

In this study, Huang et al. compared the chromosomal architectures of fetal and adult human erythroblasts and found that, globally, chromatin structures and compartments A/B are highly similar at both developmental stages. Their results uncover a new critical regulatory region as a potential target for therapeutic genome editing for hemoglobinopathies and highlight the power of chromosome conformation analysis in discovering new cis control elements.
Keywords: chromatin structure, transcription, globin switching, fetal hemoglobin
Abstract
Chromatin structure is tightly intertwined with transcription regulation. Here we compared the chromosomal architectures of fetal and adult human erythroblasts and found that, globally, chromatin structures and compartments A/B are highly similar at both developmental stages. At a finer scale, we detected distinct folding patterns at the developmentally controlled β-globin locus. Specifically, new fetal stage-specific contacts were uncovered between a region separating the fetal (γ) and adult (δ and β) globin genes (encompassing the HBBP1 and BGLT3 noncoding genes) and two distal chromosomal sites (HS5 and 3′HS1) that flank the locus. In contrast, in adult cells, the HBBP1–BGLT3 region contacts the embryonic ε-globin gene, physically separating the fetal globin genes from the enhancer (locus control region [LCR]). Deletion of the HBBP1 region in adult cells alters contact landscapes in ways more closely resembling those of fetal cells, including increased LCR–γ-globin contacts. These changes are accompanied by strong increases in γ-globin transcription. Notably, the effects of HBBP1 removal on chromatin architecture and gene expression closely mimic those of deleting the fetal globin repressor BCL11A, implicating BCL11A in the function of the HBBP1 region. Our results uncover a new critical regulatory region as a potential target for therapeutic genome editing for hemoglobinopathies and highlight the power of chromosome conformation analysis in discovering new cis control elements.
Chromosomal folding patterns differ between tissues, reflecting distinct gene expression patterns. How chromatin architecture is modified and interconnects with gene expression as cells of a given lineage traverse developmental stages has not been studied in great depth. Erythropoiesis offers an ideal model system to examine the molecular mechanisms of gene regulation within the same cell lineage during development. Erythroid precursors from fetal or adult stages have been characterized in much detail, and it was discovered that in spite of their phenotypical and functional similarities, they differ in their gene expression, enhancer utilization, DNA methylation, and chromatin signatures (Xu et al. 2012; Lessard et al. 2015; Huang et al. 2016).
Among the genes that undergo well-known developmental stage-specific changes in transcription are the β-type globin genes. The human β-globin locus comprises five coding genes that are specifically expressed in erythroid cells; namely, the embryonic-specific ε (HBE1), fetal-restricted Gγ and Aγ (HBG2 and HBG1), and adult expressed δ and β (HBD and HBB) globin genes. Expression of all of these genes requires a strong upstream enhancer known as the locus control region (LCR), which encompasses five DNaseI-hypersensitive sites (HSs). The LCR is in physical proximity to the relevant β-type globin gene promoters in a developmental stage-specific manner (Palstra et al. 2003). The switch from fetal to adult globin gene expression, which normally occurs around the time of birth, is of particular importance, as its impairment (e.g., due to mutations) or reversal (e.g., via therapeutic intervention) benefits patients with sickle cell disease (SCD) and some forms of β-thalassemia (Smith and Orkin 2016).
The intergenic region between the Aγ- and δ-globin genes contains a pseudogene (HBBP1) and a noncoding gene (BGLT3) (Fig. 1A; Kiefer et al. 2011). Genome-wide association studies (GWASs) identified two single-nucleotide polymorphisms (SNPs), rs10128556 and rs2071348, both of which reside within the second intron of HBBP1 and are associated with elevated fetal hemoglobin levels (Galarneau et al. 2010; Nuinoon et al. 2010), suggesting the presence of developmental regulatory elements in this region.
Figure 1.
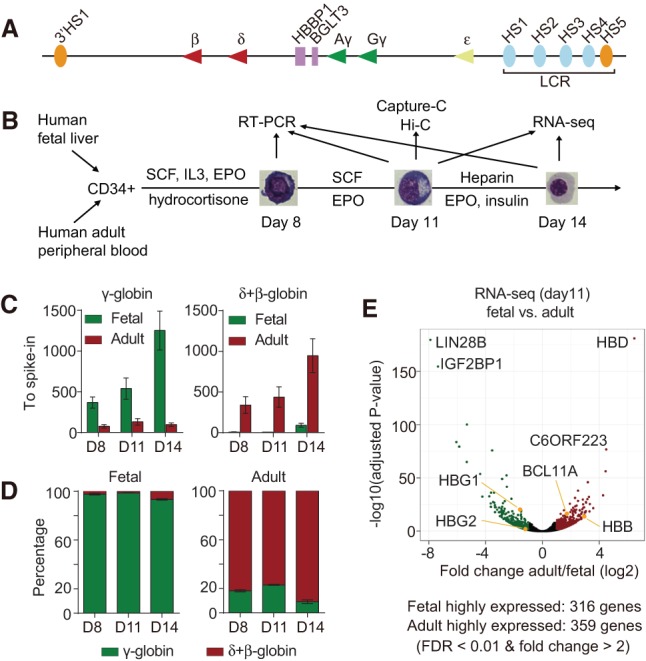
In vitro culture of primary human erythroid cells and gene expression profiles. (A) Diagram of the human β-globin locus. (B) Three-phase in vitro culture system of primary human erythroid cells and experimental design. (C,D) The expression levels (C) and percentages (D) of δ- plus β-globin and γ-globin genes in fetal and adult cells. Transcripts were normalized to spike-in ERCC-00042. Results are shown as mean ± SD. n = 4 from two donors. (E) Volcano plot of differentially expressed genes at day 11.
The major trans-acting regulatory factors of erythroid development and gene expression have been identified, and it is well-established that changes in chromatin structure are tightly coordinated with the control of gene expression. However, little is known about the dynamics of chromosomal architectural features in erythroblasts at different developmental stages. Here we carried out comparative Hi-C (chromosome conformation capture [3C] followed by high-throughput sequencing) and Capture-C (a multiplexed derivative of 3C) experiments in primary human fetal- and adult-type erythroblasts. We describe global and local similarities and distinctions in chromatin folding patterns between these stages. We show that, globally, chromatin structures of human erythroblast are largely preserved during development. Notably, at the β-globin locus, the HBBP1 region forms fetal stage-specific contacts with 3′HS1 and HS5, two DNaseI-sensitive regions bound by the architectural nuclear factor CTCF that flank the locus (Fig. 1A). In contrast, the HBBP1 region forms adult stage-specific contacts with the ε-globin region. CRISPR/Cas9-mediated deletion of the HBBP1 region in an adult-type human erythroid cell line strongly reactivates γ-globin transcription, suggesting that HBBP1 chromatin contacts contribute to the developmental control of β-globin expression. BCL11A is a well-characterized transcriptional repressor of γ-globin transcription (Sankaran et al. 2008). Deletion of BCL11A or the HBBP1 region triggers similar chromatin conformational changes at the β-globin locus, implicating BCL11A in the function of the HBBP1 region.
Finally, our study illustrates that fine-scale studies of chromosomal folding patterns enable identification of critical regulatory elements.
Results
Gene expression profiles of primary fetal and adult human erythroblasts
To study human erythroblasts at different developmental stages, we used a three-phase in vitro culture system to expand and differentiate fetal and adult CD34+ hematopoietic stem/progenitor cells (Fig. 1B). Cell morphology and surface marker phenotyping confirmed that fetal and adult erythroid cells were at similar maturation stages (Supplemental Fig. S1A,B,E), allowing comparisons between these two populations. As expected, RT-qPCR results revealed that fetal and adult erythroblasts expressed predominantly γ-globin and β-globin genes, respectively (Fig. 1C,D). Hence, the in vitro culture system does not perturb normal developmental stage specificity of gene expression.
We determined transcription profiles of fetal and adult erythroid cells each at two stages of differentiation by RNA sequencing (RNA-seq). Spike-in controls were added for normalization purposes, since global changes in gene expression during erythroid differentiation can distort measurements of RNA abundance (Stonestrom et al. 2015). The results revealed highly similar transcription patterns between fetal and adult stages when analyzed at matched time points of maturation (Supplemental Fig. S1C). Progression from day 11 to day 14 was accompanied by a down-regulation of most genes at both the fetal and adult stages. Specifically, 82.0% (21,450 out of 26,155) and 84.0% (21,690 out of 25,822) of expressed genes (FPKM [fragments per kilobase per million mapped fragments] > 0.01 in at least one replicate) were down-regulated more than twofold in mature fetal and adult cells, respectively (Supplemental Fig. S1D). At day 11, 316 and 359 genes were differentially expressed in fetal and adult erythroblasts, respectively (Fig. 1E). In addition to the β-type globin genes, transcripts that previously were known to be highly developmental stage-specific—e.g., LIN28B (Yuan et al. 2012) and BCL11A (Sankaran et al. 2008)—were expressed at the fetal and adult stage, respectively, as expected, validating the suitability of the culture systems for further investigation (Fig. 1E).
Similar chromatin structures of human fetal and adult erythroblasts
To compare chromatin architectural features between fetal and adult stages, we generated genomic contact maps by Hi-C using primary erythroblasts (two independent donors each) at day 11 of maturation (Figs. 1B, 2A). That time point was chosen because cells had activated erythroid gene expression programs, enabling us to study the relationships between chromatin structure and gene regulation. The high concordance of interaction frequencies among the biological replicates allowed us to combine them for a higher-resolution analysis (Supplemental Fig. S2A; Supplemental Table S1). Globally, the chromatin interaction frequencies were strongly correlated between fetal and adult erythroblasts (Fig. 2B). Submegabase-sized topologically associating domains (TADs) are thought to reflect a fundamental principle of chromatin organization (Dixon et al. 2012; Nora et al. 2012). To define TAD structures during erythroid development, we calculated the directionality indices (DIs) and identified 4633 and 4897 TADs in fetal and adult erythroblasts, respectively. We directly compared the DIs of fetal and adult cells and observed that they were strongly correlated (Fig. 2A,C). These results suggest that, genome-wide, chromatin interactions and TAD structures are conserved between fetal and adult erythroblasts.
Figure 2.
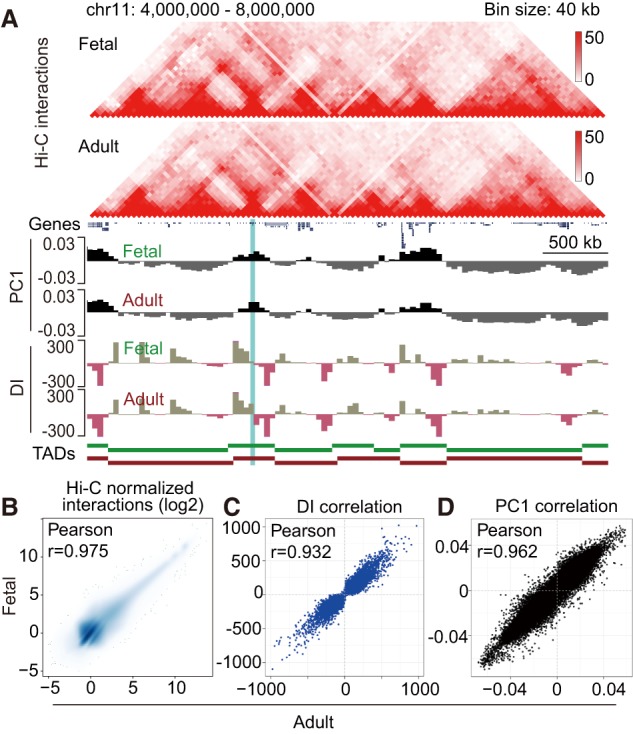
Chromatin structure comparisons of human erythroblasts during development. (A) Representative Hi-C results of fetal and adult erythroblasts. Heat maps show the cis–chromatin interactions of a 4-Mb region on chromosome 11. Compartments A and B are shown as positive (black) and negative (gray) first principal component (PC1) values, respectively. TADs are shown as green (fetal) and red (adult) bars. The β-globin locus is highlighted in blue. (B) Scatter plot of Hi-C interactions between fetal and adult erythroblasts. Hi-C bin size was 100 kb. (C) Scatter plot of DIs between fetal and adult erythroblasts. Hi-C bin size was 40 kb. (D) Scatter plot of PC1 values between fetal and adult erythroblasts. Hi-C bin size was 40 kb.
The genome is partitioned into compartments A and B, representing active and repressed regions, respectively (Lieberman-Aiden et al. 2009). To define the chromatin compartments that are subject to change during development, we performed principal component analysis (PCA) on the Hi-C data and compared the first principal component (PC1) values. Overall, the PC1 values were highly correlated between fetal and adult erythroblasts (Fig. 2A,D). Approximately 5% of the genome switched compartments between the fetal and adult stages (Supplemental Fig. S2B), and gene expression changes correlated with the compartment switches (Supplemental Fig. S2C). By comparison, compartment transitions during embryonic stem cell (ESC) differentiation (Dixon et al. 2015) or somatic cell reprogramming (Krijger et al. 2016) are more dramatic (36% and 28%, respectively), suggesting that progression among developmental stages within a single lineage, such as erythroid cells, entails more subtle changes than those during lineage alterations. As examples, the genes LIN28B and THRB are highly expressed at the fetal and adult stages, respectively (Supplemental Fig. S2D,E), and the compartments within which they reside underwent the corresponding switches during the fetal-to-adult transition (Supplemental Fig. S2F,G). Moreover, histone modifications showed that the LIN28B and THRB were regulated by developmental stage-specific enhancers (Huang et al. 2016). Our Hi-C results further revealed that the promoters of LIN28B and THRB engaged in stage-specific chromatin contacts with their putative stage-specific enhancers (Supplemental Fig. S2F,G). Together, these results suggest that the chromatin structures and chromatin compartments A/B of fetal and adult erythroblasts are highly similar.
The β-globin TAD folds into three erythroid-specific sub-TADs
The β-type globin genes are expressed exclusively in erythroid cells. To investigate possible links between erythroid-specific regulation and higher-order chromatin structure, we first examined the TAD structures and chromatin states of the β-globin locus using Hi-C. The β-globin gene cluster resides centrally within an ∼300-kb TAD that is conserved between fetal and adult cells (Fig. 3A). Consistent with the high transcriptional activity, the β-globin TAD is located in compartment A, while both flanking TADs fall into compartment B in both fetal and adult erythroblasts (Fig. 3A).
Figure 3.
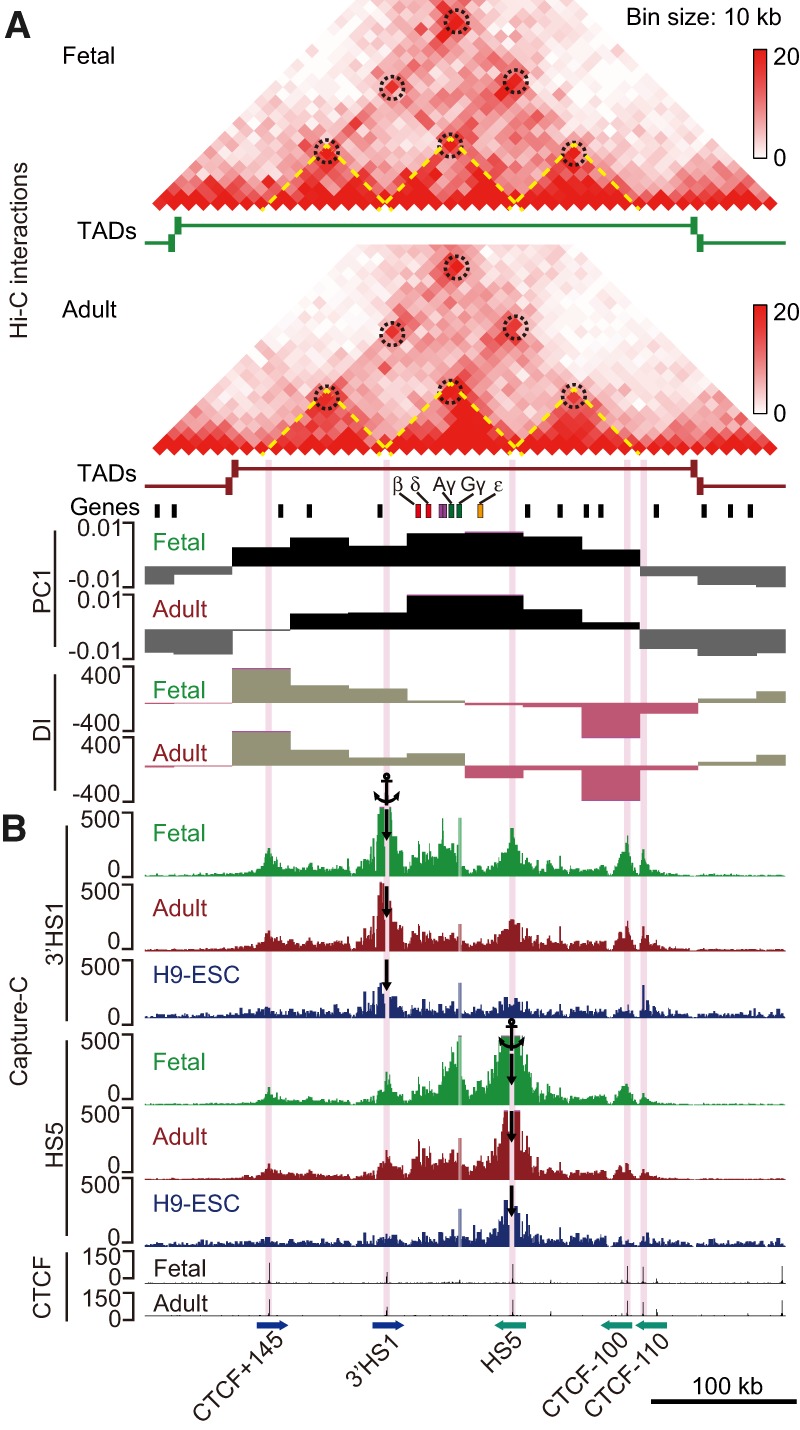
Chromatin structure of the β-globin locus in primary human erythroblasts. (A) Hi-C results of the β-globin locus in human fetal and adult erythroblasts. The interactions between CTCF cites are highlighted by dashed circles. Olfactory receptor genes are shown as black boxes. Compartments A (black) and B (gray) are shown by the PC1 values. (B) Capture-C interaction profiles of 3′HS1 and HS5 in fetal and adult erythroblasts and H9-ESCs. The anchor positions are indicated by black arrows. CTCF-binding sites are highlighted in light purple. The orientations of the CTCF motifs are indicated by blue and green arrows. Note that a single DpnII fragment at the Gγ-globin promoter showed unspecific signals in all Capture-C experiments in both fetal and adult erythroblasts and H9-ESCs. The signals of that particular fragment are shown in lighter colors in all of the tracks, as in Figures 4 and 6 (below).
Since TAD boundaries are enriched for binding sites of the architectural transcription factor CTCF (Dixon et al. 2012), we performed CTCF ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) experiments in primary fetal and adult erythroblast. Overall, we identified 50,036 and 63,518 peaks in fetal and adult cells, respectively. Among them, 48,364 peaks were shared among both stages (Supplemental Fig. S3A). However, the number of stage-specific CTCF peaks might be an overestimate owing to the stringent peak-calling algorithm, since, upon inspection, signals could be detected at sites not called at a given developmental stage (Supplemental Fig. S3B). Regardless, these results suggest that, globally, the CTCF-binding profiles are highly overlapping between the two developmental stages. Also, CTCF peaks were enriched at the TAD boundaries at both stages, as expected (Supplemental Fig. S3C). Within the β-globin TAD, CTCF strongly occupied five sites, including HS5 and 3′HS1 as well as other sites referred to as CTCF + 145, CTCF − 100, and CTCF − 110 (the numbers refer to kilobase distances relative to the ε-globin gene) (Fig. 3B).
When analyzed at 10-kb resolution, the Hi-C heat maps revealed that the boundaries of the β-globin TAD coincide with the CTCF + 145 and CTCF − 100/110 sites. The available algorithms for calling TADs and sub-TADs can yield variable results (Forcato et al. 2017). To further determine the substructure of the β-globin TAD at finer scale, we manually defined sub-TADs according to the Hi-C interactions and CTCF ChIP-seq profiles. The TAD containing the β-globin locus appears to be partitioned into three sub-TADs demarcated by the five CTCF-binding sites. The left sub-TAD is delineated by contacts between tandem CTCF-binding sites CTCF + 145 and 3′HS1, and the right sub-TAD is delineated by CTCF sites at HS5 and CTCF − 100/110 (Fig. 3A,B). The β-globin genes and LCR are located in the middle sub-TAD, which is flanked by the CTCF sites at 3′HS1 and HS5 (Fig. 3A). To examine the chromatin interactions between these CTCF-binding sites at higher resolution, we performed Capture-C using HS5 and 3′HS1 as anchors. The contact maps clearly delineate the three sub-TADs and confirm that both 3′HS1 and HS5 interact with CTCF + 145 and CTCF − 100/110 sites in both fetal and adult cells (Fig. 3B). Few if any of these contacts were detected in control H9-ESCs (Fig. 3B).
To determine whether the TAD and sub-TAD structures of the β-globin locus are conserved among different tissues, we compared published Hi-C results from eight cell lines and the corresponding CTCF ChIP-seq data from the ENCODE Consortium (Supplemental Fig. S4). The CTCF-binding profiles across the β-globin TAD are nearly identical across all lineages (Supplemental Fig. S4B). Notably, the three sub-TAD structures demarcated by the CTCF sites are variable and tissue-specific. Only K562 cells that have erythroid traits showed a chromatin conformation similar to that of primary erythroblasts (Supplemental Fig. S4A). This is consistent with a recent report showing that these CTCF sites interact with each other in K562 cells in a manner dependent on the erythroid transcription factor GATA1 (Kang et al. 2017). Taken together, these data suggest that the three sub-TAD structures of the β-globin locus are erythroid-specific, and their formation requires erythroid-specific factors in addition to CTCF (Hou et al. 2010).
The HBBP1 region engages in developmental stage-specific chromatin contacts in the β-globin locus
To assess possible relationships between architectural features of the β-globin locus and the fetal-to-adult globin switch, we analyzed the Hi-C data of the β-globin sub-TADs at a finer scale. Heat maps at 10-kb resolution illustrate developmental stage-specific interactions of the LCR with the relevant β-globin genes. Additionally, we observed developmentally dynamic chromatin contacts involving the HBBP1 region (Supplemental Fig. S5A). Interactions between the HS5 and the HBBP1 region have been observed in 5C (3C carbon copy) experiments in K562 cells, but the functional consequences have not been studied (Dostie et al. 2006). To explore these interactions at a resolution higher than that afforded by our Hi-C experiments, we carried out Capture-C using HS3 and the β-globin gene promoters as anchors. In agreement with the Hi-C results, we observed that HS3 dynamically interacted with the Gγ-globin and Aγ-globin genes and with the δ-globin and β-globin genes in fetal and adult erythroblasts, respectively, but not in H9-ESCs (Fig. 4A). These stage-specific contacts were confirmed when the Gγ-globin and β-globin promoters were used for Capture-C (Supplemental Fig. S5B). Subtraction of the adult from the fetal HS3 Capture-C contacts revealed that the region spanning HBBP1 seems to demarcate a prominent transition in fetal-specific versus adult-specific HS3 interactions (Fig. 4A). This transition in architectural features is reflected in developmental changes in gene expression that occur in this region. Thus, RNA-seq data showed that BGLT3 and genes upstream (ε-globin, Gγ-globin, and Aγ-globin) are selectively expressed in fetal cells, whereas HBBP1 and genes downstream (δ-globin and β-globin) are highly expressed in adult cells (Supplemental Fig. S5C). Detection of fetal-specific gene expression as well as low levels of fetal-type long-range chromatin contacts in adult cells are at least partly explained by the acquisition by adult cells of fetal traits during exposure to culture condition (Deng et al. 2014). Together, these results suggest that the HBBP1 region spatially and functionally separates fetal from adult gene regulation.
Figure 4.
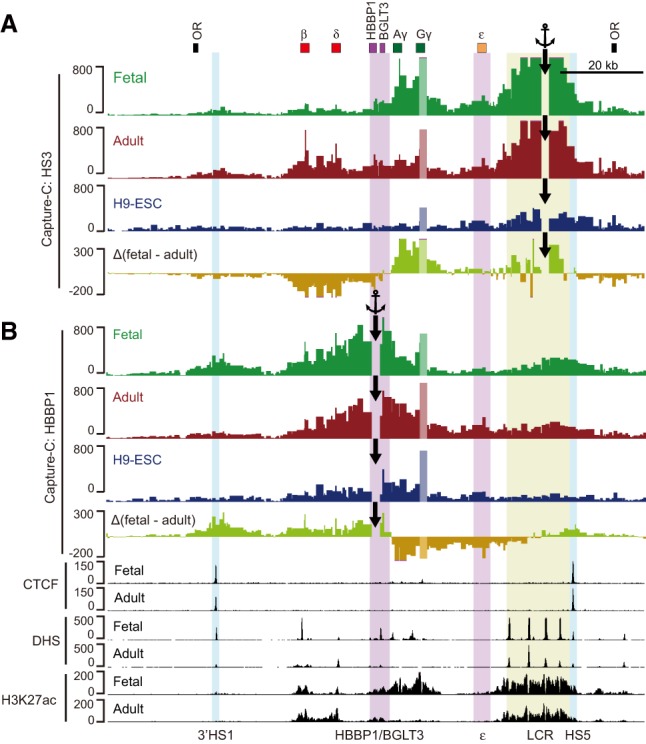
Chromatin interaction profiles of the β-globin locus. (A,B) Capture-C anchored on HS3 (A) and HBBP1 (B) in fetal and adult erythroblasts or H9-ESCs. The anchor positions are indicated by black arrows. The Δ(fetal − adult) tracks show the subtraction of normalized fetal and adult Capture-C interactions. (OR) Olfactory receptor genes.
To explore more deeply the chromatin contacts formed by the HBBP1 region in the formation of developmentally dynamic chromatin structures, we carried out Capture-C using HBBP1 as anchor. We found that the HBBP1 region strongly interacted with 3′HS1 and HS5 in fetal cells. In adult erythroblasts, these contacts were reduced greatly (with 3′HS1) or moderately (with HS5), and increased contacts were formed with a region encompassing the ε-globin gene. None of these interactions were observed in H9-ESCs (Fig. 4B).
CTCF plays a role in configuring the β-globin locus in human erythroid cells, including formation of contacts between 3′HS1 and HS5 (Hou et al. 2010). We considered the possibility that CTCF might be involved in the long-range contacts of HBBP1 with 3′HS1 and HS5 in fetal erythroid cells. However, the ChIP-seq failed to detect CTCF at the HBBP1 region in both fetal and adult erythroblasts (Fig. 4B), suggesting that the HBBP1 chromatin contacts are CTCF-independent. Together, these results demonstrate that the HBBP1 region engages in dynamic chromatin contacts within the β-globin locus by contacting HS5/3′HS1 and ε-globin regions at the fetal and adult stage, respectively.
The HBBP1 region is required for γ-globin silencing
The developmentally controlled contacts of the HBBP1 region suggest a role in hemoglobin switching. Using CRISPR/Cas9-mediated genome editing, we deleted an ∼2.3-kb fragment encompassing this region in the adult erythroblast cell line HUDEP2 (Fig. 5A; Kurita et al. 2013). Four independent clones with deletions on both alleles were selected (Supplemental Fig. S6B) and found to display a fourfold to 20-fold increase of γ-globin transcription compared with control cells (Fig. 5B). Transcription of γ-globin accounted for ∼10%–30% of total β-globin plus γ-globin production (Fig. 5C), implicating the HBBP1 region in γ-globin silencing in adult erythroid cells. The reason for the discrepancy in effect size between clones is unknown but is not due to variation in maturation efficiency (P Huang and GA Blobel, unpubl.). Other erythroid genes were expressed at levels similar to control cells in spite of variation between clones, suggesting that genome editing did not cause off-target effects that might have nonspecifically altered cell maturation (Supplemental Fig. S5D).
Figure 5.

The HBBP1 region contributes to γ-globin silencing in HUDEP2 cells. (A) Strategy of CRISPR/Cas9-mediated genome editing at the HBBP1 region. (B,C) The expression levels (B) and percentages (C) of γ-globin in the control and HUDEP2 mutant clones. (D) Expression levels of δ-globin and β-globin in the control and HUDEP2 mutant clones. Results are shown as mean ± SD. n ≥ 3. (*) P < 0.05; (**) P < 0.01; (n.s.) not significant by one-way ANOVA test.
The HBBP1 pseudogene is transcribed in adult erythroblasts (Supplemental Fig. S5C). To discern whether the effect of the HBBP1 deletion is due to loss of the transcript (Supplemental Fig. S6D) or the disruption of regulatory elements, we deleted the HBBP1 transcription start site (TSS) via CRISPR/Cas9. We generated two independent clones with biallelic deletions, and in both of them, HBBP1 expression was lost (Supplemental Fig. S6C,D). However, β-globin and γ-globin transcription remained at control levels (Fig. 5B–D), suggesting that the HBBP1 genomic region, but not the HBBP1 transcript per se, is necessary for maintaining γ-globin silencing in adult erythroid cells.
Deletion of the HBBP1 region reconfigures the conformation of the β-globin locus to enable LCR–γ-globin contacts
Our architectural studies in adult erythroid cells along with the HBBP1 deletional experiments raise the possibility that the HBBP1 region via interaction with the ε-globin gene prevents the LCR from contacting and activating the fetal globin genes (see also the model in Fig. 7). To test directly whether the HBBP1–ε-globin interaction limits LCR–γ-globin contacts, we carried out Capture-C in two HBBP1 mutant clones. First, we asked whether the HBBP1 region is required for the contacts with the ε-globin genes. Capture-C probes were designed to anchor the BGLT3 gene, which resides adjacent to the HBBP1 gene but was spared by the HBBP1 deletion. Removal of HBBP1 markedly reduced the BGLT3–ε-globin contacts when compared with controls (Fig. 6B; Supplemental Fig. S7C). These results point to a requirement for the HBBP1 region in forming contacts with the ε-globin gene and for γ-globin silencing (Fig. 5). We next investigated whether disruption of the HBBP1–ε-globin contacts reactivates γ-globin expression by enabling de novo LCR–γ-globin interactions. To this end, we performed Capture-C using HS3 of the LCR as an anchor. In the absence of HBBP1, HS3 formed increased contacts with the γ-globin genes (Fig. 6A). Moreover, the interactions of HS3–β-globin were diminished in the HBBP1 deletion clones (Fig. 6A), possibly due to competition of β-globin and γ-globin genes for LCR interactions. In concert, these results support the idea that the HBBP1–ε-globin interaction insulates the LCR from the fetal globin genes at the adult stage.
Figure 7.
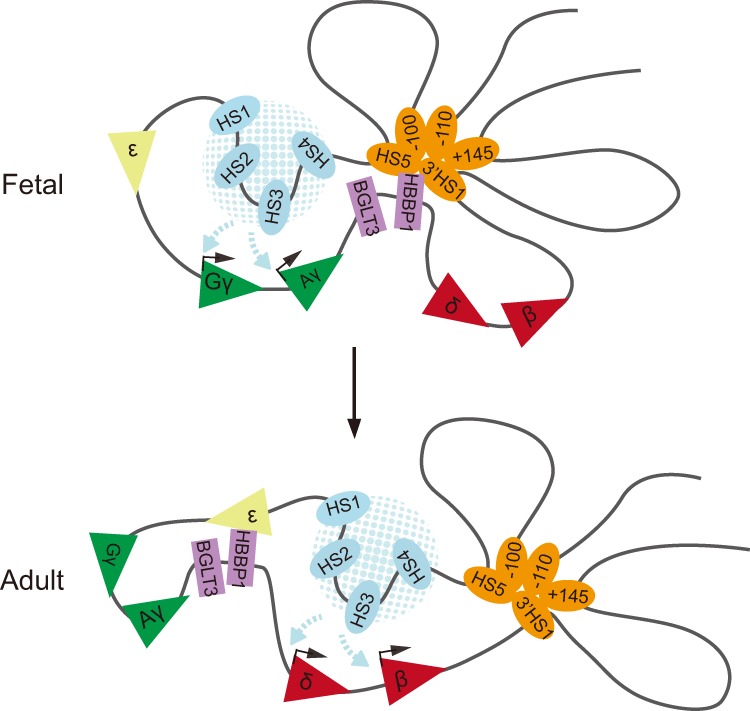
Model of chromatin configurations of the human β-globin locus in fetal and adult erythroblasts. The HBBP1 region mediates dynamic chromatin contacts with 3′HS1/HS5 and ε-globin at fetal and adult stages, respectively.
Figure 6.
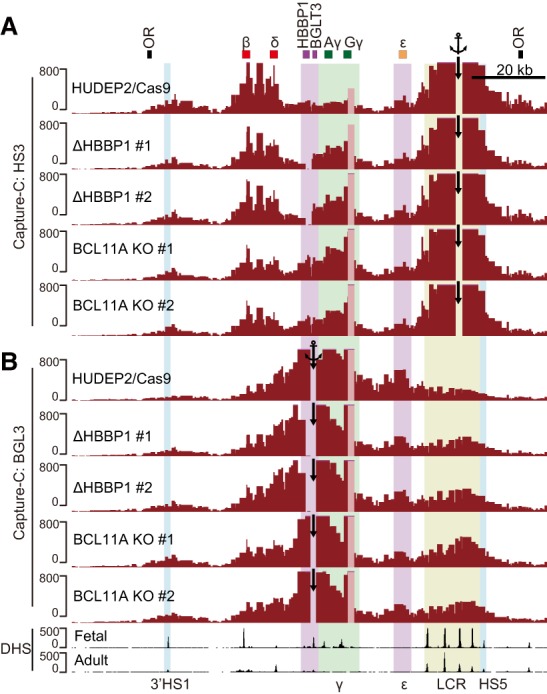
Chromatin interaction profiles of the β-globin locus in HBBP1-deleted and BCL11A knockout clones. (A,B) Capture-C interaction profiles of HS3 (A) and BGLT3 (B) in HBBP1-deleted and BCL11A knockout clones. The anchor positions are indicated by black arrows. (OR) Olfactory receptor genes.
Loss of BCL11A elevates fetal hemoglobin production in adult erythroid cells and leads to increased LCR–γ-globin gene contacts (Xu et al. 2012). Therefore, we explored a possible role of BCL11A in mediating the function of the HBBP1 region. Using CRISPR–Cas9, we generated two independent BCL11A knockout clones in HUDEP2 cells (Supplemental Fig. S7A). As expected, both clones showed elevated γ-globin gene expression (Supplemental Fig. S7B). To investigate a possible role of BCL11A in the HBBP1 region, we performed Capture-C experiments in both BCL11A-deficient lines. HS3 Capture-C revealed increased LCR–γ-globin and decreased LCR–β-globin contacts (Fig. 6A), as expected (Xu et al. 2010). These conformational changes were highly similar to those observed in the HBBP1-deleted lines (Fig. 6A). Strikingly, when using BGLT3 as an anchor, the contacts with ε-globin decreased to levels similar to those with the HBBP1 deletion (Fig. 6B; Supplemental Fig. S7C). The high similarity of chromatin conformation changes between BCL11A knockout and the HBBP1 deletion implicates BCL11A in the activity of the HBBP1 region.
Discussion
Our results provide a comparative view of genome-wide chromatin contacts in fetal and adult erythroid precursors. Hi-C experiments showed that, globally, TAD structures as well as the transcriptionally active A and inactive B compartments are highly similar between these populations. Sub-TAD structures surrounding the β-globin locus that are demarcated by CTCF-occupied sites are erythroid-specific but invariant between developmental stages. High-resolution Capture-C experiments detected previously known but also new stage-specific chromatin contacts involving the HBBP1 region that were not observed by low-resolution 3C (Chang et al. 2013). Deletion of this region led to pronounced increases in γ-globin transcription. Our results underpin a refined model of dynamic chromatin architectural features that contribute to the regulation of the fetal-to-adult globin switch during development (Fig. 7). In this model, the HBBP1 region engages in long-range contacts that configure the locus in a manner that promotes LCR interactions with its target genes at specific developmental stages. Thus, in fetal cells, the HBBP1 region contacts both HS5 and 3′HS1 to insulate the δ-globin and β-globin genes from the LCR, facilitating LCR–γ-globin contacts. In adult cells, the HBBP1 region interacts with the ε-globin gene to insulate the γ-globin genes from the LCR and enable LCR contacts with the δ-globin and β-globin genes. However, this model does not exclude the possibility that γ-globin silencing by the HBBP1 region involves direct contacts. Resolution limits of the method preclude us from definitively addressing this point.
Deletion of the HBBP1 TSS had no measurable effect on γ-globin production, indicating that transcription of HBBP1 is dispensable for γ-globin silencing. This suggests the existence of regulatory elements in this region that contribute to the structural organization of the locus. The considerable induction of γ-globin transcription upon HBBP1 deletion was surprising, since this region has not been implicated previously in the regulation of fetal hemoglobin expression. In this regard, it is noteworthy that two sequence variants (rs10128556 and rs2071348) within the second intron of the HBBP1 gene are associated with elevated fetal hemoglobin expression. However, whether these variants are causative or only implicated via association remains to be determined.
The identity of the factors governing the conformational switch of the HBBP1 contacts from HS5/3′HS1 to the ε-globin region remains unknown, but it is tempting to invoke proteins with known roles in fetal-to-adult globin switching. For example, published ChIP experiments suggest that BCL11A might occupy both the HBBP1/BGLT3 and ε-globin regions (Jawaid et al. 2010; Xu et al. 2010). However, these and other genome-wide location analyses of BCL11A are not entirely consistent with each other, which might be due to differences in the antibodies used or technical limitations of the ChIP assay. Indeed, using a method called CUT&RUN (cleavage under targets and release using nuclease) (Skene and Henikoff 2017), which can map protein-binding sites and their long-range genomic contact sites, BCL11A can be detected not only at the fetal globin gene promoters but, notably, also at multiple sites in the HBBP1 region (N Liu, Q Zhu, and SH Orkin, pers. comm.), suggesting a direct involvement of BCL11A in mediating the function of the HBBP1 region. Indeed, our Capture-C results revealed that BCL11A loss and HBBP1 deletion alter the locus in similar ways. Both modifications lowered the interactions of the HBBP1 region (or, in the case of the HBBP1 deletion, the adjacent BGLT3 region) with the ε-globin gene, increased LCR–γ-globin contacts with an accompanying reduction in LCR-β-globin contacts, and augmented γ-globin transcription. Therefore, we speculate that BCL11A contributes to the formation of the chromatin contacts of the HBBP1 region and conceivably also of the ε-globin gene. Proof of BCL11A involvement in this process will await mutation of individual or all BCL11A-binding sites in this area. It remains unknown how HBBP1 engages HS5 and 3′HS1 specifically in fetal cells. A simple survey of ENCODE ChIP-seq data suggests the possibility of numerous transcription factors binding to the HBBP1 region in K562 cells and human primary erythroblasts. Hence, a combination of nuclear factors might forge site- and stage-specific long-range contacts of the HBBP1 region.
The HBBP1 region that we identified as important for γ-globin silencing is partially or fully lost in several types of deletional hereditary persistence of fetal hemoglobin (HPFH; HPFH-2, HPFH-3, and HPFH-7) (Supplemental Fig. S6A). However, it does not overlap with a previously reported 3.5-kb region that is also located between the Aγ-globin and δ-globin genes and is required for γ-globin silencing (Sankaran et al. 2011). This suggests that multiple regulatory elements exist in the Aγ–δ intergenic region that function additively or redundantly to silence γ-globin.
Patients with SCD or some forms of β-thalassemia stand to benefit from elevated fetal hemoglobin levels. Besides gene correction or gene addition, genome editing as a tool to raise fetal hemoglobin levels has come within reach (Smith and Orkin 2016). Potential targets for genome editing include the erythroid enhancer of the BCL11A gene (Hardison and Blobel 2013; Smith et al. 2016; Chang et al. 2017), regions contained within HPFH deletions (Ye et al. 2016), and sequences upstream of the γ-globin promoters (Traxler et al. 2016). The Corfu deletion (Supplemental Fig. S6A) that is associated with HPFH might also be a potential target for CRISPR–Cas9-directed genome editing (Chakalova et al. 2005). Our results show that the HBBP1 region stands as an additional target for therapeutic genome editing with two potential advantages. First, the region is relatively small in size compared with the known HPFH deletions, which might make removal more efficient. Second, this region is not close to the promoters or coding regions of the globin genes, lowering the risk of their inadvertent disruption during genome manipulations.
In sum, our study provides a global view of developmental stage-specific chromatin architectural features in primary erythroid cells. We uncovered a requirement for the HBBP1 region for γ-globin silencing via formation of developmental stage-specific long-range chromosomal contacts and present the HBBP1 region as a potential target for therapeutic genome editing in SCD and β-thalassemia.
Materials and methods
Cells
CD34+ cells from human fetal livers were obtained from the University of Pennsylvania Stem Cell and Xenograft Core. Human adult peripheral blood mononuclear cells were obtained from the University of Pennsylvania Human Immunology Core. Adult CD34+ cells were isolated using CD34 microbead kit (Miltenyi Biotec). CD34+ cells were expanded and differentiated using a three-phase culture system as described (Bartman et al. 2016). H9-ESCs were obtained from the Human Pluripotent Stem Cell Core of the Children's Hospital of Philadelphia. HUDEP2 cells were cultured and differentiated as described (Traxler et al. 2016).
Hi-C
In situ Hi-C was carried out using DpnII enzyme as described (Rao et al. 2014; Hsu et al. 2017). Hi-C reads were processed and normalized using HiC-Pro (Servant et al. 2015) and ICE (Imakaev et al. 2012). TADs were called at 40-kb resolution as described (Dixon et al. 2012), except that the 0.5-Mb upstream/downstream regions were used to calculate the DI. PCA was performed on the observed/expected correlation matrices at 40-kb resolution. Compartments A and B were determined by the positive and negative PC1 values, respectively. The published Hi-C data were obtained using Juicebox software (Durand et al. 2016).
Capture-C
Capture-C was performed using DpnII enzyme with biotin-labeled DNA oligos (Supplemental Table S2) as described (Davies et al. 2016). The reads were processed using published scripts (Davies et al. 2016). Capture-C interactions were normalized to total interactions. Two biological replicates were carried out for all Capture-C experiments (Supplemental Table S3).
RNA-seq and RT-qPCR
Cells were counted manually and homogenized using TRIzol (Life Technologies). ERCC spike-in controls (Life Technologies) were diluted 1:10 and then added to the TRIzol lysates using 1 µL per million cells. Two biological replicates were carried out for all RNA-seq experiments, and RNA-seq libraries were constructed using the ScriptSeq Complete kit (Illumina) according to manufacturer's instructions. RNA-seq reads were normalized to the ERCC spike-in controls as described (Loven et al. 2012). Differentially expressed genes were identified using DEseq2 (Love et al. 2014). Primers for RT-qPCR are listed in Supplemental Table S4.
ChIP-seq
CTCF (Millipore, 07-729) ChIP-seq was performed in two biological replicates as described (Hsu et al. 2017). Peaks were called using MACS2 (Zhang et al. 2008) with default parameters.
CRISPR genome editing
A HUDEP2 cell line stably expressing SpCas9 was generated using lentivirus. Guide RNA plasmids (Supplemental Table S5) were transiently transfected into HUDEP2 cells as described (Traxler et al. 2016). Single-cell clones were screened and genotyped by Sanger sequencing.
Accession numbers
Published data sets used in this study are listed in Supplemental Table S6. The Hi-C, ChIP-seq, RNA-seq, and Capture-C data of this study have been deposited in the NCBI Gene Expression Omnibus (GEO) database (GSE102201).
Supplementary Material
Acknowledgments
We thank Jeremy Rupon for the help with cell cultures, Elisabetta Manduchi for assistance with RNA-seq analysis, and Kai Tan, Changya Chen, Wenbao Yu, Andrew Wells, Struan Grant, and members of the laboratory for helpful discussions. This work was supported by the National Institutes of Health (grants UO1HL129998A to G.A.B., and R24 DK106766-01 to R.C.H. and G.A.B.) and the Children's Hospital of Philadelphia Center for Spatial and Functional Genomics (G.A.B.).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.303461.117.
References
- Bartman CR, Hsu SC, Hsiung CC, Raj A, Blobel GA. 2016. Enhancer regulation of transcriptional bursting parameters revealed by forced chromatin looping. Mol Cell 62: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakalova L, Osborne CS, Dai YF, Goyenechea B, Metaxotou-Mavromati A, Kattamis A, Kattamis C, Fraser P. 2005. The Corfu δβ thalassemia deletion disrupts γ-globin gene silencing and reveals post-transcriptional regulation of HbF expression. Blood 105: 2154–2160. [DOI] [PubMed] [Google Scholar]
- Chang KH, Fang X, Wang H, Huang A, Cao H, Yang Y, Bonig H, Stamatoyannopoulos JA, Papayannopoulou T. 2013. Epigenetic modifications and chromosome conformations of the β globin locus throughout development. Stem Cell Rev 9: 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, Smith SE, Sullivan T, Chen K, Zhou Q, West JA, Liu M, Liu Y, Vieira BF, Sun C, et al. 2017. Long-term engraftment and fetal globin induction upon BCL11A gene editing in bone-marrow-derived CD34+ hematopoietic stem and progenitor cells. Mol Ther Methods Clin Dev 4: 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JO, Telenius JM, McGowan SJ, Roberts NA, Taylor S, Higgs DR, Hughes JR. 2016. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat Methods 13: 74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, et al. 2014. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell 158: 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. 2015. Chromatin architecture reorganization during stem cell differentiation. Nature 518: 331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. 2006. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16: 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. 2016. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3: 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forcato M, Nicoletti C, Pal K, Livi CM, Ferrari F, Bicciato S. 2017. Comparison of computational methods for Hi-C data analysis. Nat Methods 14: 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. 2010. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet 42: 1049–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison RC, Blobel GA. 2013. Genetics. GWAS to therapy by genome edits? Science 342: 206–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Dale R, Dean A. 2010. Cell type specificity of chromatin organization mediated by CTCF and cohesin. Proc Natl Acad Sci 107: 3651–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SC, Gilgenast TG, Bartman CR, Edwards CR, Stonestrom AJ, Huang P, Emerson DJ, Evans P, Werner MT, Keller CA, et al. 2017. The BET protein BRD2 cooperates with CTCF to enforce transcriptional and architectural boundaries. Mol Cell 66: 102–116.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Liu X, Li D, Shao Z, Cao H, Zhang Y, Trompouki E, Bowman TV, Zon LI, Yuan GC, et al. 2016. Dynamic control of enhancer repertoires drives lineage and stage-specific transcription during hematopoiesis. Dev Cell 36: 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. 2012. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods 9: 999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jawaid K, Wahlberg K, Thein SL, Best S. 2010. Binding patterns of BCL11A in the globin and GATA1 loci and characterization of the BCL11A fetal hemoglobin locus. Blood Cells Mol Dis 45: 140–146. [DOI] [PubMed] [Google Scholar]
- Kang Y, Kim YW, Kang J, Yun WJ, Kim A. 2017. Erythroid specific activator GATA-1-dependent interactions between CTCF sites around the β-globin locus. Biochim Biophys Acta 1860: 416–426. [DOI] [PubMed] [Google Scholar]
- Kiefer CM, Lee J, Hou C, Dale RK, Lee YT, Meier ER, Miller JL, Dean A. 2011. Distinct Ldb1/NLI complexes orchestrate γ-globin repression and reactivation through ETO2 in human adult erythroid cells. Blood 118: 6200–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, Di Stefano B, de Wit E, Limone F, van Oevelen C, de Laat W, Graf T. 2016. Cell-of-origin-specific 3D genome structure acquired during somatic cell reprogramming. Cell Stem Cell 18: 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita R, Suda N, Sudo K, Miharada K, Hiroyama T, Miyoshi H, Tani K, Nakamura Y. 2013. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8: e59890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard S, Beaudoin M, Benkirane K, Lettre G. 2015. Comparison of DNA methylation profiles in human fetal and adult red blood cell progenitors. Genome Med 7: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. 2009. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326: 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA. 2012. Revisiting global gene expression analysis. Cell 151: 476–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485: 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuinoon M, Makarasara W, Mushiroda T, Setianingsih I, Wahidiyat PA, Sripichai O, Kumasaka N, Takahashi A, Svasti S, Munkongdee T, et al. 2010. A genome-wide association identified the common genetic variants influence disease severity in β0-thalassemia/hemoglobin E. Hum Genet 127: 303–314. [DOI] [PubMed] [Google Scholar]
- Palstra RJ, Tolhuis B, Splinter E, Nijmeijer R, Grosveld F, de Laat W. 2003. The β-globin nuclear compartment in development and erythroid differentiation. Nat Genet 35: 190–194. [DOI] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH. 2008. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322: 1839–1842. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, Sabath DE, Groudine M, Orkin SH, Premawardhena A, et al. 2011. A functional element necessary for fetal hemoglobin silencing. N Engl J Med 365: 807–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Servant N, Varoquaux N, Lajoie BR, Viara E, Chen CJ, Vert JP, Heard E, Dekker J, Barillot E. 2015. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol 16: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S. 2017. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EC, Orkin SH. 2016. Hemoglobin genetics: recent contributions of GWAS and gene editing. Hum Mol Genet 25: R99–R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EC, Luc S, Croney DM, Woodworth MB, Greig LC, Fujiwara Y, Nguyen M, Sher F, Macklis JD, Bauer DE, et al. 2016. Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 128: 2338–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stonestrom AJ, Hsu SC, Jahn KS, Huang P, Keller CA, Giardine BM, Kadauke S, Campbell AE, Evans P, Hardison RC, et al. 2015. Functions of BET proteins in erythroid gene expression. Blood 125: 2825–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C, et al. 2016. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 22: 987–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Sankaran VG, Ni M, Menne TF, Puram RV, Kim W, Orkin SH. 2010. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev 24: 783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shao Z, Glass K, Bauer DE, Pinello L, Van Handel B, Hou S, Stamatoyannopoulos JA, Mikkola HK, Yuan GC, et al. 2012. Combinatorial assembly of developmental stage-specific enhancers controls gene expression programs during human erythropoiesis. Dev Cell 23: 796–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye L, Wang J, Tan Y, Beyer AI, Xie F, Muench MO, Kan YW. 2016. Genome editing using CRISPR–Cas9 to create the HPFH genotype in HSPCs: an approach for treating sickle cell disease and β-thalassemia. Proc Natl Acad Sci 113: 10661–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. 2012. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science 335: 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.