

Nakagawa et al. show that the maintenance of appropriate β-catenin-mediated Wnt tone is necessary for the orderly differentiation of cortical progenitors and the resultant formation of the cerebral cortex.
Keywords: cortical progenitors, APC, β-catenin, cerebral cortical development, Wnt signaling, primary cilia, autism
Abstract
Adenomatous polyposis coli (APC) regulates the activity of β-catenin, an integral component of Wnt signaling. However, the selective role of the APC–β-catenin pathway in cerebral cortical development is unknown. Here we genetically dissected the relative contributions of APC-regulated β-catenin signaling in cortical progenitor development, a necessary early step in cerebral cortical formation. Radial progenitor-specific inactivation of the APC–β-catenin pathway indicates that the maintenance of appropriate β-catenin-mediated Wnt tone is necessary for the orderly differentiation of cortical progenitors and the resultant formation of the cerebral cortex. APC deletion deregulates β-catenin, leads to high Wnt tone, and disrupts Notch1 signaling and primary cilium maintenance necessary for radial progenitor functions. β-Catenin deregulation directly disrupts cilium maintenance and signaling via Tulp3, essential for intraflagellar transport of ciliary signaling receptors. Surprisingly, deletion of β-catenin or inhibition of β-catenin activity in APC-null progenitors rescues the APC-null phenotype. These results reveal that APC-regulated β-catenin activity in cortical progenitors sets the appropriate Wnt tone necessary for normal cerebral cortical development.
Radial glial cells (RGCs) in the inner and outer ventricular zones (VZs) are progenitors of the mammalian neocortex (Hansen et al. 2010; Evsyukova et al. 2013). Symmetric RGC divisions result in two daughter RGCs and the expansion of the progenitor pool. Asymmetric divisions of RGCs can give rise to either a RGC and a daughter neuron or a transit-amplifying intermediate progenitor (IP). IPs subsequently divide symmetrically to generate more neurons. Ventricular RGCs are highly polarized, with a short apical process that forms an end foot attached to the ventricular surface and a long radial process attached to the pial basement membrane that serves as a scaffold to guide the radial migration of newborn neurons (Rakic 1988; Evsyukova et al. 2013). A subpopulation of RGCs, outer RGCs, does not have apical attachments to the ventricular surface (Hansen et al. 2010). The appropriate organization, balance of self-renewal, and differentiation of radial progenitors are central to the construction of the cerebral cortex.
Multiple signaling pathways, including Wnts, GSK-3, CDC42, APC (adenomatous polyposis coli), Arl13b, Notch, retinoic acid, neuregulins, IGF, PDGF, and Sonic Hedgehog (Shh), have been identified as regulators of radial progenitor development in the cerebral cortex (Chenn and Walsh 2002; Machon et al. 2003; Hirabayashi et al. 2004; Cappello et al. 2006; Mizutani et al. 2007; Rasin et al. 2007; Kim et al. 2009; Yokota et al. 2009, 2010; Kusek et al. 2012; Fang et al. 2013; Higginbotham et al. 2013; Imayoshi et al. 2013; Lui et al. 2014; Chen et al. 2015; Guo et al. 2015; Kong et al. 2015; Lun et al. 2015; Durak et al. 2016; Wang et al. 2016a,b). Recent evidence suggests that the primary cilium, a polarized microtubule (MT)-based organelle that projects from the cell membrane, functions as a signaling center in progenitors during development (Hildebrandt et al. 2011; Higginbotham et al. 2013). Components of the Shh pathway are enriched in primary cilia, and mutations that disrupt Shh signaling result in defective patterning and proliferation of neural progenitors (Wilson et al. 2012; Guo et al. 2015). A reciprocal relationship between the primary cilium and Wnt signal transduction exists. Components of the noncanonical Wnt/planar cell polarity pathway are important for ciliogenesis, and primary cilium signaling can either contextually restrain or activate canonical Wnt/β-catenin signaling (Lancaster et al. 2011; Kong et al. 2015). Furthermore, a role for the primary cilium in Notch signaling and progenitor differentiation has also been identified (Ezratty et al. 2011). Importantly, ciliopathies caused primarily by defects in primary ciliary structure and/or function have pronounced neurodevelopmental deficits, including disrupted progenitor development (Hildebrandt et al. 2011; Guo et al. 2015). The development and differentiation of radial progenitors are controlled by the temporal and spatial integration of these various signaling pathways and centers. However, the underlying molecular and cellular mechanisms necessary to achieve this integration in neural progenitor cells remain largely unknown.
Here, we demonstrate that APC–β-catenin-regulated Wnt signaling balance is required to integrate the developmental signaling pathways necessary for appropriate progenitor development and thus cerebral cortical formation. APC deletion up-regulates β-catenin, causing loss of primary cilia, defective Shh and Notch signaling, and complete disruption of cerebral cortical formation. However, down-modulation of β-catenin and thus Wnt signaling in APC-null progenitors dramatically restores cortical progenitor development and cerebral cortical formation in APC-null brains. These results suggest that the APC–β-catenin pathway sets the appropriate Wnt tone in neuronal progenitors necessary for cerebral cortical formation.
Results
APC is essential for cortical progenitor proliferation and neurogenesis
To examine the role of APC in the proliferation and differentiation of cortical progenitors, we generated APCLox/Lox; hGFAP-Cre (APC conditional knockout [cKO]) mice in which Cre-mediated inactivation of APC occurs in dorsal cortical progenitors from embryonic day 13.5 (E13.5) (Supplemental Fig. 1A; Zhuo et al. 2001; Yokota et al. 2009). Proliferating radial progenitors in S phase were pulse-labeled with bromodeoxyuridine (BrdU) and visualized by immunolabeling with anti-Pax6 antibodies. Loss of APC leads to reduction of BrdU+/Pax6+ radial progenitors (Fig. 1A–I). Consistently, phosphorylated histone H3-positive (PH3+) M-phase radial progenitors were also reduced in APC cKO (Supplemental Fig. 2A,B). Many of the APC-deficient radial progenitors were located ectopically in the cortical plate (CP) or intermediate zone (IZ) instead of in the VZ (Fig. 1D,H). We then determined the effect of altered proliferation of radial progenitors in the generation of IPs and neurons. Loss of APC in radial progenitors led to significantly fewer Tbr2+ IPs, which also showed reduced proliferation (Fig. 1J–R). In spite of altered generation and proliferation of cortical progenitors, neurons were generated from these APC-deficient progenitors, albeit in fewer numbers (Fig. 1S,T). However, the emergence of neuronal identity in these newborn neurons is compromised. Unlike normal newborn neurons, a significant number of APC-deficient neurons coexpressed both the radial progenitor marker Pax6 and neuron-specific Tuj1 (Fig. 1U–Y). Furthermore, BrdU pulse followed 24 h later by Ki67 antibody colabeling indicates an increase in BrdU+/Ki67+ progenitors in the APC cKO cortex (Supplemental Fig. 2F,G,K,L), suggesting affected cell cycle progression in APC cKO progenitors. Examination of neurogenic niches such as the subventricular zone (SVZ) and hippocampus in the postnatal brains also indicates compromised proliferation of neural progenitors in APC cKO (data not shown). Together, these observations indicate that APC is necessary for normal cortical progenitor proliferation, appropriate neurogenesis, and the emergence of neuronal identity in the developing cerebral cortex.
Figure 1.

APC deletion disrupts cortical progenitor proliferation and the generation of cortical neurons. (A–H) Proliferating radial progenitors in control (A,B,E,F) and APC cKO (C,D,G,H) cortices at E14.5 (A–D) and E16.5 (E–H) were immunolabeled with anti-Pax6 and anti-BrdU antibodies. (D,H) APC deletion significantly reduced the number of proliferating radial progenitors and led to ectopic collection (arrowheads) of progenitors. (I) Quantification of the number of actively proliferating radial progenitors. (J–Q) Proliferating IPs in control (J,K,N,O) and APC cKO (L,M,P,Q) cortices at E14.5 (J–M) and E16.5 (N–Q) were immunolabeled with anti-Tbr2 and BrdU antibodies. (M,Q) APC deletion resulted in reduced numbers of proliferating IPs and ectopias (arrowheads). (R) Quantification of the number of actively proliferating IPs. A, C, E, G, J, L, N, and P are low-magnification images of cerebral cortices. B, D, F, H, K, M, O, and Q are high-magnification images of dorsal cerebral walls (outlined regions in A,C,E,G,J,L,N,P). (S,T) Generation and placement of Tuj1+ cortical neurons are disrupted in the APC cKO cortex. (U–X) Coimmunolabeling with Pax6 and Tuj1 antibodies indicates an increase in the number of cells that express both Pax6 and Tuj1 (asterisks in V,X) in APC cKO cortex. Images of the cerebral wall (U,V) and high-magnification images of the outlined (box) VZ region (W,X) are shown. (Y) Quantification of the number of Pax6+ Tuj1+ cells. Data shown are mean ± SEM. n = 3 for each genotype. (**) P < 0.01; (***) P < 0.001, Student's t-test. Bars: A,C,E,G,J,L,N,P, 100 µm; U,V, 20 µm; B,D,F,H,K,M,O,Q, 12 µm; S,T, 10 µm; W,X, 5 µm.
The role of APC in progenitor organization and function is dependent on β-catenin
APC's modulation of the β-catenin signaling pathway is considered to be an essential component of its function (Nelson and Nathke 2013). However, the relative contribution of APC–β-catenin interactions to radial progenitor development in the cerebral cortex is unknown. To examine how APC/β-catenin signaling regulates cortical progenitor development, we conditionally deleted APC and β-catenin in radial progenitors by generating APCLox/Lox; Ctnnb1Lox/Lox; hGFAP-Cre (APC/β-cat cKO) mice. Deletion of APC and/or β-catenin in APC cKO, Ctnnb1Lox/Lox; hGFAP-Cre (β-cat cKO), or APC/β-cat cKO brains was confirmed by immunoblotting analysis (Supplemental Fig. 1A). Loss of APC in APC cKO RGCs drastically disrupted RGC organization and function in the embryonic cortex (Fig. 2). APC cKO RGCs are short, have misoriented radial fibers (Fig. 2A,B), and do not proliferate appropriately (Fig. 2F,G; Supplemental Fig. 2A,B). Importantly, deletion of β-catenin in APC-deficient RGCs rescued the defects in APC cKO RGC organization. APC/β-catenin-deficient RGCs formed normally polarized RG scaffolds, and these progenitors proliferated similar to controls (Fig. 2C,H; Supplemental Fig. 2C), suggesting that APC-modulated β-catenin signaling is essential for APC's pivotal role in cortical radial progenitor development. APC deletion in RGCs eventually leads to apoptosis of these progenitors (Fig. 2K,L). This enhanced apoptosis was also rescued by β-catenin deletion in APC-deficient RGCs (Fig. 2M). In contrast, β-catenin deletion alone did not severely affect the general organization of the RG scaffold, except for minor ectopias within the SVZ/IZ (Fig. 2D).
Figure 2.

APC-mediated β-catenin signaling is essential for cortical progenitor development and cerebral cortical formation. Loss of APC in RGCs impairs polarized organization and function of RGCs and the resultant neuronal layer formation in the developing cortex (E16). These APC-null defects were rescued by the codeletion of β-catenin and phenocopied by constitutive activation of β-catenin, indicating that β-catenin is a central effector of APC's role in cerebral cortical development. (A–E) RGCs were immunolabeled with radial glial-specific RC2 antibodies. APC loss completely disrupts RGC organization (cf. A and B), which was rescued by concomitant deletion of β-catenin (C). (D) β-Catenin deletion alone did not affect RGC organization, except for the occasional ectopias within the SVZ/IZ in β-catenin cKO cortices (asterisk). (E) Induction of active β-catenin in progenitors phenocopies the APC-null phenotype. (E′) Quantification of the number of polarized RC2+ basal fibers across unit width (100 µm) in the CP. (F–J) Proliferating cortical progenitors were pulse-labeled with BrdU. (J′) Quantification of the number of BrdU+ cells in the VZ. (K–O) Progenitors undergoing apoptosis were labeled with anti-cleaved caspase-3 antibodies. (O′) Quantification of changes in progenitor cell death. (P–T) IPs were immunolabeled with anti-Tbr2 antibodies. (T′) Quantification of Tbr2+ IPs. (U–Y) The emerging laminar organization of neurons was analyzed with layer-specific markers Ctip2 and Tbr1. (Y′) Changes in neuronal layer formation. Representative neuronal distribution plots from each genotype. Data shown are mean ± SEM. n = 3 for each genotype. (*) P < 0.05; (***) P < 0.001, one-way ANOVA with Tukey's multiple comparison test. Bars: A–E, 50 µm; F–J,K–O,P–T,U–Y, 60 µm.
RGCs function as neural precursors during corticogenesis. RGCs divide asymmetrically and give rise to IPs and neurons. APC deletion in RGCs resulted in markedly reduced generation of Tbr2+ IPs (Fig. 2P,Q) and Tuj1+ neurons (Fig. 1S,T). Labeling with layer-specific neuronal markers (Ctip2 and Tbr1) indicates total dysregulation of neuronal placement and layer formation in APC cKO cortices (Fig. 2U,V, Y′). However, deletion of β-catenin in APC-deficient RGCs rescued the defects in IP and neuronal generation and laminar organization of neurons (Fig. 2R,T′,W,Y′). β-Catenin deletion alone did not affect neuronal layer formation (Fig. 2X,Y′). The same pattern of rescue was observed at E16 and E18. Together, these observations indicate that APC plays an essential role in cortical radial progenitor organization and function as neural precursors. These functions of APC in cortical progenitors depend on appropriate β-catenin signaling.
To further delineate the role of APC and β-catenin in cortical progenitors, we activated β-catenin in normal cortical progenitors. This is analogous to the increased β-catenin activity that occurs in APC-deficient progenitors. We crossed Ctnnb1LoxEx3/LoxEx3 mice, in which exon 3 of the endogenous β-catenin gene is flanked by loxP sites (Harada et al. 1999), with hGFAP-Cre mice. The excision of exon 3 by Cre recombinase leads to the endogenous enhancer/promoter-driven expression of a stabilized form of β-catenin lacking the N-terminal region necessary for phosphorylation and ubiquitination-mediated degradation. The induction of stabilized β-catenin was confirmed by immunoblotting analysis (Supplemental Fig. 1B). We found that activation of β-catenin in RGCs in Ctnnb1LoxEx3/+; hGFAP-Cre (β-catEx3/+) mice phenocopied APC deletion effects seen in APC cKO mice (Fig. 2). Induction of active β-catenin alone in normal radial progenitors leads to disrupted radial scaffold organization (Fig. 2E), defective RGC proliferation and cell cycle kinetics (Fig. 2J; Supplemental Fig. 2E,J,O), increased apoptosis (Fig. 2O), decreased generation of Tbr2+ IPs (Fig. 2T), and loss of laminar organization of neurons (Fig. 2Y)—all phenotypes similar to that seen in APC cKO. These observations further support our findings that appropriate Wnt/β-catenin signaling is essential for APC-mediated progenitor development and function in the cerebral cortex.
To further examine the role of deregulated β-catenin signaling in APC-deficient radial progenitors, we expressed dominant-negative T-cell factor 4 (DNTCF; a repressor of β-catenin/TCF4-regulated transcriptional activity) or inhibitor of β-catenin and TCF4 (ICAT; an inhibitor of binding of β-catenin to TCF4 necessary for Wnt signal transduction) in these cells (Tago et al. 2000). We found that expression of either DNTCF or ICAT rescued the defects in RGC morphology in APC-deficient progenitors. We observed an increase in the percentage of polarized APC-deficient RGCs with radially oriented basal processes attached to the pial surface (Fig. 3A–E). DNTCF and ICAT also rescued proliferation defects of APC-deficient radial progenitors (Fig. 3F,G). Together, these data clearly indicate that the defects observed in APC-deficient progenitors are dependent on deregulated β-catenin signaling, and this disrupted Wnt tone underlies cortical malformation in APC cKO mice. Consistent with this, APC cKO mice do not live beyond postnatal day 15. They display severe hydrocephaly and are inactive (Supplemental Movie 1). In contrast, APC/β-cat cKO mice with the rescued cortical phenotype survive to adulthood and show normal cage exploration activity (Supplemental Movie 1). Survival time and basal behavior of β-catEx3/+ mice are similar to those of APC cKO mice (Supplemental Movie 1).
Figure 3.

Inhibition of β-catenin activity in APC-deficient RGCs rescues morphological and proliferation defects. (A) E14.5 cortices from APCLox/+; hGFAP-Cre or APCLox/Lox; hGFAP-Cre mice were focally electroporated with BLBP-DsRed and DNTCF or ICAT and analyzed 48 h later. Arrowheads point to polarized radial progenitor processes attached to the pial surface. Inhibition of β-catenin signaling in APC-null RGCs with DNTCF or ICAT leads to increased numbers of polarized RGCs attached to the pial surface. (B–E) Quantification of polarized radial progenitor cell morphology (apical attachment to the ventricular surface, apical process angle toward the ventricular surface, basal process length, and pial attachment of the basal process) following inhibition of β-catenin signaling in APC cKO. (F) Electroporated BLBP-DsRed+ RGCs in the VZ were coimmunolabeled with anti-BrdU or anti-PH3 to label actively proliferating progenitors. Arrowheads point to colabeled RGCs. The dotted line indicates the ventricular surface. (G) Quantification of changes in progenitor proliferation following inhibition of β-catenin signaling. Data shown are mean ± SEM. n = 3 for each group. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, one-way ANOVA with Tukey's multiple comparison test. Bars: A, 50 µm; F, 10 µm.
Disrupted Notch1 and Shh signaling in APC-deficient cortical progenitors
To define how APC might regulate appropriate radial progenitor development, we first examined the Notch signaling pathway, known to be essential for the development and identity of radial progenitors (Mizutani et al. 2007), in APC cKO cortices. Notch1 and its downstream effectors, Hes1 and Hes5, were significantly up-regulated and mislocalized in the APC cKO cortex (Fig. 4A–N). Processed Notch1, NICD, was also dramatically up-regulated (Fig. 4O). Although RBPJ, a constitutive repressor of Notch signaling, was not affected by APC deletion, the cell cycle inhibitor p21 was drastically up-regulated in the APC cKO cortex (Fig. 4O). These results suggest that deregulation of Notch1 activity in APC-deficient radial progenitors contributes to its disrupted development.
Figure 4.

Notch and Shh signaling pathways are deregulated in the APC-deficient cerebral cortex. (A–H). In situ localization of Notch1 and Hes5 in control and APC cKO cortices indicates disrupted Notch1 and Hes5 expression patterns in APC-deficient cortices (E16.5). (A,B,E,F) Notch1 and Hes5 are highly expressed in the VZ of the cerebral cortex (arrows). Low-magnification (A,C,E,G) and high-magnification (B,D,F,H) images of the cortex are shown. (I–N) Colabeling with Pax6 and Hes1 antibodies shows aberrant expression of Hes1 in APC cKO progenitors. (O) Immunoblot analyses of control and APC cKO cortices reveal up-regulation of Notch1, NICD, Hes1, and p21 levels in APC cKO. (P) An increase in repressor forms of Gli3 and Gli2 and a decrease in Gli1 level are evident in APC cKO. (Q–S) Abnormal Hes1 expression is rescued in the APC/β-catenin double-cKO cortex. (T) Defects in Notch signaling are rescued in the APC/β-catenin double-cKO cortex and recapitulated in the β-catEx3/+ cortex. (U) Abnormal processing of Gli proteins in APC cKO was rescued in the APC/β-catenin double-cKO cortex but not in the β-catEx3/+ cortex. Representative immunoblot images from three biological replicates are shown. Bars: A,C,E,G, 300 µm; B,D,F,H, 100 µm; I–N,Q–S, 40 µm.
Notch1 activity is influenced by Shh signaling (Kong et al. 2015), and Shh is necessary for RGC development (Wang et al. 2016a). Transcriptional regulation in response to Shh is driven primarily via the Gli transcription factors 1, 2, and 3, which can be processed to Gli activator (GliA) or Gli repressor (GliR) forms. Shh signaling normally leads to an increase of GliA; in the absence of Shh ligand, GliR is increased, which inhibits Shh target gene transcription. We noticed a significant increase in repressor forms of Gli3 and Gli2 and a corresponding decrease in Gli1 in APC mutants (Fig. 4P), suggesting disrupted Shh signaling and primary cilium functions.
APC is necessary for primary cilium maintenance and signaling
Since Shh signaling depends on primary cilia, we examined primary cilia in APC-deficient progenitors. APC is coexpressed with Arl13b, a primary cilium marker (Fig. 5A). Deletion of APC disrupts the maintenance of primary cilia in progenitors, and APC mutant cilia are significantly shorter (Fig. 5B–F). As cortical development proceeds, at late embryonic stages, APC-null cortical progenitors lose their primary cilia (Fig. 5G–K). To investigate whether the signaling function of cilia is disrupted in APC-deficient progenitors, we examined the requirement for APC in Shh signaling. Stimulating Shh signaling with SAG, a Smo agonist, leads to the rapid accumulation of Gli3 at the tips of the primary cilia in cortical progenitors (Fig. 5L,M,P). In contrast, treatment of APC-deficient progenitors with SAG did not lead to Gli3 translocation to the tips of the cilia (Fig. 5N–P). Importantly, re-expression of APC in APC-deficient progenitors rescued the defects in cilium length, RGC proliferation, and morphology (Supplemental Fig. 3). These observations suggest that APC is essential to maintain primary cilium function in radial progenitors. Deletion of APC leads to aberrant primary cilium signaling and maintenance, which may contribute to compromised radial progenitor development in APC cKO.
Figure 5.
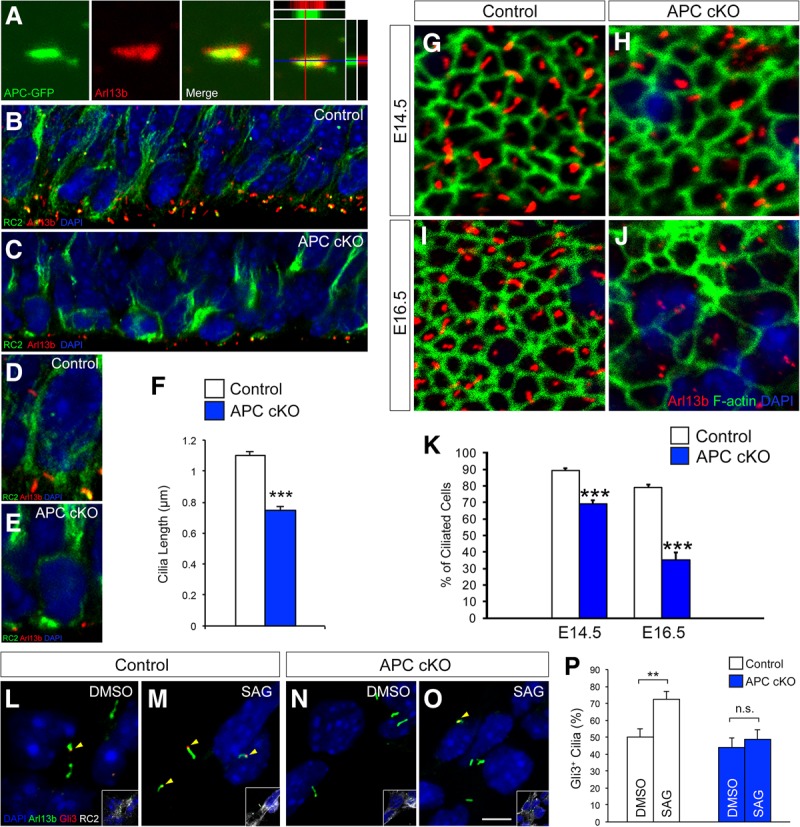
Primary cilium defects in APC-deficient radial progenitors. (A) Electroporated APC-GFP colocalizes with Arl13b, a primary cilium marker, in radial progenitors. (Right) Optical line scans (X–Z and Y–Z) through this image show colocalization. (B–E) Deletion of APC initially leads to shorter primary cilia in RGCs. Low-magnification images of primary cilia at the ventricular surface (B,C) and high-magnification images of RC2+ RGCs (D,E) are shown. (F) Quantification of primary cilium length in control and APC cKO cortices (E16.5). Data shown are mean ± SEM. n = 4 for each genotype. (G–J) Loss of APC leads to primary cilium maintenance defects. En face view of progenitor cilia on the ventricular surface at E14.5 (G,H) and E16.5 (I,J). By E16.5, substantial loss of primary cilia is evident in APC cKO. (K) Quantification of changes in the number of ciliated progenitors. Data shown are mean ± SEM. n = 3 for each genotype. (L–P) Shh signaling is perturbed in APC cKO progenitors. RC2+ cortical progenitors from control and APC cKO were treated with 200 nM SAG. SAG treatment leads to an increase in Gli3 localization to primary cilium tips (arrowheads) in control, but not in APC cKO, progenitors. Insets show RC2 labeling of radial progenitors shown in each panel. (P) Quantification of primary cilia with Gli3 at tips. Data shown are mean ± SEM. n = 3 for each condition. (**) P < 0.01; (***) P < 0.001, Student's t-test. Bars: A, 1 µm; B,C, 6 µm; D,E,G,H, 3 µm; I,J, 4 µm; L–O, 5 µm.
Aberrant APC–β-catenin signaling disrupts Notch activity and primary cilium function in cortical progenitors
Since our in vivo genetic analysis indicates that APC–β-catenin signaling is essential for radial progenitor development, we next examined whether APC-regulated β-catenin signaling underlies altered Notch activity and the primary cilium dysfunction in APC-deficient progenitors. Deletion of β-catenin in APC-deficient progenitors rescued Notch signaling deficiency and the ciliary defects (Figs. 4Q–T, 6A–C). In RGCs without APC and β-catenin, primary cilium length and number were similar to those of controls (Fig. 6A–C). Activation of β-catenin alone in wild-type progenitors resulted in shorter cilia and cilium maintenance defects similar to those observed in APC-deficient progenitors (Fig. 6A–C). Consistent with these observations, focal down-regulation of β-catenin signaling in APC-deficient progenitors using either DNTCF or ICAT rescued the defects in cilium length in APC-deficient progenitors (Fig. 6D–F). Furthermore, APC–β-catenin activity is essential for ciliary Shh signaling. Deletion of β-catenin improved the ability of APC-null RGCs to respond to SAG, whereas induction of constitutively active β-catenin in RGCs resulted in defective Shh signaling, similar to that observed in APC-deficient RGCs (Fig. 6G,H). Aberrant processing patterns of the Gli family of transcription factors were also rescued in APC/β-catenin double-cKO cortices (Fig. 4U). Together, these data strongly suggest that deregulated β-catenin signaling underlies primary cilium defects in APC-deficient RGCs.
Figure 6.

Primary cilium defects in APC-deficient cortices are dependent on β-catenin signaling. (A) VZs of cortices (E18.5) from control, APC cKO, APC/β-cat cKO, and β-catEx3/+ were labeled with anti-Arl13b or anti-β-catenin and phalloidin. Loss of primary cilia in APC cKO was rescued by codeletion of APC and β-catenin. Constitutive activation of β-catenin led to APC cKO-like ciliary loss. (B,C) Quantification of primary cilium length and percentage of ciliated cells. Data shown are mean ± SEM. n = 3 for each group. (*) P < 0.05; (***) P < 0.001, one-way ANOVA with Tukey's multiple comparison test. (D) Embryonic cortices from control or APC cKO were focally electroporated with BLBP-DsRed and DNTCF or ICAT. After 48 h, primary cilia in electroporated RGCs were immunolabeled with anti-Arl13b antibodies. Insets show primary cilia of electroporated cells. Inhibition of β-catenin signaling rescues APC cKO ciliary defects. (E,F) Quantification of cilium length and percentage of ciliated cells. Data shown are mean ± SEM. n = 3 for each group. (***) P < 0.001, one-way ANOVA with Tukey's multiple comparison test. (G) Loss of Shh signaling in APC cKO progenitors is rescued in APC/β-catenin double-null cells. Induction of active β-catenin in progenitors leads to APC cKO-like defects. RC2+ progenitors were treated with SAG and then immunolabeled with anti-Arl13b and anti-Gli3 antibodies. Insets show RC2 labeling of radial progenitors shown in each panel. (H) Quantification of primary cilia with Gli3 accumulation at tips (arrowheads in G). Data shown are mean ± SEM. n = 3 for each genotype. (**) P < 0.01; (***) P < 0.001, Student's t-test). Bars: A, 5 µm; D,G, 4 µm.
β-Catenin-mediated regulation of primary cilia
To identify cilium-related genes directly regulated by the APC–β-catenin pathway, we screened for ciliary genes that have high-affinity β-catenin–TCF4/LEF-binding sites in their regulatory regions (Schuijers et al. 2014), compared them with the SysCilia database (http://syscilia.org) (van Dam et al. 2013), and identified 34 putative targets of β-catenin–TCF4/LEF with known cilium functions (Fig. 7A). Of these genes, 24 encode proteins that are highly enriched in primary cilia and play key roles in primary cilium maintenance and signaling (Supplemental Table 1). To validate the relevant β-catenin ciliary targets in APC-deficient progenitors, we examined their expression in control and APC cKO cortices using quantitative RT–PCR. We observed a significant increase in the expression of Gpr98, Pard3, Pcdh15, and Tulp3 in APC cKO cortices (Fig. 7B). These genes were also up-regulated in β-catenin-activated cortices (β-catEx3/+), while these increases were abolished in the APC/β-catenin-double-null cortex (Fig. 7C). Identification of these ciliary genes controlled by β-catenin/TCF4 activity in APC-deficient cortices provides a potential mechanism by which APC-regulated β-catenin may directly modulate primary cilium structure and function.
Figure 7.

Identification of ciliary gene targets of β-catenin in the developing cortex and the effect of Tulp3 down-regulation in APC cKO progenitors. (A) The identification of ciliary genes regulated by canonical Wnt/β-catenin signaling. Genes with β-catenin/TCF4/LEF-binding sites were filtered through the SysCilia database to identify potential β-catenin-regulated ciliary genes. (B) Quantitative RT–PCR analysis of β-catenin-regulated ciliary gene expression in the APC-deficient cortices. Data shown are mean ± SEM. n = 3 for each genotype. (*) P < 0.05, Student's t-test. (C) Quantitative RT–PCR analysis of ciliary genes in APC/β-catenin double-cKO and β-catEx3/+ cortices. Codeletion of β-catenin rescued the deregulation of ciliary genes evident in APC cKO, whereas activation of β-catenin leads to APC cKO-like ciliary gene deregulation. Data shown are mean ± SEM. n = 3 for each genotype. (*) P < 0.05; (***) P < 0.001, one-way ANOVA with Tukey's multiple comparison test. (D–N) Down-regulation of Tulp3 ameliorates morphological, proliferation, and primary cilium defects in APC cKO progenitors. (D) Control or APC cKO cortices (E14.5) were focally electroporated with BLBP-DsRed and control or Tulp3 shRNAs and analyzed 48 h later. Arrowheads point to polarized radial progenitor processes attached to the pial surface. Down-regulation of Tulp3 rescued APC cKO defects in polarized RGCs. Coexpression of human Tulp3 (which is not targeted by murine shRNAs) did not rescue APC cKO defects. (E–H) Quantification of radial progenitor cell morphology following Tulp3 knockdown (apical attachment to the ventricular surface, apical process angle toward the ventricular surface, basal process length, and pial attachment of the basal process). (I) Primary cilia in shRNA electroporated RGCs were immunolabeled with anti-Arl13b antibodies. Insets show high-magnification images of primary cilia of electroporated cells. (J,K) Quantification of cilium length (J) and percentage of ciliated cells (K). (L) Electroporated BLBP-DsRed+ RGCs were coimmunolabeled with anti-BrdU or anti-PH3 to analyze progenitor proliferation. Down-regulation of Tulp3 rescued APC cKO RGC proliferation defects. Arrowheads point to colabeled RGCs. The dotted line indicates the ventricular surface. (M,N) Quantification of changes in radial progenitor proliferation following Tulp3 down-regulation. Data shown are mean ± SEM. n = 3 for each condition. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001, one-way ANOVA with Tukey's multiple comparison test. Bars: D, 50 µm; I, 2 µm; L, 10 µm.
Of the APC–β-catenin-regulated ciliary genes, Tulp3 is known to play a critical role in cilium maintenance and targeting of signaling receptors to primary cilia (Badgandi et al. 2017). To examine whether the Tulp3 up-regulation in APC cKO cortices contributes to APC-null phenotypes of RGCs, we expressed validated shRNA against Tulp3 in APC-deficient RGCs (Supplemental Fig. 4). Knockdown of Tulp3 in APC-deficient RGCs increased the percentage of polarized RGCs with pial-attached basal processes (Fig. 7D–H) and ameliorated cilium defects (Fig. 7I–K). Down-regulation of Tulp3 activity also rescued proliferation defects in APC cKO radial progenitors (Fig. 7L–N). Coelectroporation of shRNA-resistant human Tulp3 did not rescue APC cKO defects (Fig. 7). These results indicate that Tulp3 is a ciliary effector functioning downstream from the Wnt/β-catenin pathway, whose deregulation leads to ciliary defects associated with radial progenitor malformation in the APC-deficient neocortex.
Discussion
Here, we elucidated the functional contributions of APC-mediated β-catenin signaling in the development of cortical progenitors and cerebral cortical formation. Loss of APC in radial progenitors deregulates β-catenin activity and disrupts Notch pathway and primary cilium signaling, thus contributing to aberrant progenitor organization and differentiation. APC's effects on cortical progenitors and the resultant cerebral cortical formation depend on precise regulation of β-catenin activity and associated Wnt tone.
APC/β-catenin signaling in cortical progenitor development
APC is essential for the formation and maintenance of polarized radial progenitors during corticogenesis (Yokota et al. 2009). APC affects the outcome and patterns of proliferation of radial glia (Figs 1, 2; Supplemental Fig. 2). β-Catenin is a central component in the canonical Wnt pathway and acts downstream from APC. In the absence of Wnt ligand, the destruction complex, containing APC, phosphorylates and ubiquitinates β-catenin, leading to its proteasomal degradation. Wnt ligand binding leads to the formation of a Frizzled receptor and low-density lipoprotein receptor-related protein (LRP) coreceptor complex, which stabilizes β-catenin and thus enables its translocation to the nucleus, where it acts as a transcriptional coactivator with members of the TCF/LEF family of transcription factors (Logan and Nusse 2004; Nelson and Nathke 2013). Deletion of APC and the resultant disruption of the destruction complex leads to accumulation of β-catenin and unrestricted activation of Wnt signaling. APC in its role as an RNA-binding protein can also directly bind to the transcripts of several canonical Wnt signaling pathway members (Preitner et al. 2014). During cortical development, overexpression of active β-catenin in neuroepithelial cells promotes progenitor self-renewal (Chenn and Walsh 2002), whereas deletion of β-catenin depletes the basal progenitor pool and promotes premature neuronal differentiation (Machon et al. 2003). Increased β-catenin levels in IPs promotes neurogenesis (Mutch et al. 2010; Munji et al. 2011). Thus, modulation of optimal β-catenin signaling is critical for the appropriate spatiotemporal balance of proliferation and differentiation of progenitors during corticogenesis. Precise context-dependent regulation of β-catenin signaling levels in different progenitors at different time points along embryonic development appears to be an essential feature of cerebral cortical formation. Considering the pivotal role played by APC in β-catenin activity (Rusan et al. 2008; Nelson and Nathke 2013), we genetically dissected the effect of APC-regulated β-catenin signaling on cortical progenitor development. APC's effect on cortical progenitor development depends on an appropriate balance of downstream β-catenin activity. In the absence of β-catenin, APC deletion did not significantly affect cortical progenitor development and cerebral cortical formation. APC appears to be crucial in setting the appropriate Wnt tone in cortical progenitors during cortical development. Increased β-catenin-mediated Wnt tone and the disrupted Notch and primary cilium signaling in the absence of APC appear to prevent the appropriate developmental differentiation of radial progenitors in the cerebral cortex, thus leading to disrupted corticogenesis in APC cKO.
APC–β-catenin versus APC–MT interactions during cortical development
APC is transported along MTs and associates with MT plus ends. APC regulates cytoskeletal dynamics as a MT plus end-tracking protein by binding and recruiting β2B-tubulin mRNA to dynamic MTs and as a mediator of actin nucleation (Preitner et al. 2014). However, the rescue of the APC cKO phenotype when β-catenin is deleted in APC-null progenitors (APCLox/Lox; Ctnnb1Lox/Lox; hGFAP-Cre) suggests that APC's direct effect on MTs independently of β-catenin activity does not appear to be crucial for progenitor development. However, deregulated normal and active β-catenin levels in distinct cellular compartments (e. g., centrosomes, adhesion contacts, and nuclei) may indirectly affect MTs. Furthermore, β-catenin regulates adherens junctions between progenitors in the VZ, and the integrity of cell contacts between progenitors in the ventricular surface is disrupted in APC cKO but rescued in APC/β-cat cKO. Deregulated distribution of β-catenin in different cellular compartments (e.g., adhesion contacts vs. nuclei) in APC cKO may have further destabilized the adherens junctions between radial progenitors that are necessary for their function and the integrity of the ventricular surface (Rasin et al. 2007).
β-Catenin signaling and primary cilium function in cortical progenitors
Early radial progenitor development and adult neural progenitors are dependent on primary cilium function (Higginbotham et al. 2013; Tong et al. 2014; Guo et al. 2015). Primary cilium signaling regulates major developmental pathways important for cerebral cortex development, including Shh and Wnt/β-catenin signaling (Chenn and Walsh 2002; Houart et al. 2002; Hirabayashi et al. 2004; Logan and Nusse 2004; Shimogori et al. 2004; Wang et al. 2016a). Shh, Wnts, and other ligands necessary for progenitor development and cortical patterning are present in the embryonic brain cerebrospinal fluid (CSF) and cerebral wall (Lun et al. 2015; Wang et al. 2016b). We found that primary cilium maintenance and function are severely disrupted in APC-deficient progenitors with deregulated β-catenin signaling. Down-regulation of β-catenin activity is sufficient to rescue primary cilium deficits in APC-deficient progenitors. Furthermore, we show that ectopic induction of active β-catenin in progenitors, similar to that seen in APC-null cells, impairs Shh signaling in radial progenitors. Primary cilium and Shh signaling is thought to promote appropriate cell cycle exit (Phua et al. 2017) and generation and renewal of outer RGCs and IPs (Wang et al. 2016a). Excision of cilium tips and the resultant cilium disassembly may drive cell cycle (Phua et al. 2017). Furthermore, the asymmetric inheritance of primary ciliary membrane and thus earlier appearance of a primary cilium in a daughter cell during asymmetric cortical progenitor division may help it to retain stem cell characteristics (Paridaen et al. 2013). Together, these observations suggest that defective primary cilium/Shh signaling may contribute to the proliferation and differentiation defects observed in APC-deficient progenitors.
Understanding of how primary cilia integrate signaling critical for neural progenitor development is only beginning to emerge. For example, in the ventral spinal cord progenitors, Notch activity is required for the efficient trafficking of Smo receptors to primary cilia, necessary for Shh responsiveness and progenitor cell fate determination (Kong et al. 2015). Overactivation of Notch signaling in APC cKO may have perturbed the appropriate trafficking of Smo and Patched 1 to and from primary cilia, thus contributing to the compromised ciliary function and maintenance in APC cKO. However, the Notch pathway is also likely to play a cilium-independent role in radial progenitor maintenance (Imayoshi et al. 2013). How APC regulates both of these modes of Notch signaling and the interplay between them in progenitors remains to be elucidated. Furthermore, Staufen2, a dsRNA-binding protein and a determinant of asymmetric cortical progenitor cell divisions, preferentially associates with transcripts of genes necessary for primary cilium formation and function (Kusek et al. 2012). Our studies show that APC-regulated β-catenin modulates primary cilium maintenance and signaling. We identified that several cilium-related genes, including Gpr98, Pard3, Pcdh15, and Tulp3, are direct targets of canonical β-catenin signaling and are misexpressed in the developing cortex when β-catenin is overactivated in APC-null progenitors (Fig. 7). Tulp3, a tubby-like family member, is highly enriched in primary cilia and has been shown to both negatively regulate Shh activity (Patterson et al. 2009) and interact with IFT machinery to recruit specific GPCRs into primary cilia (Badgandi et al. 2017). Overactive Tulp3 may enable increased ciliary transport of GPR161 (Garcia-Gonzalo et al. 2015), which can inhibit Shh signaling in APC cKO progenitors. Consistent with this hypothesis, we show that down-regulation of Tulp3 in APC-deficient progenitors rescues primary cilium signaling defects. Furthermore, loss of genes critical for primary cilium structure or function can also result in aberrant β-catenin responsiveness. For example, loss of ciliary genes Kif3a, Srebp1c, Ift88, and Ofd1 results in hyperactive canonical Wnt responses (Gerdes et al. 2007; Corbit et al. 2008), whereas inhibition of other cilium-related genes, such as Chibby and Seahorse, attenuates β-catenin activity (Takemaru et al. 2003). Our observations underscore the direct regulation of cilium maintenance and signaling via APC-modulated β-catenin in cortical progenitors. This pathway contributes to the appropriate patterns of radial progenitor proliferation and organization necessary for the formation of the cerebral cortex.
APC–β-catenin signaling and neurodevelopmental disorders
Dominant mutations in APC have been known to cause familial adenomatous polyposis (FAP), characterized by intestinal polyposis and colorectal cancer (Nelson and Nathke 2013; Dow et al. 2015). Extracolonic manifestations of FAP are referred to as Gardner syndrome or Turcot syndrome, and neurological outcomes include brain tumor polyposis type 2 and autism (Zhou et al. 2007; Gómez García and Knoers 2009; Mohn et al. 2014; Song et al. 2014). Recent analysis of autism spectrum disorder (ASD) risk gene networks implicate canonical Wnt signaling as a pathway disrupted in ASD. In mice, Dvl1 and Dvl3 deficiency deregulates a transcriptional program involving β-catenin, Brn2, and Tbr2, causing an expansion of the IP cell population and ASD-related aberrant social behavior (Belinson et al. 2016). Treatment of these mice with a GSK3β antagonist, lithium, which activates β-catenin-related canonical Wnt signaling, rescued these defects. Deregulated β-catenin levels and cellular localization also impact its interactions with axin, which acts as an amplification to the differentiation switch in IPs (Fang et al. 2013). Inhibition of axin degradation in IPs with a tankyrase inhibitor, XAV939, leads to excessive production of upper-layer neurons and autism-like behaviors (Fang et al. 2014). Individual ASD risk genes such as CHD8 (Durak et al. 2016), PTEN (Chen et al. 2015), UBE3A (Yi et al. 2017), DIXDC1, DDX3X, and CTNND2 and chromatin remodeling genes overrepresented in ASD risk (e.g., ARID1B) can also modulate β-catenin-mediated Wnt signaling. Importantly, several Wnt/β-catenin pathway loci are shared risk factors for schizophrenia, bipolar disorder, and ASD, including CHD8, DISC1, TCF4, and CTNNB1 (Ernst 2016; Mulligan and Cheyette 2017). Other Wnt genes, including the secreted Wnt antagonists DKK1 and DKK4 and Wnt receptors KREMEN1, are also implicated in schizophrenia. Furthermore, genomic analyses of bipolar disorder have identified disruptions in Wnt/β-catenin pathway genes, including WNT7A, WNT2B, DISC1, and TCF4 (Ernst 2016; Mulligan and Cheyette 2017). These and other neuropsychiatric disorder-associated genes may be differentially regulated (up or down) by deviations of normal Wnt signaling in the developing brain, thus leading to different disease outcomes.
Disrupted Wnt signaling pathway in progenitors is emerging as a shared convergent route to schizophrenia, bipolar disorder, and ASD. (Ernst 2016; Packer 2016; Hazlett et al. 2017; Mulligan and Cheyette 2017). Our studies demonstrating the essential role of the APC–β-catenin pathway in setting the right Wnt signaling tone necessary for cortical progenitor development provide evidence that changes in this process can lead to disruptions in cerebral cortical circuit formation that underlie these neurodevelopmental disorders.
Materials and methods
Animals
Mice were cared for according to animal protocols approved by the University of North Carolina. APC was conditionally inactivated in radial progenitors by mating mice carrying the APC allele flanked by LoxP sites (Shibata et al. 1997) with hGFAP-Cre mice. Littermate APCLox/+; hGFAP-Cre mice served as controls. A similar mating strategy with Ctnnb1Lox/Lox or Ctnnb1LoxEx3/LoxEx3 was used to inactivate or activate β-catenin in radial progenitors. APCLox/Lox; Ctnnb1Lox/Lox mice were crossed with APCLox/+; Ctnnb1Lox/+; hGFAP-Cre mice to generate APCLox/Lox; Ctnnb1Lox/Lox; hGFAP-Cre mice in which β-catenin was deleted in APC-deficient radial progenitors. The effect of the recombination in APC cKO, Ctnnb1 cKO, APC/Ctnnb1 cKO, or Ctnnb1LoxEx3/+ cortices was assessed by immunoblot analysis as described previously (Yokota et al. 2009).
Immunohistochemistry
Immunohistochemical labeling of embryonic brain sections and isolated cortical progenitors was performed as described earlier (Higginbotham et al. 2013; Guo et al. 2015). The following primary antibodies were used: Arl13b (1:1000; NeuroMabs, 75-287), APC (1:500 [Santa Cruz Biotechnology, C-20] and 1:500 [gift from Dr. I. Nathke, University of Dundee]) (Yokota et al. 2009), β-catenin (Sigma; C2206; 1:500), BrdU (BD Biosciences; 347580; 1:100), cleaved-caspase3 (Cell Signaling; 9661; 1:400), Ctip2 (1:500; Abcam, ab18465), GFP (1:1000; Abcam, ab13970), Gli3 (1:500; R&D Systems, AF3690), Hes1 (1:500; gift from Dr. R. Kageyama, Kyoto University), Pax6 (1:500; BioLegend, 901301), PH3 (1:500; Upstate Biotechnology), RC2 (1:3; Iowa Hybridoma Bank), RFP (1:500; Abcam, ab28664), Tbr1 (1:2500; Millipore, ab2261), Tbr2 (1:500; Abcam, ab23345), and Tuj1 (1:1000; Covance, MMS-435). Appropriate Cy2, Cy3, or Alexa dye-conjugated secondary antibodies (Jackson ImmunoResearch, Molecular Probes) were used to detect primary antibody binding. DAPI (Invitrogen) was used as nuclear counterstain. For routine BrdU labeling, dams were injected intraperitoneally with 50 mg of BrdU per kilogram of bodyweight (Sigma, B5002) 1 h prior to sacrifice. To assay for cell cycle pattern, BrdU was injected 24 h before tissue harvesting. Cortical sections were then colabeled with anti-BrdU and Ki67 antibodies. Ratio of Ki67−, BrdU+/BrdU+ cells was quantified and used as the cell cycle exit index. Quantification of BrdU-, PH3-, Caspase3-, Tbr2-, Tuj1-, and Arl13b-labeled cells was performed as described previously (Yokota et al. 2009; Higginbotham et al. 2013).
Immunoblot analysis
Tissues were harvested in lysis buffer (10 mM Tris at pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100) with protease and phosphatase inhibitor cocktail (Thermo Scientific). The lysates were briefly sonicated and centrifuged at 14,000 rpm for 20 min at 4°C. Protein concentrations were determined using the Bio-Rad protein assay. Samples were then mixed with Laemmli buffer (2% SDS) and boiled for 5 min. Proteins were resolved in SDS–polyacrylamide gels, transferred to nitrocellulose, and incubated with one of the following primary antibodies: APC (1:500; Santa Cruz Biotechnology, C-20), β-actin (1:1000; Abcam, ab8226), β-catenin (1:1000; Sigma, C2206), Gli1 (1:500; Cell Signaling, 2534), Gli2 and Gli3 (1:200; gift from Dr. B. Wang, Cornell University), Hes1 (gift from Dr. R. Kageyama, Kyoto University; 1:500), NICD (1:500; Abcam, ab8925), Notch1 (1:500; Santa Cruz Biotechnology, C-20), p21 (1:200; Santa Cruz Biotechnology, F-5), or RBP-jk (1:1000; Millipore, AB2284). Immunoblots were developed using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit antibodies and detected with enhanced chemiluminescence.
RT–PCR and quantitative PCR
RNA was extracted from dorsal cortex at E18.5 using RNeasy minikit (Qiagen) and cleared of genomic DNA using Turbo DNase (Life Technologies). cDNA was synthesized using iScript (Bio-Rad). Real-time PCR was performed in triplicates using Applied Biosystem Power SYBR Green PCR master mix and the appropriate forward and reverse primers (see Supplemental Table 2). For quantification of gene expression changes, three replicates from three independent experiments were averaged. Samples were normalized against β-actin expression.
Shh assay for ciliary signaling
Isolated cortical radial progenitors from E14.5 control and APC cKO cortices were maintained in DMEM/N2/B27 for 1 d prior to treatment with 200 nM SAG (Calbiochem) for 24 h. Cells were then immunolabeled with RC2, Arl13b, and Gli3 antibodies. The percentage of radial progenitor cilia with Gli3 at the tips was quantified from three independent experiments.
In utero electroporation
E14.5 embryos were electroporated as described previously (Yokota et al. 2009; Higginbotham et al. 2013). The following constructs were coelectroporated with pBLBP-DsRed: pcDNA-DNTCF4, pcDNA-FLAG-ICAT, pCIG2-GFP, and pAPC-GFP. Radial progenitors in electroporated cortices were analyzed 48 h later.
Tulp3 shRNA
Tulp3 shRNA was obtained from the University of North Carolina Gene Therapy Center. shRNA target sequences used were as follows: CCTACAGTGTACCTGAACTTA (1), CCGACA GATTTGTCTCGTGAA (2), GTGGAGAATTTAGAGGACTTT (3), CGTGTTCACACTGGACTATAA (4), and GTGTAAAG TAACCAGGGATAA (5). Two pools (1.5 µg/µL) of shRNAs [shTulp3 #1 (1–3) and shTulp3 #2 (4–5)] containing equivalent amounts of different shRNAs were used for in utero electroporation. Nonsilencing scrambled shRNA (pLKO.1 shRNA vector; University of North Carolina Gene Therapy Center) was used as control. In shRNA + cDNA rescue experiments, human TULP3 cDNA (University of North Carolina Gene Therapy Center) was used at 1 µg/µL concentration.
Supplementary Material
Acknowledgments
We thank Jiami Guo and Charlotte Plestant for helpful comments. This research was supported by National Institutes of Health grant MH060929 to E.S.A., and the confocal imaging core of a National Institute of Neurological Disorders and Stroke institutional center core grant (5P30NS045892-12). N.N. is supported by the Uehara Memorial Foundation research fellowship. E.S.A., N.N., J.L., K.Y-N., M.C., T.M., and T.-Y.E. designed the experiments. N.N., J.L., K.Y-N., T.-Y.E., M.C., and R.T. conducted the experiments and analyzed the data. N.N., J.L., K.Y-N., T.-Y.E., M.C., and E.S.A. wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.302679.117.
References
- Badgandi HB, Hwang S, Shimada IS, Loriot E, Mukhopadhyay S. 2017. Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins. J Cell Biol 216: 743–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinson H, Nakatani J, Babineau BA, Birnbaum RY, Ellegood J, Bershteyn M, McEvilly RJ, Long JM, Willert K, Klein OD, et al. 2016. Prenatal β-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol Psychiatry 21: 1417–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello S, Attardo A, Wu X, Iwasato T, Itohara S, Wilsch-Bräuninger M, Eilken HM, Rieger MA, Schroeder TT, Huttner WB, et al. 2006. The Rho-GTPase cdc42 regulates neural progenitor fate at the apical surface. Nat Neurosci 9: 1099–1107. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang W-C, Sejourne J, Clipperton-Allen AE, Page DT. 2015. Pten mutations alter brain growth trajectory and allocation of cell types through elevated β-catenin signaling. J Neurosci 35: 10252–10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. 2002. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 297: 365–369. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Shyer AE, Dowdle WE, Gaulden J, Singla V, Reiter JF. 2008. Kif3a constrains β-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol 10: 70–76. [DOI] [PubMed] [Google Scholar]
- Dow LE, O'Rourke KP, Simon J, Tschaharganeh DF, van Es JH, Clevers H, Lowe SW. 2015. Apc restoration promotes cellular differentiation and reestablishes crypt homeostasis in colorectal cancer. Cell 161: 1539–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, Liu CY, Watson LA, Tsai L-H. 2016. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci 19: 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C. 2016. Proliferation and differentiation deficits are a major convergence point for neurodevelopmental disorders. Trends Neurosci 39: 290–299. [DOI] [PubMed] [Google Scholar]
- Evsyukova I, Plestant C, Anton ES. 2013. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu Rev Cell Dev Biol 29: 299–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E. 2011. A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145: 1129–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W-Q, Chen W-W, Fu AKY, Ip NY. 2013. Axin directs the amplification and differentiation of intermediate progenitors in the developing cerebral cortex. Neuron 79: 665–679. [DOI] [PubMed] [Google Scholar]
- Fang W-Q, Chen W-W, Jiang L, Liu K, Yung W-H, Fu AKY, Ip NY. 2014. Overproduction of upper-layer neurons in the neocortex leads to autism-like features in mice. Cell Rep 9: 1635–1643. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Phua SC, Roberson EC, Garcia G, Abedin M, Schurmans S, Inoue T, Reiter JF. 2015. Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev Cell 34: 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, et al. 2007. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 39: 1350–1360. [DOI] [PubMed] [Google Scholar]
- Gómez García EB, Knoers NV. 2009. Gardner's syndrome (familial adenomatous polyposis): a cilia-related disorder. Lancet Oncol 10: 727–735. [DOI] [PubMed] [Google Scholar]
- Guo J, Higginbotham H, Li J, Nichols J, Hirt J, Ghukasyan V, Anton ES, Guemez-Gamboa A, Coufal NG, Gleeson JG, et al. 2015. Developmental disruptions underlying brain abnormalities in ciliopathies. Nat Commun 6: 7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen DV, Lui JH, Parker PRL, Kriegstein AR. 2010. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464: 554–561. [DOI] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM. 1999. Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J 18: 5931–5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazlett HC, Gu H, Munsell BC, Kim SH, Styner M, Wolff JJ, Elison JT, Swanson MR, Zhu H, Botteron KN, et al. 2017. Early brain development in infants at high risk for autism spectrum disorder. Nature 542: 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Guo J, Yokota Y, Umberger NL, Su C, Li J, Verma N, Hirt J, Ghukasyan V, Caspary T, et al. 2013. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat Neurosci 16: 1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt F, Benzing T, Katsanis N. 2011. Ciliopathies. N Engl J Med 364: 1533–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, Gotoh Y. 2004. The Wnt/β-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 131: 2791–2801. [DOI] [PubMed] [Google Scholar]
- Houart C, Caneparo L, Heisenberg C, Barth K, Take-Uchi M, Wilson S. 2002. Establishment of the telencephalon during gastrulation by local antagonism of Wnt signaling. Neuron 35: 255–265. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Isomura A, Harima Y, Kawaguchi K, Kori H, Miyachi H, Fujiwara T, Ishidate F, Kageyama R. 2013. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 342: 1203–1208. [DOI] [PubMed] [Google Scholar]
- Kim W-Y, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, Snider WD. 2009. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci 12: 1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong JH, Yang L, Dessaud E, Chuang K, Moore DM, Rohatgi R, Briscoe J, Novitch BG. 2015. Notch activity modulates the responsiveness of neural progenitors to sonic hedgehog signaling. Dev Cell 33: 373–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusek G, Campbell M, Doyle F, Tenenbaum SA, Kiebler M, Temple S. 2012. Asymmetric segregation of the double-stranded RNA binding protein Staufen2 during mammalian neural stem cell divisions promotes lineage progression. Cell Stem Cell 11: 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Schroth J, Gleeson JG. 2011. Subcellular spatial regulation of canonical Wnt signalling at the primary cilium. Nat Cell Biol 13: 700–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan CY, Nusse R. 2004. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20: 781–810. [DOI] [PubMed] [Google Scholar]
- Lui JH, Nowakowski TJ, Pollen AA, Javaherian A, Kriegstein AR, Oldham MC. 2014. Radial glia require PDGFD–PDGFRβ signalling in human but not mouse neocortex. Nature 515: 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun MP, Monuki ES, Lehtinen MK. 2015. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci 16: 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. 2003. Role of β-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience 122: 129–143. [DOI] [PubMed] [Google Scholar]
- Mizutani K, Yoon K, Dang L, Tokunaga A, Gaiano N. 2007. Differential Notch signalling distinguishes neural stem cells from intermediate progenitors. Nature 449: 351–355. [DOI] [PubMed] [Google Scholar]
- Mohn JL, Alexander J, Pirone A, Palka CD, Lee S-Y, Mebane L, Haydon PG, Jacob MH. 2014. Adenomatous polyposis coli protein deletion leads to cognitive and autism-like disabilities. Mol Psychiatry 19: 1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan KA, Cheyette BNR. 2017. Neurodevelopmental perspectives on Wnt signaling in psychiatry. Mol Neuropsychiatry 2: 219–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. 2011. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci 31: 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutch CA, Schulte JD, Olson E, Chenn A. 2010. β-Catenin signaling negatively regulates intermediate progenitor population numbers in the developing cortex. PLoS One 5: e12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S, Nathke IS. 2013. Interactions and functions of the adenomatous polyposis coli (APC) protein at a glance. J Cell Sci 126: 873–877. [DOI] [PubMed] [Google Scholar]
- Packer A. 2016. Neocortical neurogenesis and the etiology of autism spectrum disorder. Neurosci Biobehav Rev 64: 185–195. [DOI] [PubMed] [Google Scholar]
- Paridaen JTML, Wilsch-Bräuninger M, Huttner WB. 2013. Asymmetric inheritance of centrosome-associated primary cilium membrane directs ciliogenesis after cell division. Cell 155: 333–344. [DOI] [PubMed] [Google Scholar]
- Patterson VL, Damrau C, Paudyal A, Reeve B, Grimes DT, Stewart ME, Williams DJ, Siggers P, Greenfield A, Murdoch JN. 2009. Mouse hitchhiker mutants have spina bifida, dorso–ventral patterning defects and polydactyly: identification of Tulp3 as a novel negative regulator of the Sonic hedgehog pathway. Hum Mol Genet 18: 1719–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phua SC, Chiba S, Suzuki M, Su E, Roberson EC, Pusapati GV, Setou M, Rohatgi R, Reiter JF, Ikegami K, et al. 2017. Dynamic remodeling of membrane composition drives cell cycle through primary cilia excision. Cell 168: 264–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preitner N, Quan J, Nowakowski DW, Hancock ML, Shi J, Tcherkezian J, Young-Pearse TL, Flanagan JG. 2014. APC is an RNA-binding protein, and its interactome provides a link to neural development and microtubule assembly. Cell 158: 368–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. 1988. Specification of cerebral cortical areas. Science 241: 170–176. [DOI] [PubMed] [Google Scholar]
- Rasin M-R, Gazula V, Breunig JJ, Kwan KY, Johnson MB, Liu-chen S, Li H, Jan LY, Jan Y-N, Rakic P, et al. 2007. Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nat Neurosci 10: 819–827. [DOI] [PubMed] [Google Scholar]
- Rusan NM, Akong K, Peifer M. 2008. Putting the model to the test: are APC proteins essential for neuronal polarity, axon outgrowth, and axon targeting? J Cell Biol 183: 203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuijers J, Mokry M, Hatzis P, Cuppen E, Clevers H. 2014. Wnt-induced transcriptional activation is exclusively mediated by TCF/LEF. EMBO J 33: 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata H, Toyama K, Shioya H, Ito M, Hirota M, Hasegawa S, Matsumoto H, Takano H, Akiyama T, Toyoshima K, et al. 1997. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science 278: 120–123. [DOI] [PubMed] [Google Scholar]
- Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA. 2004. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development 131: 5639–5647. [DOI] [PubMed] [Google Scholar]
- Song L, Jia Y, Zhu W, Newton IP, Li Z, Li W. 2014. N-terminal truncation mutations of adenomatous polyposis coli are associated with primary cilia defects. Int J Biochem Cell Biol 55: 79–86. [DOI] [PubMed] [Google Scholar]
- Tago K, Nakamura T, Nishita M, Hyodo J, Nagai S, Murata Y, Adachi S, Ohwada S, Morishita Y, Shibuya H, et al. 2000. Inhibition of Wnt signaling by ICAT, a novel β-catenin-interacting protein. Genes Dev 14: 1741–1749. [PMC free article] [PubMed] [Google Scholar]
- Takemaru K-I, Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT. 2003. Chibby, a nuclear β-catenin-associated antagonist of the Wnt/Wingless pathway. Nature 422: 905–909. [DOI] [PubMed] [Google Scholar]
- Tong CK, Han Y-G, Shah JK, Obernier K, Guinto CD, Alvarez-Buylla A. 2014. Primary cilia are required in a unique subpopulation of neural progenitors. Proc Natl Acad Sci 111: 12438–12443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam TJ, Wheway G, Slaats GG, Huynen MA, Giles RH, Giles RH. 2013. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Hou S, Han Y-G. 2016a. Hedgehog signaling promotes basal progenitor expansion and the growth and folding of the neocortex. Nat Neurosci 19: 888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Jossin Y, Chai G, Lien W-H, Tissir F, Goffinet AM. 2016b. Feedback regulation of apical progenitor fate by immature neurons through Wnt7–Celsr3–Fzd3 signalling. Nat Commun 7: 10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SL, Wilson JP, Wang C, Wang B, McConnell SK. 2012. Primary cilia and Gli3 activity regulate cerebral cortical size. Dev Neurobiol 72: 1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Paranjape SR, Walker MP, Choudhury R, Wolter JM, Fragola G, Emanuele MJ, Major MB, Zylka MJ. 2017. The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J Biol Chem 292: 12503–12515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y, Kim WY, Chen Y, Wang X, Stanco A, Komuro Y, Snider W, Anton ES. 2009. The adenomatous polyposis coli protein is an essential regulator of radial glial polarity and construction of the cerebral cortex. Neuron 61: 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y, Eom T, Stanco A, Kim W, Rao S, Snider WD, Anton ES. 2010. Cdc42 and Gsk3 modulate the dynamics of radial glial growth, inter-radial glial interactions and polarity in the developing cerebral cortex. Development 137: 4101–4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X-L, Giacobini M, Anderlid B-M, Anckarsäter H, Omrani D, Gillberg C, Nordenskjöld M, Lindblom A. 2007. Association of adenomatous polyposis coli (APC) gene polymorphisms with autism spectrum disorder (ASD). Am J Med Genet B Neuropsychiatr Genet 144B: 351–354. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. 2001. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 31: 85–94. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.