

Here, Yue et al. investigated the mechanisms underlying p53 gain-of-function (GOF) mutations and found that mutant p53 activates small GTPase Rac1 as a critical mechanism for mutant p53 GOF to promote tumor progression. Their findings provide insight into a new mechanism for Rac1 activation in tumors and show that activation of Rac1 is an unidentified and critical mechanism for mutant p53 GOF in tumorigenesis.
Keywords: mutant p53, gain of function, Rac1, SUMOylation, tumorigenesis, metastasis
Abstract
Tumor suppressor p53 is frequently mutated in human cancer. Mutant p53 often promotes tumor progression through gain-of-function (GOF) mechanisms. However, the mechanisms underlying mutant p53 GOF are not well understood. In this study, we found that mutant p53 activates small GTPase Rac1 as a critical mechanism for mutant p53 GOF to promote tumor progression. Mechanistically, mutant p53 interacts with Rac1 and inhibits its interaction with SUMO-specific protease 1 (SENP1), which in turn inhibits SENP1-mediated de-SUMOylation of Rac1 to activate Rac1. Targeting Rac1 signaling by RNAi, expression of the dominant-negative Rac1 (Rac1 DN), or the specific Rac1 inhibitor NSC23766 greatly inhibits mutant p53 GOF in promoting tumor growth and metastasis. Furthermore, mutant p53 expression is associated with enhanced Rac1 activity in clinical tumor samples. These results uncover a new mechanism for Rac1 activation in tumors and, most importantly, reveal that activation of Rac1 is an unidentified and critical mechanism for mutant p53 GOF in tumorigenesis, which could be targeted for therapy in tumors containing mutant p53.
Tumor suppressor p53 plays a key role in maintaining genomic stability and tumor suppression (Levine et al. 2006; Vousden and Prives 2009). In response to a variety of stress signals, p53 is activated and binds to its target genes via its DNA-binding domain (DBD) to transcriptionally regulate these genes, which contributes to its tumor-suppressive function. p53 is the most commonly mutated gene in human tumors; >50% of human tumors harbor mutant p53 (Freed-Pastor and Prives 2012). The majority of tumor-associated p53 mutations is missense mutations and is predominantly clustered in the DBD, including several hot spot mutations in tumors (e.g., R175, G245, R248, R249, R273, and R282). In addition to loss of tumor-suppressive functions of wild-type p53, tumor-associated mutant p53 proteins often exhibit oncogenic activities independently of wild-type p53, termed mutant p53 gain of function (GOF) (Freed-Pastor and Prives 2012; Muller and Vousden 2014). Many mutant p53 GOF activities have been reported, including promoting cell proliferation, survival, migration, and invasion; metabolic reprogramming; and disrupting tissue architecture (Muller et al. 2009; Freed-Pastor et al. 2012; Zhang et al. 2013).
GOF mutant p53 is an attractive target for cancer therapy; however, the mechanism of mutant p53 GOF is not well understood. An important mechanism underlying mutant p53 GOF is through interaction and regulation of specific proteins, many of which are transcriptional factors. For instance, GOF mutant p53 interacts with TAp63 and TAp73 to inhibit their transcriptional activities, which contributes to mutant p53 GOF (Stindt et al. 2015). Mutant p53 also interacts with other transcriptional factors and cofactors, including NF-Y, SREBP, VDR, ETS2, and Nrf2, to affect their transcription regulation in tumor cells to promote tumorigenesis (Sampath et al. 2001; Di Agostino et al. 2006; Walerych et al. 2016).
Rac1, a small GTPase, regulates many cellular functions, including cell proliferation, cytoskeletal reorganization, and cell mobility. Rac1 cycles between the inactive GDP-bound (Rac1-GDP) and active GTP-bound (Rac1-GTP) forms in cells (Heasman and Ridley 2008; Bid et al. 2013). Rac1 signaling is frequently activated in various cancer types to promote proliferation, migration, invasion, and metastasis of cancer cells (Heasman and Ridley 2008; Bid et al. 2013). SUMOylation is an important post-translational protein modification to regulate protein stability and/or activity. SUMOylation is a dynamic process, which is catalyzed by SUMO-specific E1, E2, and E3s and reversed by SUMO-specific proteases (SENPs) (Gareau and Lima 2010; Hay 2013). Rac1 SUMOylation is critical for Rac1 to maintain the active GTP-bound form and its activity (Castillo-Lluva et al. 2010). SENP1 can bind to Rac1 to de-SUMOylate Rac1, leading to Rac1 inactivation (Castillo-Lluva et al. 2010).
In this study, using immunoprecipitation followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) assays, we identified Rac1 as a novel binding protein for mutant p53 but not wild-type p53. Mutant p53 specifically interacted with Rac1 and activated Rac1 as an important mechanism for mutant p53 GOF in promoting tumor growth and metastasis. Mechanistically, mutant p53 inhibited the interaction of SENP1 with Rac1 to inhibit Rac1 de-SUMOylation, thereby activating Rac1. These results demonstrate that mutant p53 is a novel regulator for Rac1 and reveal that Rac1 activation by mutant p53 is a novel and critical mechanism for mutant p53 GOF in tumorigenesis.
Results
Rac1 is a novel mutant p53-binding protein
To deepen our understanding of mechanisms underlying mutant p53 GOF and identify potential targets for mutant p53 targeted therapies, we screened for mutant p53-interacting proteins using coimmunoprecipitation (co-IP) with anti-p53 antibodies followed by LC-MS/MS assays in p53-null human lung cancer H1299 cells and H1299 cells with ectopic expression of a hot spot GOF mutant p53 (R175H) or wild-type p53. Several known mutant p53-interacting proteins, including heat shock proteins, BAG2, BAG5, etc. (Muller et al. 2005; Yue et al. 2015), were among the list of identified potential mutant p53-interacting proteins (Supplemental Table 1), which validated our approach. Through this approach, we identified small GTPase Rac1 as a potential mutant p53-specific binding protein (Supplemental Table 1). The mutant p53–Rac1 interaction was confirmed by co-IP followed by Western blot assays in H1299 cells transfected with vectors expressing Rac1 together with vectors expressing mutant p53 (R175H) (Fig. 1A). Rac1 preferentially bound to mutant p53 (R175H) compared with wild-type p53 in cells (Fig. 1A). In addition to R175H mutant p53, Rac1 interacted with different hot spot mutant p53s ectopically expressed in H1299 cells, including R248W and R273H (Fig. 1B). The interaction between endogenous mutant p53 and endogenous Rac1 was observed in a panel of human cancer cells containing a single allele of the mutant p53 gene (p53Mut/−), including breast SK-BR-3 (R175H) and MDA-MB468 (R273H), prostate LAPC4 (R175H), colorectal SW480 (R273H) and HT29 (R273H), and liver Huh7 (Y220C) cells (Fig. 1C). Furthermore, the mutant p53–Rac1 interaction was observed mainly in the cytoplasm, and a small fraction of this interaction was observed in the nucleus (Supplemental Fig. S1A).
Figure 1.

Rac1 is a novel mutant p53-binding protein in human cancer cells. (A) Rac1-Flag preferentially interacted with ectopic mutant p53 (R175H) compared with ectopic wild-type p53 in human p53-null H1299 cells. H1299 cells were transfected with Rac1-Flag expression vectors together with wild-type p53 or mutant p53 (R175H) expression vectors for co-IP assays. The antibodies used for immunoprecipitation assays were as follows: anti-Flag for Rac1-Flag and DO-1 for mutant p53 and wild-type p53. (B) Rac1-Flag interacted with different hot spot mutant p53s, including R175H, R248W, and R273H, in H1299 cells transfected with vectors as indicated. (C) Endogenous Rac1 interacted with endogenous mutant p53 in SK-BR-3 (R175H), MDA-MB468 (R273H), LAPC4 (R175H), SW480 (R273H), Huh7 (Y220C), and HT29 (R273H) cells. (D) Rac1 interacted with the mutant p53 (R175H) DBD. (Left panel) The domain structure of mutant p53 (R175H) and mutant p53 fragments. (Right panel) H1299 cells were transfected with expression vectors of HA-tagged mutant p53 R175H fragments together with Rac1-Flag expression vectors. The antibody used for immunoprecipitation assays was anti-Flag for Rac1-Flag. (E) Mutant p53 interacted with the central region of the Rac1 protein. (Left panel) The domain structure of Rac1 and Rac1 fragments. (Right panel) H1299 cells were transfected with vectors expressing Myc-tagged Rac1 fragments together with mutant p53 (R175H) expression vectors. The antibody used for immunoprecipitation assays was DO-1 for mutant p53.
To define the domains of mutant p53 and Rac1 required for mutant p53–Rac1 interaction, vectors expressing HA-tagged different mutant p53 fragments and vectors expressing Myc-tagged different Rac1 fragments were constructed and transfected into H1299 cells for co-IP assays (Fig. 1D,E). Co-IP assays showed that the DBD of mutant p53, which contains the mutation, is required for mutant p53–Rac1 interaction; the mutant p53 fragment lacking the DBD failed to bind to Rac1, whereas all other mutant p53 fragments containing the DBD bound to Rac1 (Fig. 1D). Co-IP assays further showed that the central region of the Rac1 protein is required for mutant p53–Rac1 interaction; the Rac1 fragment lacking the central region failed to bind to mutant p53, whereas all other Rac1 fragments containing the central region bound to mutant p53 (Fig. 1E). Taken together, these results demonstrate that Rac1 is a novel mutant p53-binding protein in human cells, and the mutant p53 DBD and the central region of Rac1 are essential for mutant p53–Rac1 interaction.
Mutant p53 activates Rac1
Rac1 cycles between an inactive Rac1-GDP form and an active Rac1-GTP form in cells (Heasman and Ridley 2008; Bid et al. 2013). We investigated whether mutant p53 affects Rac1 activity in cells by using a Rac1 activation assay kit to specifically pull down the active Rac1-GTP in cells followed by Western blot assays to measure the levels of Rac1-GTP, which was then normalized with the levels of total Rac1 protein in cells. Ectopic expression of mutant p53 (R175H, R248W, or R273H) greatly increased Rac1-GTP levels but did not clearly affect the levels of total Rac1 in H1299 cells, indicating that mutant p53 enhances Rac1 activity but not total Rac1 levels in H1299 cells (Fig. 2A). The levels of ectopically expressed mutant p53 protein levels in H1299 cells were comparable with endogenous mutant p53 protein levels in the majority of tumor cells that we tested (Supplemental Fig. S1B). p21-activated kinase-1 and kinase-2 (PAK1/2) are critical downstream effector kinases of Rac1. Rac1-GTP can bind to PAK1/2 and subsequently increase PAK1/2 activity and autophosphorylation at multiple sites, including enzymatic active residues Ser144 of PAK1 (p-PAK1Ser144) and Ser141 of PAK2 (p-PAK2Ser141) (Chong et al. 2001; Heasman and Ridley 2008). The levels of p-PAK1Ser144 and p-PAK2Ser141 have been widely used to reflect Rac1 activity in cells (Kumar et al. 2006; Zhang et al. 2016). As shown in Supplemental Figure S2, Rac1 bound to PAK1 in H1299 cells. Notably, mutant p53 expression did not affect the interaction between Rac1 and PAK1. Ectopic expression of different forms of mutant p53 greatly increased the levels of p-PAK1Ser144 and p-PAK2Ser141 in H1299 cells (Fig. 2B). Similar results were observed in different human cancer cells containing different forms of endogenous mutant p53, including SK-BR-3, MDA-MB468, SW480, and LAPC4 cells; much higher levels of Rac1-GTP, p-PAK1Ser144, and p-PAK2Ser141 were observed in these cells compared with their corresponding cells in which endogenous mutant p53 was knocked down by shRNA vectors (Fig. 2C). The efficient knockdown of endogenous mutant p53 by shRNA was shown at both the mRNA (Supplemental Fig. S3) and protein (Fig. 2C) levels. The promoting effect of mutant p53 on Rac1 activity is independent of wild-type p53 function, since expression of wild-type p53 in H1299 decreased the levels of Rac1-GTP (Fig. 2D). Similar observations were made in two pairs of cell lines, including human colorectal HCT116 p53+/+ and p53−/− cells and breast MCF7 cells with or without stable knockdown of endogenous wild-type p53 by shRNA vectors. Wild-type p53 expression reduced the levels of Rac1-GTP but not total Rac1 in both HCT116 and MCF7 cells (Fig. 2D). These results are consistent with previous studies, including ours, showing that wild-type p53 inhibits Rac1 activity in cells (Guo et al. 2003; Zhang et al. 2016). Collectively, these results demonstrate that mutant p53 activates Rac1 in cells.
Figure 2.
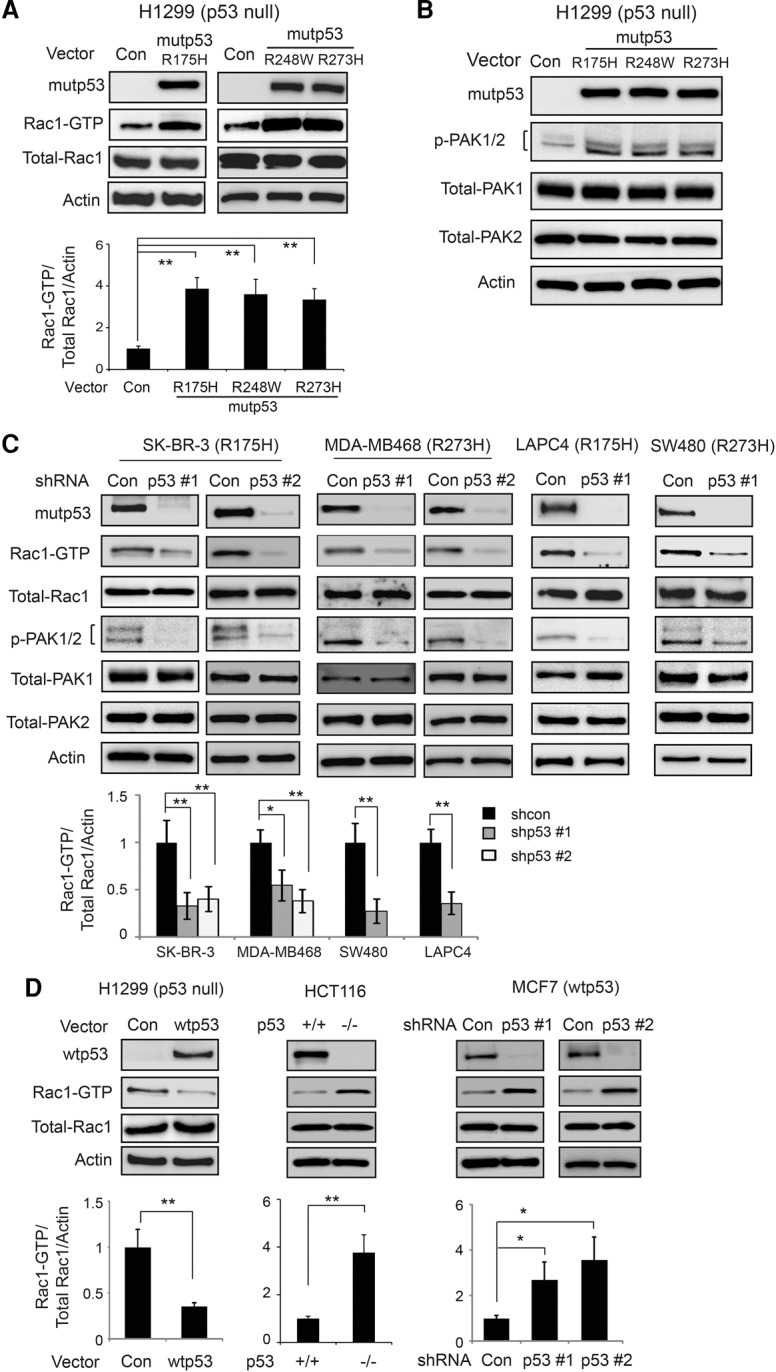
Mutant p53 activates Rac1 in human cancer cells. (A) Mutant p53 increased Rac1 activity in H1299 cells, represented by increased levels of Rac1-GTP. H1299 cells with or without ectopic mutant p53 (R175H, R248W, or R273H) expression were used to perform the GST-p21-binding domain of PAK1 pull-down assays to specifically pull down Rac1-GTP in cells followed by Western blot assays to measure the Rac1-GTP levels. (B) Ectopic mutant p53 expression increased the levels of p-PAK1Ser144 and p-PAK2Ser141 (p-PAK1/2) in H1299 cells. (C) Knockdown of endogenous mutant p53 by shRNA vectors decreased the levels of Rac1-GTP and p-PAK1/2 in SK-BR-3, MDA-MB468, LAPC4, and SW480 cells. Two shRNA vectors against p53 were used for all cell lines, and very similar results were observed. For the sake of clarity, results of one shRNA vector are presented for SW480 and LAPC4 cells. (D) Wild-type p53 decreased Rac1 activity, represented by decreased Rac1-GTP levels in human cancer cells. H1299 cells with or without ectopic wild-type p53 expression, human colorectal cancer HCT116 cells with or without wild-type p53, and MCF7 cells with or without wild-type p53 knockdown were used. For A, C, and D, the top panels show representative Western blot images for Rac1 activity analysis, and the bottom panels show quantitation of relative Rac1-GTP/total Rac1/Actin levels. Data are presented as mean ± SD. n = 3. (*) P < 0.01; (**) P < 0.001, Student's t-test.
We further investigated whether mutant p53 increases Rac1 activity in a cohort of human colorectal cancer samples with known p53 mutation status (n = 98) (Zheng et al. 2013). Rac1 activity was determined by measuring the levels of p-PAK1/2 (p-PAK1Ser144 and p-PAK2Ser141) using immunohistochemistry (IHC) staining. In tumors containing mutant p53, 72% of tumors (36 out of 50) had high levels of p-PAK1/2 (>30% of the cells were stained), whereas only 31% of the tumors (15 out of 48) with wild-type p53 had high levels of p-PAK1/2, indicating a significant correlation (P = 0.0001) between p53 mutation and Rac1 activity (Fig. 3A). The association of p53 mutation with Rac1 activity was further analyzed in the metastases from castration-resistant prostate cancer patients (n = 14) at rapid autopsy (Rubin et al. 2000). We were able to obtain both the p53 mutation status and the p-PAK2Ser141 status from these samples (Grasso et al. 2012; Drake et al. 2016). Compared with tumors without p53 missense mutations (including wild-type p53 or p53 deletion; n = 6), tumors containing missense p53 mutations (n = 8) expressed higher levels of p-PAK2Ser141 (Fig. 3B). These results suggest that mutant p53 status is significantly correlated with increased Rac1 activity in human cancers.
Figure 3.
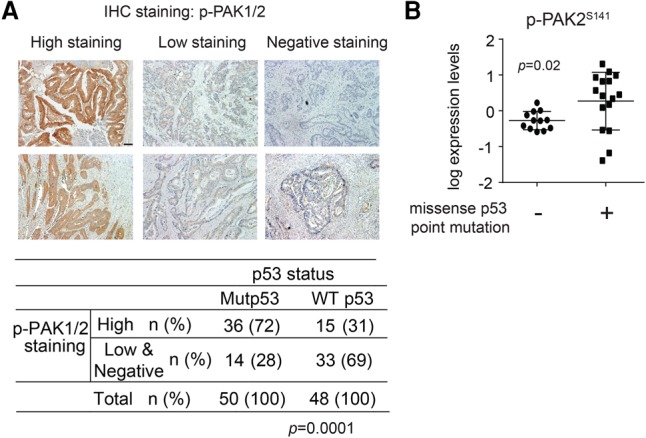
Mutant p53 is associated with high Rac1 activity in human tumor samples. (A) p53 mutations are associated with high Rac1 activity in human colorectal tumors. The levels of p-PAK1/2 were analyzed by IHC staining using an anti-p-PAK1/2 antibody. (Top panel) Representative images of p-PAK1/2 IHC staining in human colorectal tumor tissues. (High staining) More than 30% of cells stained with p-PAK1/2; (low staining) 10%–30% of cells stained with p-PAK1/2; (negative staining) <10% of cells stained with p-PAK1/2. Bar, 100 µm. (Bottom panel) Correlation between p53 mutations and Rac1 activity in human colorectal tumors. n = 98. P = 0.0001, χ2 test. (B) The high levels of p-PAK2Ser141 in metastatic castration-resistant prostate cancer lesions obtained at rapid autopsy (n = 8) with missense p53 point mutations as compared with tumors containing wild-type p53 or p53 deletions (n = 6). P = 0.02. Each sample was evaluated via the mass spectrometer with technical replicates, and both data points are displayed. Unpaired t-test with Welch's correction was used to compare the two groups.
Mutant p53 enhances Rac1 SUMOylation
SUMOylation is an important protein post-translational modification to regulate protein stability and/or activity (Hay 2013). SUMO1, SUMO2, and SUMO3 are the most common SUMO isoforms in mammalian cells. SUMO1 is very different from SUMO2 and SUMO3 in sequence and function, while SUMO2 and SUMO3 are almost identical in sequence and function (Hay 2013). It has been reported that Rac1 can be conjugated to SUMO-1, and this Rac1 SUMOylation is critical to maintain the active Rac1-GTP form and enhance Rac1 activity in cells (Castillo-Lluva et al. 2010). Indeed, ectopic SUMO-1 expression increased Rac1-GTP but not total Rac1 levels, whereas knockdown of endogenous SUMO-1 by siRNA oligos reduced Rac1-GTP but not total Rac1 levels in H1299 cells (Fig. 4A). The efficient knockdown of SUMO-1 by siRNA was shown at both the mRNA (Supplemental Fig. S4) and protein (Fig. 4A) levels. To investigate whether mutant p53 activates Rac1 through increasing Rac1 SUMOylation levels, H1299 cells were cotransfected with vectors expressing mutant p53 (R175H or R273H), Myc-Rac1, and HA-SUMO-1, respectively. The Myc-Rac1 SUMOylation levels in cells were determined by Myc-Rac1 pull-down followed by Western blot assays using an anti-HA antibody. Both R175H and R273H mutant p53 greatly increased the SUMOylation levels of Myc-Rac1 but not the levels of Myc-Rac1 protein in H1299 cells (Fig. 4B,C). Ectopic expression of mutant p53 (R175H) increased the SUMOylation levels of endogenous Rac1 but not the levels of endogenous Rac1 protein in H1299 cells (Fig. 4D). Knockdown of endogenous p53 (R175H and R273H, respectively) in SK-BR-3 and MDA-MB468 cells greatly reduced the SUMOylation levels of both ectopically expressed Rac1 and endogenous Rac1 (Fig. 4E,F). These results demonstrate that mutant p53 increases Rac1 SUMOylation, which could be a critical mechanism for Rac1 activation by mutant p53.
Figure 4.

Mutant p53 increases Rac1 SUMOylation levels in human cancer cells. (A) SUMOylation increased Rac1 activity in H1299 cells. (Left panels) Ectopic expression of HA-SUMO-1 in H1299 cells increased the levels of Rac1-GTP. (Right panels) Knockdown of endogenous SUMO-1 by two different siRNA oligos decreased the levels of Rac1-GTP. (Top panels) Representative Western blot images for Rac1 activity analysis. (Bottom panels) Quantitation of relative Rac1-GTP/total Rac1/Actin levels. Data are presented as mean ± SD. n = 3. (*) P < 0.01; (**) P < 0.001, Student's t-test. (B,C) Ectopic mutant p53 (R175H in B and R273H in C) increased SUMOylation levels of Myc-Rac1 in H1299 cells. H1299 cells were transfected with expression vectors of mutant p53 (R175H and R273H, respectively), Myc-Rac1, and HA-SUMO-1. The Myc-Rac1 SUMOylation levels were determined by Myc-Rac1 pull-down using an anti-Myc antibody followed by Western blot assays using an anti-HA antibody. (D) Mutant p53 (R175H) increased SUMOylaiton levels of endogenous Rac1 in H1299 cells. (E,F) Knockdown of endogenous mutant p53 in SK-BR-3 (E) and MDA-MB468 (F) cells, respectively, decreased SUMOylation levels of both ectopic Myc-Rac1 (top panels) and endogenous Rac1 (bottom panels). SK-BR-3 and MDA-MB468 cells with or without endogenous mutant p53 knockdown were transfected with expression vectors of HA-SUMO-1 along with or without Myc-Rac1.
Mutant p53 inhibits SENP1-mediated Rac1 de-SUMOylation to enhance Rac1 SUMOylation and Rac1 activity
SUMOylation is a dynamic process, which can be reversed by SENPs (Gareau and Lima 2010; Hay 2013). SENP1 has been reported to interact with Rac1 and de-SUMOylate Rac1 to down-regulate Rac1 activity in cells (Castillo-Lluva et al. 2010). Consistent with a previous report, ectopic expression of SENP1 reduced Rac1 SUMOylation levels in H1299 cells (Fig. 5A). Interestingly, we found that the central region of the Rac1 protein, which is required for mutant p53–Rac1 interaction, is also required for SENP1–Rac1 interaction; the Rac1 fragment lacking the central region failed to interact with SENP1 as determined by co-IP assays in cells (Fig. 5B). This result raises the possibility that mutant p53 may inhibit SENP1–Rac1 interaction through competing with SENP1 for the same interaction region in Rac1 protein. To test this possibility, H1299 cells were transfected with increasing amounts of mutant p53 (R175H or R273H) together with constant Myc-Rac1 and Flag-SENP1 expression for co-IP assays. Ectopic expression of mutant p53 (R175H and R273H, respectively) reduced the interaction of Flag-SENP1 with Myc-Rac1 in H1299 cells in a mutant p53 protein level-dependent manner as determined by co-IP assays (Fig. 5C). Furthermore, knockdown of endogenous mutant p53 (R175H and R273H, respectively) in SK-BR-3 and MDA-MB468 cells promoted the interaction of Flag-SENP1 with Myc-Rac1 in cells (Fig. 5D).
Figure 5.

Mutant p53 inhibits SENP1–Rac1 interaction and SENP1-mediated Rac1 de-SUMOylation in human cancer cells. (A) SENP1 decreased Rac1 SUMOylation levels in H1299 cells. H1299 cells were transfected with vectors expressing Flag-SENP1, Myc-Rac1, and HA-SUMO-1, respectively. (B) The central region of the Rac1 protein is required for SENP1–Rac1 interaction in cells. H1299 cells were transfected with vectors expressing Flag-SENP1 and Myc-Rac1 (full length or with the deletion of the central region), respectively, for co-IP assays. (FL) Full length. (C) Mutant p53 inhibited SENP1–Rac1 interaction in H1299 cells. H1299 cells were transfected with increasing amounts of mutant p53 (R175H or R273H) together with constant Flag-SENP1 and Myc-Rac1 expression for co-IP assays. (D) Knockdown of endogenous mutant p53 (R175H and R273H, respectively) in SK-BR-3 and MDA-MB468 cells increased SENP1–Rac1 interaction in cells. SK-BR-3 and MDA-MB468 cells with or without endogenous mutant p53 knockdown were transfected with vectors expressing Flag-SENP1 and Myc-Rac1, respectively, for co-IP assays. (E) Knockdown of endogenous SENP1 largely abolished the promoting effect of mutant p53 on Rac1 SUMOylation in H1299 cells. H1299 cells with or without endogenous SENP1 knockdown by siRNA oligos were transfected with the indicated combination of vectors expressing mutant p53 (R175H), Myc-Rac1, and HA-SUMO-1, respectively. (F) Knockdown of endogenous SENP1 largely abolished the promoting effect of mutant p53 on Rac1 activation in H1299 cells, which can be restored by expression of SENP1 but not SENP1C603S resistant to the SENP1 siRNA targeting endogenous SENP1 at its 3′ untranslated region (UTR). H1299 cells with or without endogenous SENP1 knockdown by siRNA targeting endogenous SENP1 at its 3′ UTR were transfected with the indicated combination of vectors. (G) Knockdown of SENP1 by siRNA in SK-BR-3 and MDA-MB468 cells largely abolished the promoting effect of endogenous mutant p53 (R175H and R273H) on Rac1 activity, which can be largely restored by expression of SENP1 but not SENP1C603S resistant to the SENP1 siRNA. In F and G, data are presented as mean ± SD. n = 3. (**) P < 0.001, Student's t-test.
We further investigated whether mutant p53 increases Rac1 SUMOylation through inhibiting SENP1-mediated de-SUMOylation. Knockdown of SENP1 by siRNA oligos in H1299 cells increased the SUMOylation levels of both ectopically expressed Myc-Rac1 and endogenous Rac1 (Fig. 5E; Supplemental Fig. S5A) and their activities (Fig. 5F; Supplemental Fig. S5B) in cells, which validated the role of SENP1 in Rac1 de-SUMOylation and inhibition of Rac1 activity. Notably, the promoting effect of mutant p53 on Rac1 SUMOylation and Rac1 activity was largely abolished in H1299 cells with SENP1 knockdown (Fig. 5E,F; Supplemental Fig. S5A,B). In SK-BR-3 and MDA-MB468 cells, knockdown of SENP1 by siRNA greatly increased Rac1 activity in cells with mutant p53 knockdown but had a much less pronounced effect in cells transduced with control shRNA and, furthermore, greatly abolished the promoting effect of endogenous mutant p53 (R175H and R273H, respectively) on Rac1 activity in cells (Fig. 5G; Supplemental Fig. S5C). The efficient knockdown of SENP1 by siRNA was shown at both the mRNA (Supplemental Fig. S5D) and protein (Fig. 5E–G) levels. A single missense mutation (C603S) in the catalytic domain of SENP1 was reported to create a catalytic inactive mutant SENP1 (Bailey and O'Hare 2004; Xu et al. 2006). To investigate whether the de-SUMOylation activity of SENP1 is critical for the promoting effect of mutant p53 on Rac1 activity, vectors expressing wild-type SENP1 or SENP1C603S were transfected into H1299, SK-BR-3, and MDA-MB468 cells in which endogenous SENP1 was knocked down by siRNA targeting the 3′ untranslated region (UTR) of endogenous SENP1. As shown in Figure 5F,G, expression of SENP1 but not SENP1C603S restored the promoting effect of mutant p53 on Rac1 activity, suggesting that the de-SUMOylation activity of SENP1 is critical for the promoting effect of mutant p53 on Rac1 activity. Collectively, these results suggest that mutant p53 interacts with Rac1 to inhibit SENP1-mediated Rac1 de-SUMOylation, which in turn increases Rac1 SUMOylation to activate Rac1 in cells.
Mutant p53 activates Rac1 to promote anchorage-independent cell growth and xenograft tumor growth
Promoting proliferation of tumor cells is an important mutant p53 GOF (Duan et al. 2008; Adorno et al. 2009). Rac1 activation plays an important role in promoting proliferation of tumor cells (Moore et al. 1997; Myant et al. 2013). Here, we investigated whether Rac1 activation by mutant p53 contributes to mutant p53 GOF in promoting anchorage-independent cell growth in soft agar. Expression of mutant p53 (R175H and R273H, respectively) in H1299 cells significantly promoted anchorage-independent cell growth in soft agar (Fig. 6A). Blocking Rac1 function by knockdown of endogenous Rac1 or expression of dominant-negative Rac1 (Rac1 DN; Rac1-T17N) inhibited anchorage-independent cell growth in soft agar. Notably, blocking Rac1 function largely abolished mutant p53 GOF in promoting anchorage-independent cell growth in H1299 cells (Fig. 6A,B). The efficient knockdown of Rac1 by shRNA at the mRNA level is shown in Supplemental Figure S6. Similar results were obtained when Rac1 function was blocked by NSC23766 (a specific Rac1 inhibitor) in H1299 cells (Fig. 6C).
Figure 6.

Blocking Rac1 function largely abolishes mutant p53 GOF in promoting anchorage-independent growth of tumor cells and growth of xenograft tumors. (A–C) Blocking Rac1 activity greatly abolished mutant p53 (R175H and R273H) GOF in promoting anchorage-independent growth of H1299 cells in soft agar. (A) H1299 cells expressing mutant p53 (R175H and R273H) and control H1299-Con cells were transfected with shRNA vectors against Rac1. (Left panels) Representative images. Bar, 200 µm. (Right panels) Average colony numbers in soft agar. (B) Cells were transfected with vectors expressing Rac1 DN. (C) Cells were treated with 10 µM NSC23766 or PBS. (D) Expression of Rac1-R but not Rac1ΔSUMO-R in H1299 cells with knockdown of endogenous Rac1 largely restored mutant p53 GOF in promoting anchorage-independent cell growth in soft agar. (E,F) Blocking Rac1 activity by shRNA vectors against Rac1 (E) or 10 µM NSC23766 treatment (F) greatly inhibited anchorage-independent cell growth in soft agar for SK-BR-3 (left panels) and MDA-MB468 (right panels) cells but had a less pronounced effect on SK-BR-3 and MDA-MB468 cells with knockdown of endogenous mutant p53. For A–F, data are presented as mean ± SD. n = 6. (#) P < 0.05; (*) P < 0.01; (**) P < 0.001, Student's t-test. (G) Rac1 knockdown by shRNA vectors greatly inhibited the growth of H1299 mutant p53 (R175H and R273H) xenograft tumors but had a less pronounced effect on H1299-Con tumors. (Left panels) Representative images of xenograft tumors. Bar, 1 cm. (Right panels) Growth curves of xenograft tumors. (H) Blocking Rac1 activity by expression of Rac1 DN largely abolished mutant p53 (R175H) GOF in promoting the growth of H1299 xenograft tumors. (I) Expression of Rac1-R but not Rac1ΔSUMO-R in H1299 cells with knockdown of endogenous Rac1 largely restored mutant p53 (R175H and R273H) GOF in promoting xenograft tumor growth. (J) Ki-67 IHC staining of H1299-Con and H1299 mutant p53 (R175H and R273H) xenograft tumors with or without Rac1 knockdown. (Left panels) Representative images. Bar, 20 µm. (K) The levels of Rac1 and p-PAK1/2 in H1299 mutant p53 (R175H) xenograft tumors as examined by Western blot assays. (L) Blocking Rac1 activity by NSC23766 treatment largely abolished mutant p53 (R175H) GOF in promoting the growth of H1299 xenograft tumors. Mice were treated with 2.5 mg/kg NSC23766 intraperitoneally once every 2 d for 2 wk starting from day 6 after cell inoculation. For G–I, and L, tumor volumes are presented as mean ± SD. n = 6 per group. (**) P < 0.001, ANOVA followed by Student's t-test.
It has been reported that almost 95% of the SUMO-1-modified Rac1 was conjugated at Lys188, Lys183, Lys184, and Lys186. Replacing lysine at these sites with arginine abolished most of Rac1 SUMOylation (Castillo-Lluva et al. 2010). To study whether the regulation of Rac1 SUMOylation by mutant p53 contributes to mutant p53 GOF in promoting anchorage-independent cell growth, vectors expressing wild-type Rac1 (Rac1-R) and SUMOylation-resistant Rac1 (Rac1ΔSUMO-R) that are resistant to Rac1 shRNA were constructed and transfected into H1299 cells with knockdown of endogenous Rac1. The expression levels of Rac1-R and Rac1ΔSUMO-R were comparable with endogenous Rac1 levels in cells (Supplemental Fig. S7A). Notably, expressing Rac1-R but not Rac1ΔSUMO-R in H1299 cells with knockdown of endogenous Rac1 largely restored mutant p53 GOF in promoting anchorage-independent cell growth (Fig. 6D). Mutant p53 GOF in promoting anchorage-independent cell growth was also observed in SK-BR-3 and MDA-MB468 cells. Knockdown of endogenous mutant p53 (R175H and R273H, respectively) in SK-BR-3 and MDA-MB468 cells significantly inhibited anchorage-independent cell growth in soft agar (Fig. 6E,F). Notably, knockdown of endogenous Rac1 in SK-BR-3 and MDA-MB468 cells largely abolished mutant p53 GOF in promoting anchorage-independent cell growth in these cells (Fig. 6E). Similar results were obtained when Rac1 function was blocked by NSC23766 in SK-BR-3 and MDA-MB468 cells (Fig. 6F). These results suggest that Rac1 activation by mutant p53 greatly contributes to mutant p53 GOF in promoting anchorage-independent cell growth.
We further investigated whether Rac1activation by mutant p53 contributes to mutant p53 GOF in tumorigenesis by using subcutaneous xenograft tumorigenesis assays in nude mice. Mutant p53 significantly promoted the growth of xenograft tumors; the growth rate of xenograft tumors formed by H1299 cells expressing R175H and R273 mutant p53 (H1299-R175H and H1299-R273H cells), respectively, was significantly faster than that formed by control cells transfected with empty vectors (H1299-Con cells) (Fig. 6G,H). Notably, knockdown of endogenous Rac1 or expression of Rac1 DN (Rac1-T17N) greatly abolished mutant p53 GOF in promoting xenograft tumor growth in H1299 cells (Fig. 6G,H). Expression of Rac1-R but not Rac1ΔSUMO-R in H1299 cells with knockdown of endogenous Rac1 largely restored mutant p53 GOF in promoting xenograft tumor growth (Fig. 6I), suggesting that up-regulation of Rac1 SUMOylation by mutant p53 contributes to mutant p53 GOF in promoting xenograft tumor growth. IHC staining of Ki-67, a cell proliferation marker, in xenograft tumors showed that mutant p53 (R175H and R273H) significantly promoted tumor cell proliferation in H1299 xenograft tumors (Fig. 6J). Furthermore, this promoting effect of mutant p53 was largely abolished in H1299 tumors with endogenous Rac1 knockdown (Fig. 6J). The promoting effect of mutant p53 on Rac1 activity was confirmed in xenograft tumors formed by H1299-Con and H1299-R175H cells by measuring the levels of p-PAK1/2 in tumors (Fig. 6K).
We further tested whether blocking Rac1 by the Rac1 inhibitor NSC23766 inhibits mutant p53 GOF in tumorigenesis. Mice were subcutaneously inoculated with tumor cells for xenograft tumor formation and then treated with 2.5 mg/kg NSC23766 or vehicle intraperitioneally once every 2 d for 2 wk starting at day 6. NSC23766 treatment significantly inhibited the growth of H1299 xenograft tumors and largely abolished the mutant p53 GOF in promoting xenograft tumor growth (Fig. 6L). Together, these results demonstrate that mutant p53 activates Rac1 to promote its GOF in anchorage-independent growth of cancer cells and growth of xenograft tumors.
Mutant p53 activates Rac1 to promote tumor metastasis
Promoting migration, invasion, and metastasis of tumor cells is one of important mutant p53 GOFs (Freed-Pastor and Prives 2012; Zheng et al. 2013; Muller and Vousden 2014). Rac1 activation plays a critical role in promoting migration, invasion, and metastasis of tumor cells (Heasman and Ridley 2008; Bid et al. 2013). Here, we investigated whether Rac1 activation is an important mechanism underlying mutant p53 GOF in promoting cancer metastasis. In H1299, SK-BR-3, and MDA-MB468 cells, mutant p53 (R175H and R273H, respectively) significantly promoted cell migration and invasion as determined by transwell assays (Fig. 7A–D [for migration in H1299 cells], E–H [for migration in SK-BR-3 and MDA-MB468 cells]; Supplemental Fig. 7B–K for invasion). Blocking Rac1 function by knockdown of endogenous Rac1, expression of Rac1 DN, or NSC23766 treatment in these cells significantly inhibited cell migration and invasion (Fig. 7A–H for migration; Supplemental Fig. S7B–K for invasion). Notably, blocking Rac1 function largely abolished mutant p53 GOF in promoting cell migration and invasion in these cells (Fig. 7A–H for migration; Supplemental Fig. S7B–K for invasion). Furthermore, this effect on cell migration and invasion was not due to the changes of rates of cell proliferation or survival in these cells (Supplemental Fig. S8). Notably, expression of Rac1-R but not Rac1ΔSUMO-R in H1299 and SK-BR-3 cells with knockdown of endogenous Rac1 largely restored mutant p53 GOF in promoting cell migration and invasion in cells (Fig. 7D,H; Supplemental Fig. S7E), suggesting that the regulation of Rac1 SUMOylation by mutant p53 contributes to mutant p53 GOF in promoting migration and invasion.
Figure 7.

Blocking Rac1 function largely abolishes mutant p53 GOF in promoting migration and metastasis of tumor cells. (A–C) Blocking Rac1 activity greatly abolished mutant p53 (R175H and R273H) GOF in promoting migration of H1299 cells as measured by transwell assays. (A) H1299 mutant p53 (R175H and R273H) and H1299-Con cells were transfected with shRNA vectors against Rac1. (B) Cells were transfected with vectors expressing Rac1 DN. (C) Cells were treated with 10 µM NSC23766 or PBS. (D) Expression of Rac1-R but not Rac1ΔSUMO-R largely restored mutant p53 GOF in promoting migration in H1299 cells. (E–G) Blocking Rac1 by shRNA vectors against Rac1 (E), expression of Rac1 DN (F), or treatment with 10 µM NSC23766 (G) greatly abolished mutant p53 GOF in promoting migration of SK-BR-3 and MDA-MB468 cells as measured by transwell assays. (H) Expression of Rac1-R but not Rac1ΔSUMO-R largely restored mutant p53 GOF in promoting migration in SK-BR-3 cells with knockdown of endogenous Rac1. The left panels in A and E show representative images. Bar, 200 µm. (Right panel) Quantification of the average number of migrated cells per field. In A–H, data are presented as mean ± SD. n = 6. (#) P < 0.05; (*) P < 0.01; (**) P < 0.001, Student's t-test. (I,J) Blocking Rac1 activity by shRNA vectors against Rac1 (I) or expression of Rac1 DN (J) largely abolished mutant p53 GOF in promoting lung metastasis of H1299 cells in mice. H1299-Con and H1299 mutant p53 (R175H and R273) cells with or without Rac1 knockdown (I) or expression of Rac1 DN (J) were injected into nude mice via the tail vein. The number of lung tumors was determined after 6 wk. n = 8 mice per group. (Left panel) Representative H&E images of lung sections. Bar, 100 µm. (K) A schematic model depicting that mutant p53 inhibits SENP1-meidated Rac1 de-SUMOylation to activate Rac1 as an important mechanism of mutant p53 GOF in tumorigenesis.
We further investigated whether mutant p53 activates Rac1 to promote metastasis in vivo. To this end, H1299-Con and H1299-R175H cells with Rac1 knockdown or Rac1 DN expression were injected into mice through the tail vein for in vivo lung metastasis assays. Mutant p53 expression in H1299 cells significantly promoted lung metastatic tumor formation, demonstrating mutant p53 GOF in promoting tumor metastasis (Fig. 7I,J). Notably, Rac1 knockdown or Rac1 DN expression largely abolished GOF of R175H mutant p53 in promoting lung tumor metastasis (Fig. 7I,J). Similar results were observed for R273H mutant p53; Rac1 knockdown in H1299-R273H cells largely abolished mutant p53 GOF in promoting lung metastasis (Fig. 7I, right panel). These results strongly suggest that Rac1 activation by mutant p53 is an important mechanism underlying mutant p53 GOF in metastasis.
Discussion
p53 is frequently mutated in cancer, and mutant p53 often exhibits GOF in tumorigenesis, which makes mutant p53 an attractive target for cancer therapy. While the concept of mutant p53 GOF in tumorigenesis has been well established and many GOF activities of mutant p53 have been demonstrated, the mechanisms underlying mutant p53 GOF in tumorigenesis are not well understood, which hinders the development of efficient therapeutic strategies for cancer containing mutant p53 (Freed-Pastor and Prives 2012; Muller and Vousden 2013; Gurpinar and Vousden 2015). Currently, different strategies are being tested for mutant p53 targeted cancer therapy. One of the strategies is to restore the conformation of mutant p53 protein to wild-type p53 with pharmacological agents (Selivanova and Wiman 2007; Gurpinar and Vousden 2015). Mutant p53 proteins often become stable and accumulate to high levels in tumors to exert mutant p53 GOF (Freed-Pastor and Prives 2012; Muller and Vousden 2013). Recent studies, including ours, identified several proteins—including HSP90, BAG2, BAG5, and cancer-associated MDM2 short isoforms—that can interact with mutant p53 and inhibit its degradation, contributing to mutant p53 stabilization and accumulation in tumors (Muller et al. 2005; Zheng et al. 2013; Yue et al. 2015, 2016). Therefore, inducing mutant p53 protein degradation has been tested as an alternative strategy in cancer therapy (Alexandrova et al. 2015; Gurpinar and Vousden 2015). However, these strategies have certain limitations, in part due to the heterogeneity of mutant p53 proteins in cancer. Targeting signaling pathways that mediate mutant p53 GOF could be another effective therapeutic strategy for tumors containing mutant p53 (Gurpinar and Vousden 2015). In this study, we identified Rac1 signaling as an unidentified and important pathway for mutant p53 GOF to promote tumorigenesis and metastasis. Results from this study showed that Rac1 activation by mutant p53 contributes greatly to mutant p53 GOF in promoting tumor growth and metastasis. Blocking Rac1 function by knockdown of Rac1 or expression of Rac1 DN largely abolished mutant p53 GOFs in promoting tumor growth and metastasis (Figs. 6, 7). Importantly, the pharmacological inhibitor of Rac1 can effectively block mutant p53 GOF in tumor growth, migration, and invasion (Figs. 6, 7). These results strongly suggest that targeting Rac1 could be a feasible therapeutic strategy for cancer cells containing mutant p53.
Rac1 signaling plays a crucial role in a number of important cellular functions, including the assembly and disassembly of cytoskeletal elements, cell adhesion, migration, invasion, cell growth, and cell cycle regulation (Heasman and Ridley 2008; Bid et al. 2013). The increase of Rac1 activity is observed in many human cancers and contributes to tumor development and progression (Thomas et al. 2007; Kamai et al. 2010; Chen et al. 2014). In this study, we found that different GOF mutant p53s bind to and activate Rac1 in different human cancer cells. It remains unclear how these mutant p53 proteins with different mutations in the DBD that we tested all acquire the ability to bind to Rac1, which will be of interest to investigate in future studies. In human colorectal cancer and prostate cancer samples that we examined, p53 mutation is significantly associated with increased Rac1 activity, strongly suggesting the activation of Rac1 signaling by mutant p53 in human tumors. Furthermore, we found that mutant p53 inhibits the interaction of Rac1 with SENP1, which in turn inhibits SENP1-mediated Rac1 de-SUMOylation to activate Rac1. These results demonstrate that mutant p53 is a novel regulator of Rac1, which reveals a novel mechanism for Rac1 activation in human cancer.
Mutant p53 can exert its GOF through interacting with several different transcription factors to regulate gene expression. The results in Figure 2, A and B, show that mutant p53 did not affect total Rac1 levels. We examined the effect of mutant p53 on the expression levels of a list of Rac1 regulators (Hodge and Ridley 2016) in three pairs of human cancer cell lines with or without knockdown of endogenous mutant p53 by analyzing the Affymetrix microarray gene expression data from three Genomic Spatial Event (GSE) data sets from published studies, including ours (Girardini et al. 2011; Freed-Pastor et al. 2012; Zhao et al. 2015). Mutant p53 did not clearly affect the expression of these Rac1 regulators (Supplemental Table 2). These results excluded the possibility that the promoting effect of mutant p53 on Rac1 activity is contributed by the expression changes of these genes, although there is the possibility that the expression regulation of some unknown Rac1 regulators by mutant p53 contributes to the promoting effect of mutant p53 on Rac1 activity. It is worth noting that mutant p53 and wild-type p53 often exert opposite effects on the same signaling pathway and biological processes through different mechanisms. It appears that wild-type p53 and mutant p53 exert opposite effects on Rac1 signaling. As shown in Figure 2D, wild-type p53 inhibits Rac1 activity in human cancer cells. Previous studies, including ours, showed that glutaminase 2 (GLS2) is a target gene of wild-type p53 and that wild-type p53 induces GLS2 expression in cells (Hu et al. 2010; Suzuki et al. 2010). Our recent study further showed that GLS2 mediates the inhibitory function of wild-type p53 on Rac1 activity; GLS2 binds to Rac1 at its switch I and II regions to inhibit Rac1 activation (Zhang et al. 2016). Results in this study showed that mutant p53 protein binds to Rac1 at its central region (Fig. 1E), which is different from the region for GLS2–Rac1 interaction. Unlike wild-type p53, mutant p53 does not induce GLS2 expression in H1299 cells with ectopic expression of mutant p53 (R175H, R248W, or R273H) (Supplemental Fig. S9A). GLS2 can bind to the mutant p53/Rac1 complex; the interaction between mutant p53 and GLS2 was observed by co-IP assays using the DO-1 antibody against p53 in H1299 cells transfected with vectors expressing GLS2, Rac1, and mutant p53 (R175H) (Supplemental Fig. S9B). Knockdown of endogenous Rac1 abolished the interaction of mutant p53 with GLS2 in H1299 cells (Supplemental Fig. S9B), suggesting that mutant p53 interacts with GLS2 through Rac1. However, knockdown of endogenous GLS2 by shRNA vectors in H1299 cells with and without ectopic mutant p53 expression and in SK-BR-3 and MDA-MB468 cells with and without mutant p53 knockdown did not have an obvious effect on basal Rac1 activity or the Rac1 activation by mutant p53 (Supplemental Fig. S9C,D). These results suggest that GLS2, which expresses at a low level in cells without wild-type p53, plays a very limited role in regulating Rac1 activity in cells without wild-type p53.
In summary, results in this study demonstrate that mutant p53 activates Rac1 through binding to Rac1 protein to inhibit SENP1-mediated Rac1 de-SUMOylation, which in turn promotes mutant p53 GOF in tumor cell growth and metastasis (Fig. 7K). Results from this study uncover an unidentified and important mechanism underlying mutant p53 GOF in tumorigenesis and reveal a novel mechanism for Rac1 activation in human cancer. These results suggest that targeting Rac1 and its signaling is a potential therapeutic strategy for cancer with mutant p53.
Materials and methods
Cells, vectors, siRNAs, shRNAs, and reagents for cell treatment
H1299, SK-BR-3, MDA-MB468, LAPC4, SW480, HT29, and Huh7 cell lines were obtained from American Type Culture Collection. H1299 cells with stable ectopic mutant p53 (R175H, R248W, and R273H) overexpression were established as described previously (Zhang et al. 2013). Vectors expressing mutant p53 (R175H, R248W, and R273H), HA-tagged mutant p53 fragments, Myc-Rac1, Myc-Rac1-DN, and Myc-tagged Rac1 fragments were constructed as described previously (Zheng et al. 2013; Zhang et al. 2016). The pcDNA3-HA-SUMO-1 vector was a gift from Dr. Junying Yuan (Addgene plasmid no. 21154) (Terui et al. 2004). The Flag-SENP1 vector was constructed by PCR cloning with the primers listed in Supplemental Table 3. The Flag-SENP1C603S vector was constructed by using QuikChange IIXL site-directed mutagenesis kit (Agilent) with the primers listed in Supplemental Table 3. The lentiviral shRNA vectors against Rac1 (#1: V3LHS-317667, and #2: V3LHS_317664) were obtained from Thermo Scientific. siRNA oligos were purchased from IDT, and their sequences are listed in Supplemental Table 3. Myc-Rac1 and GFP-Rac1ΔSUMO (a gift from Dr. Angeliki Malliri at the University of Manchester) were cloned into pLVX-IRES-tdTomat vectors to construct Rac1-R and Rac1ΔSUMO-R that are resistant to Rac1 shRNA vector #1 using QuikChange IIXL site-directed mutagenesis kit with the primers listed in Supplemental Table 3. The Rac1 inhibitor NSC23766 was purchased from Sigma.
Co-IP assays and LC-MS/MS assays
To determine potential mutant p53-binding proteins, H1299 cells transfected with the empty vector and H1299 cells with ectopic expression of mutant p53 (R175H) or wild-type p53 were used for co-IP by using anti-p53 (DO-1) beads. The p53 protein complex was eluted by 0.1 M blycine solution, separated in a SDS gel, visualized by silver staining using the silver staining kit (Invitrogen), and analyzed by LC-MS/MS at the Biological Mass Spectrometry Facility of Rutgers University as described previously (Yue et al. 2015). Co-IP assays were performed as described previously (Yue et al. 2015). Anti-p53 (DO-1) and anti-Flag (M2, Sigma) agarose beads were used to pull down p53, Rac1-Flag, and Flag-SENP1 proteins, respectively.
Subcellular fractionation and Western blot assays
Cytoplasmic and nuclear fractions were separated by using a Qproteome cell compartment kit (Qiagen). Standard Western blot assays were used to analyze protein expression in cells. The following antibodies were used for assays: anti-p53 (1:2000 dilution; Santa Cruz Biotechnology, FL393), anti-Flag (1:40,000 dilution; Sigma, F7425), anti-Rac1 (1:4000 dilution; Millipore, 05-389), anti-HA (1:2000 dilution; Roche, 12013819001), anti-Myc (1:2000 dilution; Santa Cruz Biotechnology, SC-40 HRP), anti-SENP1 (1:2000 dilution; Cell Signaling Technology, 11929), anti-α-tubulin (1:2000 dilution; Santa Cruz Biotechnology, sc8035), anti-Histone H3 (1:1000 dilution; Cell Signaling Technology, 9715), anti-SUMO-1 (1:2000 dilution; Cell Signaling Technology, 4930), anti-p-PAK1/2 (1:1000 dilution; Cell Signaling Technology, 2606), anti-PAK1 (1:2000 dilution; Cell Signaling Technology, 2602), and anti-PAK2 (1:2000 dilution; Cell Signaling Technology, 2615).
Rac1 activity analysis
The p21-binding domain of the Rac1 effector protein PAK1 specifically binds to Rac1-GTP in cells (Hayashi-Takagi et al. 2010). Rac1 activity was analyzed by using a Rac1 activation assay kit (Millipore) to perform the GST-p21-binding domain of PAK1 pull-down assays, which specifically pull down Rac1-GTP in cells as described previously (Krauthammer et al. 2012; Zhang et al. 2016). The levels of precipitated Rac1-GTP were measured by Western blot assays and normalized to total Rac1 levels in cells.
Quantitative real-time PCR
Total RNAs of cells were isolated and reverse-transcribed to cDNA by using a cDNA reverse transcription kit (Thermo Scientific). The expression of genes was detected by TaqMan real-time PCR (Applied Biosystems) and normalized with actin gene as described (Hu et al. 2010).
IHC staining
A cohort of deidentified colorectal cancer tissues with known p53 mutation status was obtained from the database of the First Affiliated Hospital of Harbin Medical University (Harbin, China) with institutional review board approval (Zheng et al. 2013). None of these patients received presurgical chemotherapy. IHC staining was performed as described previously (Zheng et al. 2013). Anti-p-PAK1/2 (Abcam, ab40795) antibodies were used to detect the levels of p-PAK1/2 in tissues. Anti-Ki-67 (BD Biosciences, 556003) antibodies were used to detect the levels of Ki67 in xenograft tumor tissues.
Rac1 SUMOylation assays
SUMOylation assays were performed as described previously (Castillo-Lluva et al. 2010). In brief, cells were harvested 24 h after transfection with vectors expressing HA-SUMO-1, and cell lysates were prepared in NP40 buffer containing proteinase inhibitors and 10 mM NEM (N-ethylmaleimide). The SUMOylation levels of Myc-Rac1 and endogenous Rac1 were determined by immunoprecipitation using anti-Myc and anti-Rac1 antibodies, respectively, followed by Western blot assays using an anti-HA antibody.
Anchorage-independent growth assays
Anchorage-independent growth assays were performed as described previously (Li et al. 2014; Liu et al. 2014). In brief, cells were seeded in six-well plates coated with medium containing 0.6% agarose and cultured in medium containing 0.3% agarose. Colonies were stained by violet crystal for counting after 2–3 wk.
Xenograft tumorigenesis assays
Assays were performed as described previously (Zheng et al. 2013). In brief, 5 × 106 cells in 0.2 mL of PBS were injected subcutaneously into 6-wk-old BALB/c athymic male nude mice (Taconic). Tumor volumes were measured every 3 d for 3 wk. Tumor volume = 1/2 (length × width2) (n = 6 mice per group). The mouse experiments were performed with the approval of the Institutional Animal Care and Use Committee of the university.
Migration and invasion assays
Migration and invasion assays were performed by using the transwell system (BD Biosciences) as described previously (Zhang et al. 2016). In brief, cells in FBS-free medium were seeded into upper chambers coated with or without Matrigel (BD Biosciences) for invasion and migration assays, respectively. The lower chamber was filled with medium supplemented with 10% FBS. For SK-BR-3 cells, the lower chamber was filled with a 1:1 mix of medium supplemented with 10% FBS and NIH3T3 cell-conditioned medium. Cells on the lower surface of upper chambers were fixed by methanol and stained by violet crystal for counting after culturing for 24 h.
In vivo lung metastasis assay
In vivo lung metastasis assays were performed as described previously (Zheng et al. 2013; Zhang et al. 2016). In brief, 1 × 106 cells in 0.1mL of PBS were injected into 6-wk-old male nude mice via the tail vein (n = 8 mice per group). The mice were sacrificed at 6 wk after the inoculation to collect lung tissues. The numbers of lung tumors were counted under a dissecting microscope and confirmed by histopathological analysis.
Statistical analysis
The data are presented as mean ± SD. The differences in xenograft tumor growth among groups were analyzed for statistical significance by ANOVA followed by Student's t-tests using GraphPad Prism software. Unpaired t-test with Welch's correction was used to analyze the difference of the p-PAK2Ser141 levels between the two groups of prostate cancer samples. All other P-values were obtained using Student's t-test or χ2 test. Values of P < 0.05 were considered to be significant.
Supplementary Material
Acknowledgments
This study was supported by National Institutes of Health (NIH) grants 1R01CA160558-01 and 1R01CA203965 and Department of Defense grant W81XWH-16-1-0358 to W.H.; and NIH grant R01CA143204, the New Jersey Health Foundation, and Bush Medical Research Award to Z.F. X.Y. is supported by a New Jersey Commission on Cancer Research Post-doctoral Fellowship Award. Y.Z. is supported by NIH grant F99CA222734. W.H. and Z.F. designed experiments, analyzed the data, and wrote manuscript; X.Y., C.Z., Y.Z., J. Liu, A.W.L., M.N.B., and J. Li carried out the experiments and analyzed the data; and V.M.T., J.M.D., and L.L. carried out cancer sample data analysis.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.301564.117.
References
- Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, Solari A, Bobisse S, Rondina MB, Guzzardo V, et al. 2009. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137: 87–98. [DOI] [PubMed] [Google Scholar]
- Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, Lozano G, Dobbelstein M, Moll UM. 2015. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523: 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey D, O'Hare P. 2004. Characterization of the localization and proteolytic activity of the SUMO-specific protease, SENP1. J Biol Chem 279: 692–703. [DOI] [PubMed] [Google Scholar]
- Bid HK, Roberts RD, Manchanda PK, Houghton PJ. 2013. RAC1: an emerging therapeutic option for targeting cancer angiogenesis and metastasis. Mol Cancer Ther 12: 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo-Lluva S, Tatham MH, Jones RC, Jaffray EG, Edmondson RD, Hay RT, Malliri A. 2010. SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat Cell Biol 12: 1078–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Fu M, Zhang G, Zhou Y, Zhu S, Liu J, Wang D, Deng A, Wang Z. 2014. Rac1 regulates skin tumors by regulation of keratin 17 through recruitment and interaction with CD11b+Gr1+ cells. Oncotarget 5: 4406–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong C, Tan L, Lim L, Manser E. 2001. The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J Biol Chem 276: 17347–17353. [DOI] [PubMed] [Google Scholar]
- Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, Blandino G, Piaggio G. 2006. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10: 191–202. [DOI] [PubMed] [Google Scholar]
- Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, Stoyanova T, Faltermeier CM, Uzunangelov V, Carlin DE, et al. 2016. Phosphoproteome integration reveals patient-specific networks in prostate cancer. Cell 166: 1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Gao L, Jin D, Otterson GA, Villalona-Calero MA. 2008. Lung specific expression of a human mutant p53 affects cell proliferation in transgenic mice. Transgenic Res 17: 355–366. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. 2012. Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, et al. 2012. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148: 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gareau JR, Lima CD. 2010. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev Mol Cell Biol 11: 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, Capaci V, Jordan L, Quinlan P, Thompson A, et al. 2011. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell 20: 79–91. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. 2012. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487: 239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Gao Y, Wang L, Zheng Y. 2003. p19Arf–p53 tumor suppressor pathway regulates cell motility by suppression of phosphoinositide 3-kinase and Rac1 GTPase activities. J Biol Chem 278: 14414–14419. [DOI] [PubMed] [Google Scholar]
- Gurpinar E, Vousden KH. 2015. Hitting cancers’ weak spots: vulnerabilities imposed by p53 mutation. Trends Cell Biol 25: 486–495. [DOI] [PubMed] [Google Scholar]
- Hay RT. 2013. Decoding the SUMO signal. Biochem Soc Trans 41: 463–473. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, Makino Y, Seshadri AJ, Ishizuka K, Srivastava DP, et al. 2010. Disrupted-in-schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci 13: 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heasman SJ, Ridley AJ. 2008. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9: 690–701. [DOI] [PubMed] [Google Scholar]
- Hodge RG, Ridley AJ. 2016. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol 17: 496–510. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. 2010. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci 107: 7455–7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamai T, Shirataki H, Nakanishi K, Furuya N, Kambara T, Abe H, Oyama T, Yoshida K. 2010. Increased Rac1 activity and Pak1 overexpression are associated with lymphovascular invasion and lymph node metastasis of upper urinary tract cancer. BMC Cancer 10: 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. 2012. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet 44: 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Gururaj AE, Barnes CJ. 2006. p21-activated kinases in cancer. Nat Rev Cancer 6: 459–471. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Hu W, Feng Z. 2006. The P53 pathway: what questions remain to be explored? Cell Death Differ 13: 1027–1036. [DOI] [PubMed] [Google Scholar]
- Li X, Yang Q, Yu H, Wu L, Zhao Y, Zhang C, Yue X, Liu Z, Wu H, Haffty BG, et al. 2014. LIF promotes tumorigenesis and metastasis of breast cancer through the AKT–mTOR pathway. Oncotarget 5: 788–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Zhang C, Wang XL, Ly P, Belyi V, Xu-Monette ZY, Young KH, Hu W, Feng Z. 2014. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ 21: 1792–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KA, Sethi R, Doanes AM, Johnson TM, Pracyk JB, Kirby M, Irani K, Goldschmidt-Clermont PJ, Finkel T. 1997. Rac1 is required for cell proliferation and G2/M progression. Biochem J 326: 17–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. 2013. p53 mutations in cancer. Nat Cell Biol 15: 2–8. [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. 2014. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller P, Ceskova P, Vojtesek B. 2005. Hsp90 is essential for restoring cellular functions of temperature-sensitive p53 mutant protein but not for stabilization and activation of wild-type p53: implications for cancer therapy. J Biol Chem 280: 6682–6691. [DOI] [PubMed] [Google Scholar]
- Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, Lukashchuk N, Gillespie DA, Ludwig RL, Gosselin P, et al. 2009. Mutant p53 drives invasion by promoting integrin recycling. Cell 139: 1327–1341. [DOI] [PubMed] [Google Scholar]
- Myant KB, Cammareri P, McGhee EJ, Ridgway RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos D, et al. 2013. ROS production and NF-κB activation triggered by RAC1 facilitate WNT-driven intestinal stem cell proliferation and colorectal cancer initiation. Cell Stem Cell 12: 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin MA, Putzi M, Mucci N, Smith DC, Wojno K, Korenchuk S, Pienta KJ. 2000. Rapid (‘warm’) autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res 6: 1038–1045. [PubMed] [Google Scholar]
- Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD. 2001. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem 276: 39359–39367. [DOI] [PubMed] [Google Scholar]
- Selivanova G, Wiman KG. 2007. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene 26: 2243–2254. [DOI] [PubMed] [Google Scholar]
- Stindt MH, Muller PA, Ludwig RL, Kehrloesser S, Dotsch V, Vousden KH. 2015. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 34: 4300–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. 2010. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci 107: 7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terui Y, Saad N, Jia S, McKeon F, Yuan J. 2004. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J Biol Chem 279: 28257–28265. [DOI] [PubMed] [Google Scholar]
- Thomas EK, Cancelas JA, Chae HD, Cox AD, Keller PJ, Perrotti D, Neviani P, Druker BJ, Setchell KD, Zheng Y, et al. 2007. Rac guanosine triphosphatases represent integrating molecular therapeutic targets for BCR-ABL-induced myeloproliferative disease. Cancer Cell 12: 467–478. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, Rajkowska K, Gaweda-Walerych K, Ingallina E, Tonelli C, et al. 2016. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat Cell Biol 18: 897–909. [DOI] [PubMed] [Google Scholar]
- Xu Z, Chau SF, Lam KH, Chan HY, Ng TB, Au SW. 2006. Crystal structure of the SENP1 mutant C603S–SUMO complex reveals the hydrolytic mechanism of SUMO-specific protease. Biochem J 398: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Zhao Y, Liu J, Zhang C, Yu H, Wang J, Zheng T, Liu L, Li J, Feng Z, et al. 2015. BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. Elife 4: e08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X, Zhao Y, Huang G, Li J, Zhu J, Feng Z, Hu W. 2016. A novel mutant p53 binding partner BAG5 stabilizes mutant p53 and promotes mutant p53 GOFs in tumorigenesis. Cell Discov 2: 16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, et al. 2013. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun 4: 2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Zhao Y, Yue X, Zhu Y, Wang X, Wu H, Blanco F, Li S, Bhanot G, et al. 2016. Glutaminase 2 is a novel negative regulator of small GTPase Rac1 and mediates p53 function in suppressing metastasis. Elife 5: e10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang C, Yue X, Li X, Liu J, Yu H, Belyi VA, Yang Q, Feng Z, Hu W. 2015. Pontin, a new mutant p53-binding protein, promotes gain-of-function of mutant p53. Cell Death Differ 22: 1824–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T, Wang J, Zhao Y, Zhang C, Lin M, Wang X, Yu H, Liu L, Feng Z, Hu W. 2013. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat Commun 4: 2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.