent effect on the discrimination of threat (Duits et al. 2015). The effect of threat learning can be quantified by the amount of discrimination between the unsafe and the safe cue during threat conditioning, and subsequent presentations of the CS+ without the US induce extinction learning.
The α-1 adrenergic receptor antagonist prazosin has shown promise in the treatment of PTSD. Prazosin attenuates noradrenaline effects at central postsynaptic α-1 adrenergic receptor after peripheral administration (Menkes et al. 1981) and has been shown to reduce PTSD symptoms including nightmares, poor sleep quality, hyperarousal, and impaired global function (Raskind et al. 2000, 2002, 2003, 2007, 2013; Taylor et al. 2008; Germain et al. 2012; Ahmadpanah et al. 2014; George et al. 2016) which may involve abnormally heightened activity of the noradrenergic central nervous system (Southwick et al. 1993; Raskind et al. 2016).
Previous threat conditioning studies in humans have shown that learned threat memory resisted extinction training following noradrenergic stimulation by yohimbine before threat acquisition, while the initial learning of threat was unaffected (Soeter and Kindt 2011, 2012). Yohimbine is an α-2-adrenergic antagonist that stimulates central noradrenergic activity by blocking the α-2-adrenergic autoreceptor, and its physiological effects typically include an increase in systolic and diastolic blood pressure. Prazosin, on the other hand, has antihypertensive effects by blocking α-1-adrenergic receptors. Although the central actions of α-1-adrenergic receptors with respect to threat learning are not fully understood, the opposite physiological effects of yohimbine and prazosin suggest that prazosin may have opposite effects on extinction learning compared with yohimbine.
Specifically, threat discrimination learned under prazosin, and thus under attenuated noradrenergic effects on α-1 adrenergic receptors, may reverse the extinction effect of yohimbine: compared with placebo, threat discrimination should be unaffected during acquisition but easier to subsequently extinguish, i.e., higher responses to CS+ compared with CS− during acquisition in both the prazosin and placebo group, but a faster decay of the CS+ response during extinction and reextinction in the prazosin group compared with the placebo group, without affecting the CS−. Contrary to this prediction, however, studies in rodents found that α-1 adrenoreceptor antagonists enhanced threat acquisition and impaired threat extinction (Cain et al. 2006; Bernardi and Lattal 2010; Do-Monte et al. 2010; Lazzaro et al. 2010), raising the concerning possibility that prazosin might in fact work against extinction-based treatments (Do-Monte et al. 2010; Maren 2011). Notably, additional noradrenergic effects on β- and α-2 adrenergic receptors may have also contributed to these memory processes (Do-Monte et al. 2010).
To clarify the effects of prazosin on threat acquisition and extinction in healthy humans, we used a randomized double-blind between–within-subjects experimental design over three consecutive days, conducted in the same context (Fig. 1): Threat learning on day 1, threat extinction on day 2, and reextinction test on day 3; the reextinction test examined whether participants retrieve the threat discrimination (learned on day 1) or the extinction memory (learned on day 2). On day 1 (acquisition), participants were randomly assigned to receive either placebo or 3 mg of prazosin (1 mg capsule followed by 2 mg capsule 30 min later) 2 h before threat acquisition (at the expected peak plasma prazosin level). Immediately before and every 30 min until 90 min after the administration of the study drug, blood pressure and heart rate were measured. In addition, we measured the trait anxiety subscale of the Spielberger State-Trait Anxiety Inventory (STAI-T) (Spielberger et al. 1983) before the experiment. Forty healthy human participants provided written informed consent and were compensated. The experiment was approved by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai. The final sample consisted of 38 participants, one of which did not show up for the third day (Table 1).
Figure 1.

Experimental design and behavioral results. (Upper panel) This was a randomized double-blind experimental design in healthy volunteers involving three consecutive days: Threat learning on day 1, threat extinction on day 2, and reextinction test on day 3; the reextinction test examined whether participants retrieve the threat discrimination (learned on day 1) or the extinction memory (learned on day 2). On day 1 (acquisition), participants were randomly assigned to receive either the placebo or 3 mg of prazosin (1 mg capsule followed by 2 mg capsule 30 min later) 2 h before threat acquisition (at the expected peak plasma prazosin level). During threat conditioning, two colored squares were presented for 4 sec on each trial, one of which was paired with a mild electric shock in 43% of the trials (CS+) while the other was never paired with a shock (CS−). On day 2 (extinction) and day 3 (reextinction), no shocks were delivered, but the stimulating bar electrodes were connected to the participant's nondominant wrist. Extinction and reextinction started with a CS− presentation and thus involved one additional CS−; subsequent presentations of CS+ and CS− were counterbalanced. (CS) Conditioned stimulus; (US) unconditioned stimulus; (ITI) intertrial interval. (Bottom panel) Stimulus discrimination summarized by group and stage show successful acquisition for both groups but persisting stimulus discrimination (mediated by effects on the safe stimulus) in the prazosin group in extinction and reextinction. Mean logarithmized anticipatory sudomotor nerve activity estimates (aSNA) for acquisition, extinction, and reextinction after administration of placebo and prazosin. Error bars denote standard errors. (Plac) Placebo; (Praz) prazosin; (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
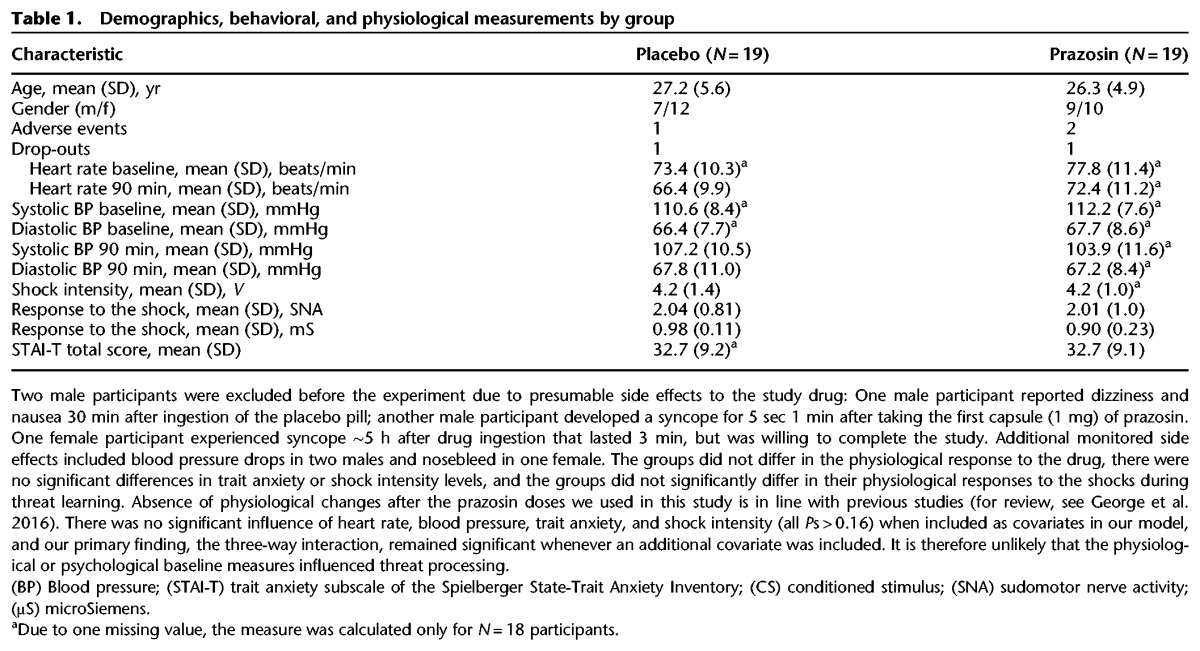
Table 1.
Demographics, behavioral, and physiological measurements by group
During threat conditioning, two colored squares were presented for 4 s on each trial, one of which was paired with a mild electric shock in 43% of the trials (CS+) while the other was never paired with a shock (CS−). On day 2 (extinction) and day 3 (reextinction) no shocks were delivered but the stimulating bar electrodes were connected to the participant's nondominant wrist. Skin conductance response (SCR) to the stimuli was measured throughout with Ag–AgCl electrodes, filled with standard NaCl electrolyte gel, and attached to the middle phalanges of the second and third fingers of the nondominant hand. SCR signal was amplified and recorded with a MP150 BIOPAC Systems skin conductance module connected to a PC. Data were continuously recorded at a rate of 200 samples per second. Shocks were delivered using a Grass Medical Instruments SD9 stimulator and stimulating bar electrodes attached to the participant's nondominant wrist. Shock intensity was calibrated up to a maximum of 60 V to reach a level described by participants as “uncomfortable, but not painful.”
The outcome measure was the psychophysiological arousal response to the CSs, indexed by the estimated anticipatory sudomotor nerve activity (aSNA) amplitude (Bach et al. 2010). Estimates of aSNA indicate the anticipation of an aversive event within the time window of stimulus presentation. These were calculated by inverting a forward model that describes how (hidden) SNA translates into an (observable) SCR using a variational Bayes approximation. A unit increase in aSNA corresponds to an increase in SCR of 1 µS. This method uses summary statistics across all available trials (i.e., average aSNA per condition and stimulus) to demonstrate successful experimental manipulations such as threat conditioning, and has shown to be more sensitive compared with a conventional SCR base-to-peak analysis (Bach 2014; Bach et al. 2010; Staib et al. 2015).
We used linear mixed models with restricted maximum likelihood estimation for all analyses, due to their well-established advantages over conventional analysis of variance (e.g., Gueorguieva and Krystal 2004). Mixed models efficiently allow for a full analysis of the data even in the presence of missing data (allowing us to include the participant that did not complete day 3). We used the estimates of aSNA as outcome measure and the R software (R Core Team 2016) and the packages lme4 (Bates 2005; Baayen et al. 2008; Bates et al. 2015) and lsmeans (Lenth 2016) for all analyses. A log-transformation was applied on the aSNA estimates to correct for unequal variances (heteroscedasticity). Fixed effects included drug (placebo, prazosin), session (acquisition, extinction, reextinction) and stimulus (CS+, CS−) as well as their interactions. In addition, a mean-centered linear term for trial was entered to account for the time-effect in each session. Significance of fixed effects was assessed using likelihood ratio tests against a χ2 distribution and maximum likelihood as estimation method. A maximal random effects structure including random intercept and slopes for session and stimulus was included to avoid inflated type-1 errors (Barr et al. 2013). Random slopes for session, stimulus, and the session × stimulus interaction for each subject were included to account for random variation between subjects’ in the effects of interest. An unstructured variance–covariance matrix was used to allow for correlation between the random effects. The significance threshold was set at 0.05, two-tailed.
To assess whether there were group differences in threat discrimination in any of the sessions, we tested for an interaction of drug (placebo, prazosin) × session (acquisition, extinction, reextinction) × stimulus (CS+, CS−) and found that the three-way interaction was significant (χ2 (2) = 7.26, P = 0.03; Fig. 1) and driven by enhanced stimulus discrimination (CS+ vs. CS−) in the prazosin group compared with placebo during extinction and reextinction but not acquisition (Fig. 2). Specifically, both groups showed successful acquisition, indicated by stronger response to the CS+ compared with the CS− and effect sizes between medium and large (placebo: t(18) = 2.9, P = 0.0097; Cohen's d = 0.66; prazosin: t(18) = 4.1, P = 0.0007; Cohen's d = 0.94). However, in the placebo group, the stimulus discrimination was not significant during extinction (t(18) = 1.55, P = 0.14) and reextinction (t(17) = 1.3, P = 0.21) whereas for the prazosin group, aSNA for CS+ was significantly higher compared with CS− during extinction (t(18) = 5.04, P = 0.0001) and reextinction (t(18) = 4.76, P = 0.0002; Fig. 2), confirming the three-way interaction. These results indicate that threat acquisition was successful with no group differences, but stimulus discrimination differed between the groups during extinction and reextinction, with augmented discrimination in the prazosin compared with the placebo group. Notably, these results were consistent with the results obtained by using the conventional manually scored base-to-peak SCR data (see Supplemental Results).
Figure 2.
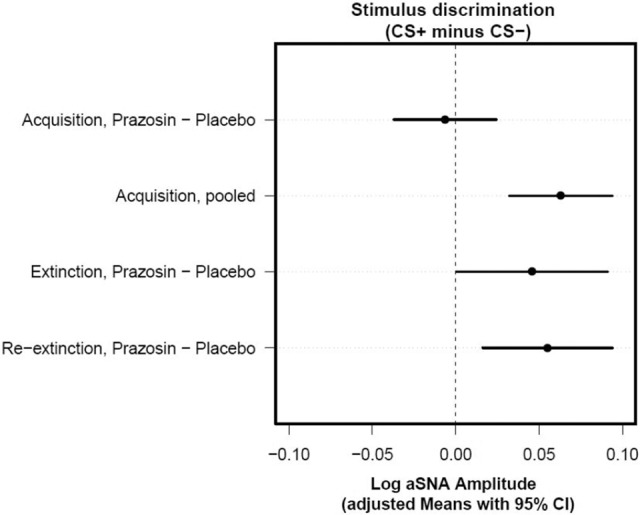
Planned contrasts for stimulus-discrimination differences between the prazosin and placebo groups for each session show effects specific for extinction and reextinction but not acquisition. Confidence intervals that do not include zero (cross the vertical dashed line) indicate that the corresponding contrast is statistically significant. Adjusted means (indicating the mean response for each factor, adjusted for any other variables in the model) with 95% confidence intervals for each drug, session, and stimulus were calculated from the multilevel model output and used to test the stimulus-discrimination (CS+ minus CS−) differences between prazosin and placebo for each session. Specifically, during acquisition, there was no significant group × stimulus interaction (t(38.42) = −0.40, P = 0.69), and there was evidence across groups for significant stimulus discrimination (t(38.42) = 4.07, P < 0.001). In contrast, when examining the subsequent stages, we found a significant group × stimulus interaction during extinction (t(39.3) = 2.06, P = 0.046) and reextinction (t(38.42) = 2.86, P = 0.006). (CS) Conditioned stimulus; (CI) confidence interval.
Next, we asked whether changes in response to one stimulus in particular drove the discrimination, i.e., we tested for differences between the placebo and the prazosin group in aSNA responses to CS+ as well as for aSNA responses to CS− (Fig. 1). We found that responses to the CS− were significantly lower in the prazosin group compared with the placebo group in extinction (t(36.04) = −2.24, P = 0.031) and reextinction (t(34.05) = −2.9, P = 0.007) but not acquisition (t(39.74) = 0.44, P = 0.66), whereas CS+ did not significantly differ between groups in any session (all P > 0.84) or phase within sessions (all P > 0.71). These results indicate that prazosin boosts threat discrimination during extinction and reextinction via effects on the safe stimulus. Detailed analysis of early and late phases in each stage confirmed that prazosin had no significant effects during acquisition. The protocol induced extinction and reextinction in both groups, with responses to both stimuli gradually decreasing from early to late phases, but responses to the CS− were significantly lower in the prazosin compared with placebo group during late extinction and throughout reextinction (see Supplemental Results).
Thus, prazosin effects during threat memory formation may change the fate of memory: threat discrimination learned under prazosin would be harder to extinguish over time. Consistent with the rodent data (i.e., potentiating threat memory), and in contrast to the expected effects of α-1 adrenergic receptor blockade in humans (i.e., reducing threat memory), we found that the prazosin group compared with placebo showed enhanced threat discrimination memory during extinction and reextinction, driven by lower responding to the safe stimuli, with no effects during acquisition.
How can prazosin interfere with extinction? Potentially, prazosin acts upon the encoding of the memory, affecting both inhibitory and excitatory plasticity, which is later recruited for extinction (Clem and Schiller 2016). Some clues may arise from evaluating the effects of prazosin on threat learning and extinction, especially since α-1 adrenergic receptors are abundant in the lateral nucleus of the amygdala (LA), a key region of synaptic plasticity in threat learning and extinction (LeDoux 2000). The LA receives inputs from the locus coeruleus that contain noradrenaline and exhibit tonic and phasic firing in response to aversive stimuli (Tully and Bolshakov 2010). However, the role of noradrenaline in the modulation of threat learning is less clear. On the one hand, it has been shown that noradrenaline may suppress feed-forward inhibition of threat conditioning thalamic pathway, thereby enhancing learning-related plasticity (Tully et al. 2007; Ehrlich et al. 2009). On the other hand, α-1 adrenergic receptors in the LA may inhibit descending output from the central nucleus of the amygdala to brain regions controlling arousal and defensive responses (Braga et al. 2004; Pape and Pare 2010). As prazosin has a short half-life of only 3 h, the results cannot be explained by direct action of prazosin during extinction or reextinction, suggesting that α-1 receptor blockade may alter the long-term consequences of newly acquired threat associations. In addition, state-dependent learning effects instead of a drug effect of prazosin appears to be an unlikely explanation, given that we found the drug effect in extinction and again in reextinction.
Rodent studies indicate that prazosin may in fact reduce the amygdala's inhibitory tone (Cain et al. 2006; Bernardi and Lattal 2010; Do-Monte et al. 2010; Lazzaro et al. 2010). Although α-1 adrenergic receptor blockade during threat learning is an unlikely model for PTSD since it should counteract symptoms induced by elevated sympathetic activity, prazosin may capture certain conditions and individual differences in α-1 adrenergic receptor activity during the experience of a traumatic event, which may affect the development of PTSD in interesting and important ways. The current results indeed suggest that learning threat discrimination under prazosin is less flexible and better remembered over time.
Lower responding to the CS− but not CS+ drove the effect of prazosin on threat discrimination. Although it may seem as if prazosin augmented memory for the safe stimulus (i.e., decreased aSNA in response to the CS−) without affecting threat memory, a few considerations should be noted. The discrimination between safe and unsafe cues is adaptive only during acquisition when threat is imminent. During extinction, both cues are safe and the learned discrimination is no longer adaptive. A recent meta-analysis (Duits et al. 2015) showed that anxiety patients exhibit less discrimination than controls in acquisition (when it is adaptive) and more discrimination in extinction (when it is maladaptive). Our results show that the prazosin group, but not placebo, persistently maintained the discrimination between the CS+ and the CS−, never fully extinguishing the difference between the stimuli despite two consecutive extinction sessions, which is maladaptive. It might be problematic, therefore, to argue that prazosin has positive effects by augmenting memory for safety learning when the net result is abnormally persistent threat discrimination. Nevertheless, to fully disentangle these competing interpretations, future studies should examine administration of prazosin prior to extinction, when the CS+ undergoes safety learning, and examine whether this would diminish or augment subsequent stimulus discrimination. Consistent with the latter possibility, rodent studies found impairments in extinction learning when prazosin was administered prior to extinction training (Do-Monte et al. 2010) and between repeated extinction sessions (Bernardi and Lattal 2010), indicating it does not enhance but rather counteracts safety learning.
The findings of this study may have clinical relevance as prazosin is often prescribed for the treatment of PTSD symptoms (Taylor et al. 2008; Raskind et al. 2000, 2002, 2003, 2007, 2013; Germain et al. 2012; Ahmadpanah et al. 2014; George et al. 2016), and prolonged exposure therapy is currently the most effective behavioral therapy in PTSD (McLean and Foa 2011). If prazosin is combined with or followed by extinction-based treatments, it might influence reexperienced or new memories in the course of therapy. In addition, as extinction was only indirectly affected in this study through memory encoding or consolidation, the clinical implications may be more relevant for individuals who are taking α-1 blockers when they are traumatized (or who receive them immediately after the trauma) or for individuals with innately reduced α-1 signaling in defensive brain circuits.
Competing interest statement
Dr. Murrough declares no conflicts related to the current study. In the past three years, Dr. Murrough has provided consultation services to Novartis, Janssen Research and Development, and Genentech; he is named on patents pending for neuropeptide Y as a treatment for mood and anxiety disorders, a patent pending for the combination of ketamine and lithium for suicidal ideation, and a patent pending for ketamine plus lithium to extend the antidepressant response of ketamine. Dr. Homan declares no conflicts related to the current study. Dr. Homan has received speaker's fees from Neurolite and Takeda Pharma in the past three years. All other authors report no conflicts of interest.
Supplementary Material
Acknowledgments
This study was funded by National Institute of Mental Health (NIMH) grant MH105515 and a Klingenstein-Simons Fellowship Award in the Neurosciences to D.S.; NIMH grant MH105414 to R.L.C.; and Swiss National Science Foundation grant SNF 161077 to P.H. The funding source had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author contributions: P.H. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. P.H., Q.L., L.S., J.W.M., R.L.C., and D.S. conceived of and designed the study. All authors contributed to acquisition, analysis, or interpretation of data. P.H. and D.S. drafted the manuscript. All authors contributed to critical revision of the manuscript for important intellectual content. P.H., D.R.B., and D.S. performed statistical analysis. P.H., R.L.C., and D.S. obtained funding. D.S. contributed to administrative, technical, or material support and supervised the study. This work was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.045898.117.
References
- Ahmadpanah M, Sabzeiee P, Hosseini SM, Torabian S, Haghighi M, Jahangard L, Bajoghli H, Holsboer-Trachsler E, Brand S. 2014. Comparing the effect of prazosin and hydroxyzine on sleep quality in patients suffering from posttraumatic stress disorder. Neuropsychobiology 69: 235–242. [DOI] [PubMed] [Google Scholar]
- Baayen R, Davidson D, Bates D. 2008. Mixed-effects modeling with crossed random effects for subjects and items. J Mem Lang 59: 390–412. [Google Scholar]
- Bach DR. 2014. A head-to-head comparison of SCRalyze and Ledalab, two model-based methods for skin conductance analysis. Biol Psychol 103: 63–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach DR, Daunizeau J, Friston KJ, Dolan RJ. 2010. Dynamic causal modelling of anticipatory skin conductance responses. Biol Psychol 85: 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr DJ, Levy R, Scheepers C, Tily HJ. 2013. Random effects structure for confirmatory hypothesis testing: keep it maximal. J Mem Lang 68: 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates DM. 2005. Fitting linear mixed models in R. R News 5: 27–30. [Google Scholar]
- Bates D, Mächler M, Bolker B, Walker S. 2015. Fitting linear mixed-effects models using lme4. J Stat Softw 67: 1–48. [Google Scholar]
- Bernardi RE, Lattal KM. 2010. A role for α1-adrenergic receptors in extinction of conditioned fear and cocaine conditioned place preference. Behav Neurosci 124: 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Manion ST, Hough CJ, Li H. 2004. Stress impairs α1a adrenoceptor-mediated noradrenergic facilitation of gabaergic transmission in the basolateral amygdala. Neuropsychopharmacology 29: 45–58. [DOI] [PubMed] [Google Scholar]
- Cain CK, Bush DE, LeDoux JE. 2006. Prazosin, an α1-adrenergic receptor antagonist, enhances acquisition and impairs extinction of Pavlovian cue fear. In Proceedings from the 2006 Neuroscience Meeting, Society for Neuroscience, Atlanta, GA. [Google Scholar]
- Clem RL, Schiller D. 2016. New learning and unlearning: strangers or accomplices in threat memory attenuation? Trends Neurosci 39: 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do-Monte FH, Allensworth M, Carobrez AP. 2010. Impairment of contextual conditioned fear extinction after microinjection of α-1-adrenergic blocker prazosin into the medial prefrontal cortex. Behav Brain Res 211: 89–95. [DOI] [PubMed] [Google Scholar]
- Duits P, Cath DC, Lissek S, Hox JJ, Hamm AO, Engelhard IM, van den Hout MA, Baas JMP. 2015. Updated meta-analysis of classical fear conditioning in the anxiety disorders. Depress Anxiety 32: 239–253. [DOI] [PubMed] [Google Scholar]
- Ehrlich I, Humeau Y, Grenier F, Ciocchi S, Herry C, Lüthi A. 2009. Amygdala inhibitory circuits and the control of fear memory. Neuron 62: 757–771. [DOI] [PubMed] [Google Scholar]
- George KC, Kebejian L, Ruth LJ, Miller CW, Himelhoch S. 2016. Meta-analysis of the efficacy and safety of prazosin versus placebo for the treatment of nightmares and sleep disturbances in adults with posttraumatic stress disorder. J Trauma Dissociation 17: 494–510. [DOI] [PubMed] [Google Scholar]
- Germain A, Richardson R, Moul DE, Mammen O, Haas G, Forman SD, Rode N, Begley A, Nofzinger EA. 2012. Placebo-controlled comparison of prazosin and cognitive-behavioral treatments for sleep disturbances in US military veterans. J Psychosom Res 72: 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueorguieva R, Krystal JH. 2004. Move over ANOVA: progress in analyzing repeated-measures data and its reflection in papers published in the Archives of General Psychiatry. Arch Gen Psychiatry 61: 310–317. [DOI] [PubMed] [Google Scholar]
- Lazzaro SC, Hou M, Cunha C, LeDoux JE, Cain CK. 2010. Antagonism of lateral amygdala alpha1-adrenergic receptors facilitates fear conditioning and long-term potentiation. Learn Mem 17: 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE. 2000. Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. [DOI] [PubMed] [Google Scholar]
- Lenth RV. 2016. Least-square means: the R package lsmean. J Stat Softw 69: 1–33. [Google Scholar]
- Maren S. 2011. Seeking a spotless mind: extinction, deconsolidation, and erasure of fear memory. Neuron 70: 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CP, Foa EB. 2011. Prolonged exposure therapy for post-traumatic stress disorder: a review of evidence and dissemination. Expert Rev Neurother 11: 1151–1163. [DOI] [PubMed] [Google Scholar]
- Menkes DB, Baraban JM, Aghajanian GK. 1981. Prazosin selectively antagonizes neuronal responses mediated by α1-adrenoceptors in brain. Naunyn Schmiedebergs Arch Pharmacol 317: 273–275. [DOI] [PubMed] [Google Scholar]
- Pape HC, Pare D. 2010. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev 90: 419–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskind MA, Dobie DJ, Kanter ED, Petrie EC, Thompson CE, Peskind ER. 2000. The alpha1-adrenergic antagonist prazosin ameliorates combat trauma nightmares in veterans with posttraumatic stress disorder: a report of 4 cases. J Clin Psychiatry 61: 129–133. [DOI] [PubMed] [Google Scholar]
- Raskind MA, Thompson C, Petrie EC, Dobie DJ, Rein RJ, Hoff DJ, McFall ME, Peskind ER. 2002. Prazosin reduces nightmares in combat veterans with posttraumatic stress disorder. J Clin Psychiatry 63: 565–568. [DOI] [PubMed] [Google Scholar]
- Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson CE, Dobie DJ, Hoff D, Rein RJ, Straits-Tröster K, et al. 2003. Reduction of nightmares and other PTSD symptoms in combat veterans by prazosin: a placebo-controlled study. Am J Psychiatry 160: 371–373. [DOI] [PubMed] [Google Scholar]
- Raskind MA, Peskind ER, Hoff DJ, Hart KL, Holmes HA, Warren D, Shofer J, O'Connell J, Taylor F, Gross C, et al. 2007. A parallel group placebo controlled study of prazosin for trauma nightmares and sleep disturbance in combat veterans with post-traumatic stress disorder. Biol Psychiatry 61: 928–934. [DOI] [PubMed] [Google Scholar]
- Raskind MA, Peterson K, Williams T, Hoff DJ, Hart K, Holmes H, Homas D, Hill J, Daniels C, Calohan J, et al. 2013. A trial of prazosin for combat trauma PTSD with nightmares in active-duty soldiers returned from Iraq and Afghanistan. Am J Psychiatry 170: 1003–1010. [DOI] [PubMed] [Google Scholar]
- Raskind MA, Millard SP, Petrie EC, Peterson K, Williams T, Hoff DJ, Hart K, Holmes H, Hill J, Daniels C, et al. 2016. Higher pretreatment blood pressure is associated with greater posttraumatic stress disorder symptom reduction in soldiers treated with prazosin. Biol Psychiatry 80: 736–742. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- Soeter M, Kindt M. 2011. Noradrenergic enhancement of associative fear memory in humans. Neurobiol Learn Mem 96: 263–271. [DOI] [PubMed] [Google Scholar]
- Soeter M, Kindt M. 2012. Stimulation of the noradrenergic system during memory formation impairs extinction learning but not the disruption of reconsolidation. Neuropsychopharmacology 37: 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southwick SM, Krystal JH, Morgan CA, Johnson D, Nagy LM, Nicolaou A, Heninger GR, Charney DS. 1993. Abnormal noradrenergic function in posttraumatic stress disorder. Arch Gen Psychiatry 50: 266–274. [DOI] [PubMed] [Google Scholar]
- Spielberger CD, Gorsuch RL, Lushene R, Vagg PR, Jacobs GA. 1983. Manual for the State-Trait Anxiety Inventory. Consulting Psychologists Press, Palo Alto, CA. [Google Scholar]
- Staib M, Castegnetti G, Bach DR. 2015. Optimising a model-based approach to inferring fear learning from skin conductance responses. J Neurosci Methods 255: 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor FB, Martin P, Thompson C, Williams J, Mellman TA, Gross C, Peskind ER, Raskind MA. 2008. Prazosin effects on objective sleep measures and clinical symptoms in civilian trauma posttraumatic stress disorder: a placebo-controlled study. Biol Psychiatry 63: 629–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Bolshakov VY. 2010. Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol Brain 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tully K, Li Y, Tsvetkov E, Bolshakov VY. 2007. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc Natl Acad Sci 104: 14146–14150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.