

Abstract
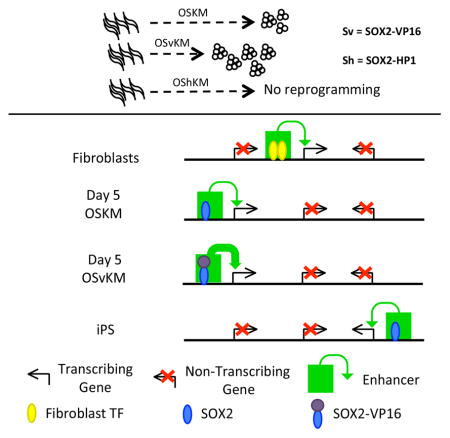
SOX2 and OCT4, in conjunction with KLF4 and cMYC, are sufficient to reprogram human fibroblasts to induced pluripotent stem cells (iPSCs) but it is unclear if they function as transcriptional activators or as repressors. We now show that, like OCT4, SOX2 functions as a transcriptional activator. We substituted SOX2-VP16 (a strong activator) for WT SOX2 and saw an increase in the efficiency and rate of reprogramming, whereas the SOX2-HP1 fusion (a strong repressor) eliminated reprogramming. We report that at an early stage of reprogramming, virtually all DNA-bound OCT4, SOX2, and SOX2-VP16 were embedded in putative enhancers, about half of which were created de novo. Those associated with SOX2-VP16 were, on average, stronger than those bearing WT SOX2. Many newly created putative enhancers were transient and many transcription factor locations on DNA changed as reprogramming progressed. These results are consistent with the idea that, during reprogramming, there is an intermediate state that is distinct from both parental and iPS cells.
eTOC blurb
Narayan et al. show that substituting SOX2 with the strong activator SOX2-VP16 increases reprogramming efficiency of human fibroblasts, especially those cultured from older donors. Thousands of enhancers are created and destroyed in the course of reprogramming, including many enhancers created at binding sites of OCT4 or SOX2.
INTRODUCTION
Ectopic expression of four transcription factors - OCT4, SOX2, KLF4 and cMYC (OSKM) - can reprogram differentiated human and murine fibroblasts to iPS (induced pluripotent stem) cells (Takahashi et al., 2007; Takahashi and Yamanaka, 2006; Yu et al., 2007). During transition to the pluripotent state many genes expressed in the differentiated state are silenced, and many others activated (Theunissen and Jaenisch, 2014). The iPS state, in turn, is maintained by feedback loops involving (at least) endogenously encoded OCT4, SOX2 and KLF4 (Boyer et al., 2005; Chew et al., 2005; Do and Scholer, 2009; Jaenisch and Young, 2008; Kim et al., 2008; Loh et al., 2006; Martello and Smith, 2014). The efficiency of reprogramming human neonatal fibroblasts is typically low (0.002–0.02%), and that for human cells from older donors even lower (Maherali et al., 2008; Park et al., 2008; Paull et al., 2015; Rohani et al., 2014; Takahashi et al., 2007; Yu et al., 2007). Elimination of any one of the factors OCT4, SOX2 or KLF4 abolishes reprogramming and in the absence of cMYC reprogramming efficiency is very low. TRIM71 and LIN28A are RNA binding proteins, ectopic expression of either of which has been reported to abrogate the requirement for ectopic cMYC during reprogramming (Worringer et al., 2014; Yu et al., 2007).
In a typical experiment, putative iPSC colonies formed by reprogrammed fibroblasts are identified as staining positive for the human embryonic stem cell surface marker TRA1-81 (Adewumi et al., 2007), and then are tested for their abilities to differentiate into three major cell lineages - ectoderm, mesoderm and endoderm (Takahashi et al., 2007; Yu et al., 2007). Recovery of TRA1-81-positive colonies is usually observed only after some 20 days following ectopic expression of OSKM, and certain proteins crucial for reprogramming (e.g. NANOG) are detectably expressed beginning only at about day 9 (Cacchiarelli et al., 2015; Polo et al., 2012).
In higher eukaryotes, genes are often controlled by transcriptional activators that bind to DNA regulatory elements to form enhancers. A typical eukaryotic transcription activator comprises two functional domains, the DNA-binding domain and an activating region. (Ptashne and Gann, 2002). VP16, a herpes viral protein, is a particularly strong activating region that works in a wide array of eukaryotic cells when tethered to DNA (Sadowski et al., 1988; Triezenberg et al., 1988). Enhancers typically bear more than one DNA-bound activator, and are sometimes positioned many thousands of base pairs from the regulated gene (Benoist and Chambon, 1981; Muller and Schaffner, 1990; Ptashne and Gann, 2002). A gene activated in one cell type by one enhancer may be regulated by a different enhancer, comprising at least in part different activators, in another cell type (Berrozpe et al., 2006; Perry et al., 2011; Stergachis et al., 2013). “Decommissioning” an enhancer, which can suffice to turn off transcription of the target gene driven by that enhancer, leaves that target gene free to respond to other enhancers, a key aspect of the regulatory logic of developing embryos (Gilbert and Barressi, 2016; Ptashne and Gann, 2002).
It has recently been shown that enhancers bear, in addition to transcription factors, Pol II and nucleosomes bearing the modifications H3K27ac and H3K4me1. We found, for example, that the enhancer that drives expression of the murine cKit gene in mast cells bears such modified nucleosomes flanking ca. 300 bp regions at which transcriptional activators have displaced nucleosomes (Berrozpe et al., 2013). The function of the nucleosome modifications is not known (Calo and Wysocka, 2013; Shlyueva et al., 2014). These various features of enhancers have prompted their identification genome-wide, and comparison of their apparent relative strengths, using Chip-Seq and related methods (Barski et al., 2007; Birney et al., 2007; Heintzman et al., 2007; Zhou et al., 2011). Enhancers identified in that fashion are “putative” until verified by functional assays in proper cell types and chromosomal contexts (Inoue et al., 2017).
We and others have reported that OCT4 fused to a strong activation domain (e.g. full length OCT4 fused to VP16), expressed in combination with native SKM proteins, reprogrammed both murine and human fibroblasts to iPS cells (Hammachi et al., 2012; Hirai et al., 2011; Wang et al., 2011). In contrast, inclusion of a fusion protein bearing the ‘repressing’ protein HP1, attached to OCT4, abolished reprogramming (Hammachi et al., 2012). We therefore surmised that OCT4 works primarily, if not solely, as an activator in reprogramming.
Here we extend that conclusion by showing that SOX2, like OCT4, works as a transcriptional activator in reprogramming human fibroblasts. Thus SOX2-VP16, substituted in the canonical OSKM mix for WT SOX2, increased the efficiency and rate of reprogramming compared to the WT mix. Moreover, consistent with the notion that these proteins work as activators, OCT4, SOX2 and SOX2-VP16 (expressed in different mixes), although often found bound to pre-existing putative enhancers, about equally frequently created new putative enhancers at an early stage. These and other new putative enhancers were present only transiently, suggesting that they reflect an intermediate stage of reprogramming that is not simply a combination of parental and iPS states.
RESULTS
Ectopic expression vectors
We designed lentiviral vectors expressing OCT4, SOX2, KLF4, or cMYC, and, separately, vectors expressing each of these factors fused to a heterologous activating (VP16) or repressing (HP1) region. The fusions, for the case of SOX2 for example, are designated Sv (SOX2-VP16) and Sh (SOX2-HP1). An analogous vector set expresses one or another of each DNA binding domain (DBD) alone. Each vector also co-expresses one or another fluorescent protein. Wild type (WT) and fusion proteins with attached fluorescent markers and other features are listed in Supplementary Table S1 (Hammachi et al., 2012; Papapetrou et al., 2009). Human fibroblasts were cultured from a 14-week old fetus, from a newborn, and from adult donors of various ages (Supplementary Table S2).
Reprogramming human fibroblasts
Fibroblasts growing on plates were infected with lentiviruses expressing various combinations of reprogramming factors, and five days later these cells were re-plated. In most cases, the re-plating was onto plates seeded with a feeder layer (i.e. mitomycin-C treated mouse embryonic fibroblasts), and some 20 days later colonies were stained for TRA1-81. Except as otherwise indicated, all TRA1-81 positive colonies tested, differentiated into the three major lineages (Supplementary Figure S1C). We therefore take the count of TRA1-81 positive colonies as a measure of the efficiency of reprogramming.
We first tested a wide array of reprogramming mixes, differing in the factors expressed (e.g. OSKM versus OSvKM), and at different relative MOIs of the individual factors in fetal fibroblasts (Supplementary Table S3). Maximal reprogramming was achieved with MOIs that differed depending upon the identity of the factors. For example, maximal reprogramming was achieved at MOIs of 4, 1, 1, 0.4 for the OSKM mix, and MOIs of 4, 4, 4, 0.4 for the OSvKM mix (Table 1, rows 1 and 4). As shown in this table, swapping these MOIs decreased reprogramming efficiency by about 3 fold for the OSKM mix and by about 2 fold for the OSvKM mix (Table 1, compare rows 1 and 2 for the OSKM mix and rows 3 and 4 for the OSvKM mix). In the following experiments we used reprogramming mixes at the MOIs that, according to these preliminary experiments, mediated maximal reprogramming. Where cells from older donors were employed for reprogramming (see below), a higher titer of all four viruses, but at the same ratio of MOIs, was used (Supplementary Table S4).
Table 1. Reprogramming of fetal fibroblasts using OSKM, OSvKM or OShKM.
TRA1-81 positive iPS cells derived from fetal lung fibroblasts that were infected with lentiviruses encoding the standard Yamanaka mix expressing OCT4, SOX2, KLF4 and cMYC (OSKM), the Yamanaka mix with SOX2 replaced with SOX2-VP16 (OSvKM, grey rows) or the Yamanaka mix with SOX2 replaced with SOX2-HP1 (OShKM). Five days following infection, cells were re-plated in the presence or absence of feeder (mitomycin-C treated mouse embryonic fibroblasts) cells. Reprogramming efficiency, as measured by the number and size of TRA1-81 positive colonies on day 25 at multiplicities of infection (MOIs) with each mix are shown. Data shown in this table is taken from Figure 1A–D. Sizes of TRA1-81 positive colonies were estimated using a phase contrast microscope as either large (ca ≥ 50 cells/colony) or small to medium-sized colonies (ca 25–50 cells/colony). ND= not determined. (also see Supplementary materials and methods).
| # | Reprogramming Mix | MOI for each lentivirus in reprogramming mix | Reprogramming Efficiency (TRA1-81 positive colonies on day 25 per ~ 2500 cells/cm2 plated on day 5) | ||
|---|---|---|---|---|---|
| Total # colonies | Large colonies | Medium/Small colonies | |||
| With Feeders | |||||
| 1 | OSKM | 4,1,1,0.4 | 20.1 +/− 5.7 | 2.5 +/− 2.2 | 17.6 +/− 4 |
| 2 | OSKM | 4,4,4,0.4 | 7.5 +/− 2 | 1 +/− 1.4 | 6.5 +/− 0.7 |
| 3 | OSvKM | 4,1,1,0.4 | 70.5 +/− 2.8 | 57.5 +/− 2.8 | 13 +/− 0 |
| 4 | OSvKM | 4,4,4,0.4 | 113.3 +/− 13.8 | 77.8 +/− 10.3 | 35.5 +/− 3.5 |
| 5 | OShKM | 4,4,4,0.4 | 0 | 0 | 0 |
| Without Feeders | |||||
| 6 | OSKM | 4,1,1,0.4 | 2.6 +/− 2.8 | 0.8 +/− 1.2 | 1.8 +/− 2.2 |
| 7 | OSvKM | 4,4,4,0.4 | 33.1 +/− 5.6 | 19.3 +/− 4.7 | 13.8 +/− 1.8 |
The effect of SOX2-VP16 on the efficiency of reprogramming
Substitution of SOX2-VP16 for WT SOX2 improved the efficiency of reprogramming of fetal fibroblasts. Ectopic expression of OSvKM in human fetal fibroblasts induced about 6 fold more iPS colonies than did expression of OSKM (Figure 1A, B). OSvKM also worked more efficiently than did OSKM on fibroblasts cultured from an array of older donors (37y, 42y, 61y, 66y, 67y, 82y and 96y), as well as on fibroblasts cultured from a new born (Figure 1E, F, and Supplementary Table S4, rows 1–18). Fibroblasts from older donors expressed elevated levels of senescence-activated beta-galactosidase (SA-βgal) as typically found for older cells (Supplementary Figure S1D). Also, as expected, most of the nuclei from older donor fibroblasts contained (as revealed by staining) lower levels of H3K9me3, H3K27me3 and LAP2α (Studer et al., 2015) than did nuclei of younger fibroblasts (Supplementary Figure S1 E–G). The mix OShKM, which includes a fusion bearing the ‘repressing’ protein HP1α to SOX2, abolished reprogramming (Figure 1A, B). SOX2-VP16 also increased the rate of reprogramming. TRA1-81-positive colonies were recovered from fetal fibroblasts earlier (day 10 versus day 15) for OSvKM- expressing cells than for cells expressing the WT mix (data not shown).
Figure 1. Substitution of SOX2 with SOX2-VP16 (Sv) improves reprogramming efficiency in human fibroblasts.

TRA1-81 positive iPS cells derived from fetal lung fibroblasts that were infected with lentiviruses encoding the standard Yamanaka mix expressing OCT4, SOX2, KLF4 and cMYC (OSKM), the Yamanaka mix with SOX2 replaced with SOX2-VP16 (OSvKM) or the Yamanaka mix with SOX2 replaced with SOX2-HP1 (OShKM) at MOIs previously determined to be optimal for reprogramming (see text and Table 1). Five days following infection, cells were re-plated with feeders at a density of approximately 2500 cells/cm2. (A) Plates representing each of the above mixes were stained to determine the number of TRA1-81 positive colonies on day 25. (B) Quantitation of total TRA1-81 positive colonies from (A). (C) TRA1-81 positive iPS cells generated using the OSKM and OSvKM mixes as described in (A) except that cells were re-plated without feeders. (D) Quantitation of total TRA1-81 positive colonies from (C). (E) TRA1-81 positive cells derived from fibroblasts cultured from donors of different ages: fetal (14 weeks), new born (NB), 42 year and 96 year old donors generated using the OSKM and OSvKM mixes as described in (A). (F) Quantitation of total TRA1-81 positive colonies from (E) along with other donors of different ages. (G) TRA1-81 positive cells derived from fibroblasts, in the absence of cMYC, from donors of different ages: fetal (14 weeks), new born (NB), and 42 and 96 year old generated in the absence of ectopic cMYC using the OSK and OSvK mixes. (H) Quantitation of total TRA1-81 positive colonies from (G) along with other donors of different ages.
Reprogramming of fibroblasts taken from both fetal and older human donors by OSvKM was less dependent on feeder cells than was reprogramming by OSKM. Thus, fetal fibroblasts were reprogrammed with OSvKM at about 3 fold lower efficiency in the absence of feeders compared to the presence of feeders. In contrast, reprogramming by OSKM decreased by least 10 fold with elimination of the feeder cells (Table 1 and Figure 1B, D). OSvKM, but not OSKM, reprogrammed fibroblasts from older donors (37y, 42y, 61y, 66y, 82y, 96y) in the absence of feeders (data not shown).
Substitution of SOX2-VP16 for SOX2 in the reprogramming mix completely relieved the requirement for cMYC in reprogramming fetal fibroblasts. Consistent with the observations of others, ectopic expression of OSK (i.e. no cMYC) failed to reprogram fibroblasts from donors of most age groups (Nakagawa et al., 2008; Wernig et al., 2008; Worringer et al., 2014). However, OSvK reprogrammed fetal fibroblasts almost as efficiently as did OSvKM (Figure 1G, H and Supplementary Table S4). TRA1-81 positive colonies derived from older donor fibroblasts reprogrammed with OSvK were small and difficult to passage (Figure 1G, H and Supplementary Table S4) and most did not differentiate in to the three major lineages. The karyotypes of one OSvKM and another OSvK- derived iPS lines (passage >10), originating from fetal fibroblasts, were analyzed and found to be normal (Supplementary Figure S1B).
A reprogramming mix with KLF4-VP16 or cMYC-VP16 in place of their WT counterparts, worked only very inefficiently. KLF4-HP1, cMYC-HP1, like OCT4-HP1 and SOX2-HP1 worked as ‘dominant negatives’, eliminating reprogramming (Supplementary Tables S3). Expression of DBDs alone resulted in no detectable reprogramming (Supplementary Table S3).
Changes in expression of specific endogenous genes
We used QPCR analysis to determine how substitution of SOX2-VP16 for WT SOX2 might have affected expression of genes NANOG, TRIM71, and LIN28A, known to be important for reprogramming. All 3 of these genes were dramatically induced by day 5 in fetal fibroblasts expressing OSvKM (Figure 2A–C). For example, by day 5, the level of NANOG mRNA was several fold higher than that induced by OSKM (Figure 2A) and was within a factor of 3 of that found in human iPS and ES cells. To confirm that NANOG protein levels were increased by substitution of SOX2-VP16, we subjected day 5 cells to FACS analysis. About 10% of cells expressing OSvKM were also NANOG positive, whereas the corresponding figure for OSKM expressing cells was some 30-fold lower (data not shown). Please recall that, as described above, reprogramming by WT mixes required ectopic cMYC, but reprogramming by mixes containing SOX2-VP16 was detectable in the absence of ectopic cMYC. The QPCR analysis of Figure 2D–F shows that elimination of ectopic cMYC decreased, but did not abolish, early expression of NANOG and TRIM71 in cells expressing SOX2-VP16. Substitution of either KLF4 or cMYC with its VP16- fusion derivative (i.e. KLF4-VP16 or cMYC-VP16) had no effect on NANOG expression, and OCT4-VP16 derivatives had only a small positive effect (data not shown).
Figure 2. Early induction of NANOG, TRIM71 and LIN28A mRNA by SOX2-VP16.

(A) QPCR measurement of NANOG mRNA levels at days 1 through 5 in fetal derived fibroblasts expressing either the OSKM (red curve), OSvKM (green curve) mixes or in mock-infected cells (blue curve). For comparison, the levels of NANOG mRNA in iPS cells is also shown (purple data point). (B, C) QPCR measurement of TRIM71 (B) and LIN28A (C) mRNA for the mixes described in (A). (D) QPCR measurement of NANOG mRNA levels at days 1 through 5 in fetal derived fibroblasts expressing either the OSK (red curve) and OSvK (green curve) mixes, i.e. in the absence of ectopically expressed cMYC. (E, F) QPCR measurement of TRIM71 (E) and LIN28A (F) mRNA for the mixes described in (D). mRNA level in mock-infected fibroblasts are shown in blue.
Whole genome microarray analysis identified other genes expressed in ES cells that were induced by OSvKM (measured at day 5), but much less so, if at all, with OSKM. These genes include SOX21, UTF1, and ALPL (Supplementary Figure S2). Several fibroblast-specific genes that were down-regulated about equally in cells expressing OSvKM or OSKM included COL3A, COL5A, GREM1, DCN, LOX and LUM (Supplementary Figure S2).
Genome wide mapping of putative enhancers and DNA-bound transcription factors
To further probe the gene regulatory events underlying reprogramming, and in particular the difference in the effects of SOX2-VP16 and WT SOX2, we used a modified ChIP-Seq assay (ChIP-Endo-Seq) to analyze transcription factor binding and putative enhancer formation genome-wide. We located, to within 100 bp DNA segments, sites of binding of each of the transcription factors OCT4, SOX2, SOX2-VP16, KLF4 and cMYC in day 5 cells, and in human iPS cells. We also searched for signals for putative enhancers and promoters and quantitated their strengths at sequential 2 Kb segments covering the genome. The putative enhancers we identify using this method can extend ca. 2 to 20 Kb in length. We applied this analysis to 6 cell types: fetal fibroblasts; fetal fibroblasts at day 5 following initiation of expression of one or the other of the four different reprogramming mixes (OSKM, OSvKM, OSK and OSvK); and iPS cells derived from fetal fibroblasts. We compared those signals, at those locations, to corresponding regions in each of the other cell types.
To illustrate the information we can obtain from this kind of survey we show, in Figure 3, a DNA segment that includes a gene, transcription of which is induced by OSKM and by OSvKM. In the parental fibroblasts (Figure 3A), there is little or no indication of enhancers in this region, and Pol II is only lightly distributed along the length of the gene. In contrast, in the OSKM-expressing cells (Figure 3B) at day 5, we detect two major putative enhancer peaks that include H3K27ac, H3K4me1, Pol II, OCT4 and SOX2 positioned some 65,000 bp upstream of the gene. The gene itself bears Pol II along its length and a significant peak of H3K4me3 at the promoter. In OSvKM-expressing cells at day 5 (Figure 3C), these enhancer appears at a location identical to that found in the OSKM cells but with stronger enhancer signals. Further experiments would be required to definitively conclude that transcription of HMGCR gene is driven by the detected enhancers. In general, we do not know the identities of the gene(s), if any, that are activated by the putative enhancers we identify in the remainder of the paper.
Figure 3. The HMGCR gene and upstream region in fetal fibroblasts and in those cells expressing OSKM or OSvKM.

The region encompassing the HMGCR gene (red bar), and ca. 65,000 bp upstream (grey bar) is shown. For each case, ChIP-Endo-Seq was used to identify the positions and identities of each of the proteins and histone modifications listed on the left side of the figure. The identified putative enhancer is delineated by the faint orange box, and the promoter region by a green box. (A) Parental fibroblasts. (B) Fibroblasts at day 5 following ectopic expression of OSKM. (C) Same as B except that the cells were expressing OSvKM.
Changing locations of DNA bound factors
The following experiments show that, as others have found, many OCT4 and SOX2 proteins change their sites of DNA binding when going from an early stage of reprogramming (Day 5 in our case) to iPS cells (Sridharan et al., 2009). We further found that this effect is mitigated for the case of the OCT4/SOX2, a pair that others have shown binds cooperatively (Chew et al., 2005; Rodda et al., 2005; Yuan et al., 1995).
We first identified sites of binding of a given transcription factor in one cell type, and then measured the occupancies of those sites by the same factor in other cell types. Part A of Figure 4 shows that, at day 5, as found by others, each of the transcription factors binds mostly (but not entirely) to a different set of sites than those occupied in iPS cells (Sridharan et al., 2009). The effect is illustrated for OCT4 in upper left panel. OCT4 was significantly bound (left most red bar) to some 5,934 sites on DNA in cells expressing OSKM. The remaining red bars are ordered heat maps showing the extent of binding of OCT4 to those sites in other cell types, including in iPS cells. The figure shows that the overlap of bound sites is higher (greater than 50 %) between OSKM day 5 cells and the other day 5 cells, but the overlap is much less so between OSKM day 5 and iPS cells. This overlap pattern is also observed when OCT4 bound sites identified in other cells e.g., day 5 cells expressing OSvKM, are probed in other day 5 and in iPS cells as shown in the figure. For sites identified in iPS cells and interrogated in various day 5 cells (last column in top row), we find that most of the OCT4 sites in iPS cells are not bound by OCT4 in the day 5 cells. Row 2 of Figure 4A shows that the pattern of OCT4 binding holds as well for binding of SOX2.
Figure 4. Transcription factor binding displayed as ordered heat maps at specific sites.

(A) Overlaps of transcription factor binding, for each of the factors OCT4, SOX2 or SOX2-VP16, KLF4 and cMYC, in day 5 and in iPS cells. Row 1, left panel: at each of the 5,934 OCT4-bearing sites in OSKM expressing cells at day 5, the ordered heat map of OCT4 signal strengths at those sites is shown for each of the scenarios indicated at the bottom of each bar. For example, in iPS cells, there is considerably less OCT4 binding at these sites than is found in any of the day 5 cells. In the top panel, second column, the same analysis was performed for 3,809 OCT4 sites identified at day 5 in OSvKM infected cells. Rows 2–4 as for Row 1 except that, as indicated, SOX2 or SOX2-VP16 (blue), KLF4 (green), and cMYC (cyan)-bound sites were identified in one or another scenario (e.g. bound KLF4 in OSvK expressing cells and then probed for KLF4 signal in other scenarios, including in iPS cells). (B) Co-binding of OCT4 and SOX2 or SOX2-VP16. The sites of OCT4 and SOX2 binding measured in the various scenarios in (A) (e.g. in OSvKM expressing cells at day 5) were further probed for the presence of the other factor (OCT4 or SOX2 or SOX2-VP16) positioned with the same 2 Kb fragment in the same cell. As in (A), strengths of OCT4 signals are in red and of SOX2 or SOX2-VP16 signals in blue. (C) Overlap in different cells between co-bound sites and singly bound sites. Top row, first panel: 1,094 positions at which OCT4 and SOX2 were co-bound were identified in OSKM-expressing day 5 cells. As before, red bars indicate OCT4 binding and the blue bars SOX2 or SOX2-VP16 binding. The signal strengths as indicated by heat maps for these proteins at the same sites in other cells (e.g. iPS) is shown. The other panels in this top row perform the same analysis for sites originally identified as bearing the pair of proteins (OCT4, SOX2 or SOX2-VP16) in other cells. The bottom two rows show the same analysis for sites bearing just one or the other of these proteins. ND = not done.
We imagined that the changing locations of OCT4 and SOX2 (day 5 vs. iPS) were caused by changes in the partners with which the factors bind cooperatively. To probe this idea we further examined binding of OCT4 and SOX2, proteins reported to be able to bind cooperatively with each other, and both of which are present in day 5 and in iPS cells. Consistent with finding that OCT4 and SOX2 can bind to DNA cooperatively, we found that OCT4 and SOX2 are bound together (i.e. in the same 2 Kb fragment) at day 5 at a much higher frequency than expected were they to always bind independently (Figure 4B). The figure (left part) shows that in all five cell types some 20–25 % of sites bound by OCT4 also bound by SOX2. The figure also shows (right part) even higher percentage of sites bound SOX2 are also bound by OCT4. Were these proteins to bind independently we would expect to find less than 1% of 2 Kb fragments bearing one protein would also bear the other.
We then compared sites co-bound by OCT4/SOX2, or singly bound by either, in day 5 and in iPS cells, and found that sites co-bound by OCT4/SOX2 at day 5 were more likely to remain occupied by these same proteins in iPS cells than were singly bound sites (Figure 4C). In the top row sites co-bound by OCT4 and SOX2 in one cell type e.g., day 5 fibroblasts expressing OSKM were interrogated for binding of both protein in other cell types. Note sites bound by both proteins in one cell type are more likely to be bound by both proteins in other cell types including iPS cells. The next two rows in figure 4C show, in contrast, singly bound sites in one cell type are less likely (compare to doubly bound sites) to be bound by the same protein in other cell types including iPS cells.
Putative enhancers at sites of bound transcription factors
OCT4, SOX2, and SOX-VP16
At most (> 80%) of the positions on DNA at which OCT4, SOX2 or SOX2-VP16 was bound at day 5 we found a putative enhancer (Figure 5A and Supplementary Figure S3–S5). This result held for all four reprogramming mixes (data for OSvKM, OSK and OSvK mixes are not shown). The phenomena is illustrated in Figure 5A (top left panel). The panel shows a distribution plot in which the proportion of OCT4 bound sites (Y axis) that have a given enhancer signal strength (log2 of Pol II + H3K27ac + H3K4me1) (X axis), for day 5 cells expressing OSKM. We arbitrarily define a putative enhancer as a 2 Kb fragment with a signal strength above 2.7 (vertical line). The grey area shows the distribution of these signals genome wide, and the green area the distribution at OCT4 bound sites. As indicated in the figure, some 13% of the 2 Kb fragments analyzed genome-wide register as putative enhancers whereas 84% of the OCT4 bound sites (green curve) register as putative enhancers. The next panel examines the same sites (i.e., OCT4 bound sites in OSKM day 5 cells) in fibroblasts. In this case only 48% of the interrogated sites registered as putative enhancers, indicating that about half of the putative enhancers bearing OCT4 in day 5 cells were newly created. The third panel in the top row shows, in addition, that at 83% of the sites of OCT4 binding in day 5 cells (whether or not there was a preexisting enhancer), the putative enhancer signal was increased compare to that of the parental fibroblasts. A similar set of results was obtained comparing SOX2 (Figure 5A) or SOX2-VP16 (not shown) bound sites in day 5 cells with corresponding regions in fibroblasts. To determine whether our definition of a putative enhancer is consistent with those of others, we subjected our data to analysis by ChromHMM (Ernst et al., 2011) (Supplementary Figure S9A–C). Over 90% of our identified OCT4- bound sites is classified as enhancers (strong, moderate, etc) by ChromHMM (Supplementary Figure S9B). A similar correspondence was found for all of the enhancers we describe here and below (Supplementary Figure S9C). For one particular gene and enhancer, i.e. that of Figure 3, the ChromHMM results are noted in Supplementary Figure S9D.
Figure 5. Putative enhancers at transcription factor binding sites.

(A) Each panel shows the distribution plot of the enhancer signal at the site of transcription factor binding (colored curves) or the distribution of the enhancer signal strength genome wide (grey curves). A distribution plot shows the proportion of sites (y-axis) that have a specified signal strength (x-axis) over the range of signal strengths. The signal strengths are plotted on the x-axis as the Log2 of the enhancer signal (Pol II + H3K27ac + H3K4me1 + 0.6). The y-axis (p) is the fraction of sites that have a Log2 signal within a range divided by the size of the range and plotted at the midpoint of the range. The enhancer signal was measured from OSKM day 5 cells, fibroblasts or the ratio of the enhancer signal from OSKM day 5 divided by the signal from fibroblasts at the sites that the transcription factor bound in Day 5 OSKM cells (colored curve) or all sites genome wide (grey curve). (B) Each panel shows distribution plots similar to those described in the first row of (A) except the top row in (B) shows the distribution of the enhancer signal at the sites of OCT4 binding in day 5 OSKM cells that also have a Log2 enhancers signal less then 2.7 in fibroblast cells (green curve) or all sites genome wide that have a Log2 enhancers signal less then 2.7 in fibroblast cells (gray curve). The bottom row is similar to the top except the sites have a Log2 enhancer signal greater then 2.7 in fibroblast cells. (C) Each panel shows a distribution plots of the enhancer signal at the sites where SOX2 and SOX2-VP16 bind at the same location at day 5 after infecting mixes that contain either SOX2 or SOX2-VP16.
We infer that about half of the putative enhancers associated with bound OCT4 (detected in any of the four mixes at day 5) were created de novo. Thus at only about half of those OCT4 locations did we find a pre-existing putative enhancer in the parental cells (Figure 5B, Supplementary Figure S5A). This result held for all four reprogramming mixes (data for the OSvKM, OSK and OSvK mixes are not shown). Where weak putative enhancers were found at the corresponding positions in the parental cells, almost invariably the enhancer signal strengths were increased by binding of OCT4. But even in the few cases where these enhancer signals decreased, the effect was small (Figure 5B). Only some 5% of the OCT4 bound sites, in day 5 cells, were associated with a high level of the promoter signal H3K4me3 (Supplementary Figure S3B). The distribution of the repressive marks H3K27me3 or H3K9me3 at regions bound by OCT4 is not different from that found genome wide (Supplementary Figures S3C and D (upper panels)). Cells infected with the other three reprogramming mixes showed a similar effect of OCT4 binding for Day 5 OSvKM, Day 5 OSK and Day 5 OSvK mixes (data not shown).
In iPS cells, as in Day 5 cells, most (>80%) of the sites of bound OCT4 or SOX2 were within putative enhancers. In only about 25% of those cases did we find an enhancer at the corresponding location in parental fibroblasts (Supplementary Figure S6A).
The effects of OCT4 binding were mimicked by binding of SOX2 and/or SOX2-VP16, and were observed with all four reprogramming mixes (OSKM, OSvKM, OSK and OSvK) (Supplementary Figure S5A) (data for other reprogramming mixes are not shown). An interesting difference between putative enhancers created by SOX2 and SOX2-VP16 was revealed by the following analysis. We compared the location and enhancer signal strengths of sites bound by SOX2 and SOX2-VP16 in four pairs of day 5 cells as shown in Figure 5C. The locations bound by SOX2 and SOX2-VP16 were similar but not identical (Figure 4A), a matter we return to in the Discussion. Where SOX2 and SOX2-VP16 bound to the same sites, the distribution of putative enhancer signals elicited by SOX2-VP16 was shifted higher in comparison to that elicited by WT SOX2 (Figure 5C).
KLF4 and cMYC
Unlike OCT4 and SOX2, only 50% of KLF4 bound sites were embedded in putative enhancers at day 5 and virtually all of these putative enhancers pre-existed in the parental cells. KLF4 binding elicited no change in signals, whether that binding occurred inside or outside of an putative enhancer (Figure 5A). This result held for all four reprogramming mixes (data for the OSvKM, OSK and OSvK mixes are not shown). About half of the locations where cMYC bound correspond to a promoter (very high H3K4me3 signal), and the other half corresponds equally to putative enhancers and unmarked regions (Supplementary Figure S5B). cMYC binding, like that of KLF4 did not change any of the signals (histone modifications or Pol II) at day 5 compared to parental fibroblasts (Figure 5A). This result also held for the OSvKM reprogramming mix (data not shown).
Putative enhancers created and destroyed genome - wide
We find approximately 110,000 2 kb genomic fragments (without regard to binding of OCT4, SOX2, SOX2-VP16, KLF4 or cMYC) fit the definition of a putative enhancer in each of the cell types: fibroblasts, cells infected with each of the four reprogramming mixes at day 5, and iPS cells (Figure 6A).
Figure 6. Putative enhancers created and destroyed.

(A) All enhancers whether or not they bear OCT4 or SOX2. Top row, first panel: 111,385 2 Kb fragments were identified as enhancers in fibroblasts the relative strengths of which are indicated by the first purple ordered heat map on the left. Enhancer strength is defined as the sum of signals of Pol II, H3K4me1 and H3K27ac. The remaining heat maps show the enhancer signal strengths at the corresponding positions in other scenarios, e.g. fibroblasts expressing OSKM at day 5. Each subsequent panel in this row identifies enhancers in various cell types, e.g. fibroblasts expressing OSvKM, and displays the enhancer signal strengths at the corresponding positions in other cells (e.g. fibroblasts expressing OSKM at day 5). Second row, second panel: 15,804 newly created enhancers were identified in OSKM-expressing fibroblasts (day 5) by signals strengths as above, and by the finding that these enhancers were not present in parental fibroblasts as shown in this panel. The remaining enhancers were identified in other cells (e.g. OSvKM expressing fibroblasts at day 5) and the positions of these enhancers were examined in other cells. Third row, second panel: The positions of some 14,768 enhancers present in fibroblasts expressing OSKM, with a distribution of signal strengths as indicated, and these positions were interrogated for enhancer signals in other cells (e.g. OSvKM expressing fibroblasts).
(B) OCT4 and SOX2 binding at created and destroyed enhancers. Top row, first panel: The 15,804 enhancers detected at day 5 in OSKM-expressing fibroblasts were further interrogated for the presence of OCT4 (red bar heat map) and for SOX2 or SOX2-VP16 (blue bar heat map). This analysis was repeated for the other cells (e.g. OSvKM expressing fibroblasts). Second row, first panel: Enhancers present in fibroblasts, but absent from day 5 cells expressing OSKM. The remaining panels compare enhancers found in fibroblasts to those found in other cells (e.g. OSvKM expressing fibroblasts). NA = not applicable.
In day 5 cells (for any of the four mixes) tens of thousands of new putative enhancers (compared with the parental cells) have appeared, and a similar number have vanished (Figure 6A). Among the newly created putative enhancers only a small fraction remain in iPS cells (Figure 6A). The vast majority of putative enhancers destroyed at day 5 are also absent from iPS cells (Figure 6A). Of the newly created putative enhancers at day 5, some 10–25% bore OCT4 and/or SOX2, and of the destroyed enhancers only a small percentage (ca. < 2%) bore OCT4 or SOX2 (Figure 6B).
DISCUSSION
OCT4 and SOX2 work as activators during reprogramming
Two different kinds of experiments indicate that OCT4 and SOX2 work as activators, not as repressors, during reprograming of human fibroblasts to iPS cells. First, as shown here, replacing SOX2 with SOX2-VP16 in the canonical OSKM mix increased both the rate and efficiency of reprogramming human fibroblasts, an effect particularly evident when cells from older donors were used. We had previously shown that OCT4-VP16 in place of OCT4 had no deleterious effect on reprogramming. In contrast, for each of the 4 factors (OSKM), expression of its fusion derivative bearing an HP1 (repressing) domain eliminated reprogramming (present paper and Hammachi et al., 2012). Second, at an early stage (day 5) of reprogramming, regardless of the mix used for reprogramming, Chip-Seq experiments found that most (>80%) of the DNA-bound OCT4, SOX2 or SOX2-VP16 were embedded in putative enhancers, about one half of which were not present in the parental fibroblasts. That is, for the majority of cases, wherever any of these proteins bound outside of an existing enhancer, a new putative enhancer was created. Wherever SOX2 and SOX2-VP16 bound to identical sites within or outside of pre-existing enhancers, (in separate experiments), the putative enhancer signals found at sites bound by SOX2-VP16 were generally greater than those found at SOX2 bound sites. We saw almost no cases where binding of OCT4 and/or SOX2 or SOX2-VP16 eliminated a pre-existing enhancer, nor did we see any change in ‘repressive’ marks – H3K27me3 and H3K9me3 – at the regions where these proteins bound. One aspect of our results differs from those of Chronis et al. (Chronis et al., 2017). Those authors suggested that OCT4 binds to pre-existing fibroblast enhancers, and in some cases at least, immediately initiates their inactivation. We find, in contrast, that in virtually all cases where OCT4 (or SOX2) binds to a pre-existing putative enhancer, that enhancer remains. We do not understand why our results differ.
Other results are consistent with the idea that SOX2-VP16 works as a strong activator in reprogramming. Thus, inclusion of SOX2-VP16 in a reprogramming mix evoked stronger and earlier expression of key genes associated with the acquisition of pluripotency. For example, NANOG, which is otherwise expressed late during reprogramming (e.g. by day 9), was expressed early, and to a higher level (by day 5). Other genes, including TRIM71, an RNA binding protein known to be involved in reprogramming, were also expressed early and to a higher level with SOX2-VP16 containing mixes. Worringer et al showed that ectopic expression of TRIM71 can replace the requirement for cMYC in reprogramming (Worringer et al., 2014). This may explain why requirement for cMYC is partially abrogated when reprogramming with SOX2-VP16. We do not know whether these effects are direct or indirect.
The effect of SOX2-VP16 on reprogramming, compared with that of SOX2, might be solely attributable to stronger transcriptional activation of an identical set of genes in one case versus the other. But there are other possibilities as well, one of which is suggested by the observation that, at day 5, SOX2-VP16 and SOX2 occupied a similar but not identical set of positions on DNA. These differences in binding locations between SOX2 and SOX2-VP16 at day 5 are presumably accounted for by different sets of partner DNA-binding proteins present in cells expressing OSKM vs. OSvKM. Even were SOX2 and SOX2-VP16 to initially bind to identical sites, thanks to different strengths of activation of genes encoding regulatory proteins, the protein milieu could be significantly different at day 5 in the two cases. Another possibility is that SOX2 attached to VP16 prefers to interact with a set of proteins that is not identical to those chosen by its WT counterpart.
Transcription factor repositioning
The finding that, at day 5, OCT4 and SOX2 are almost invariably embedded in one or another putative enhancer applies as well to endogenously encoded OCT4 and SOX2 in iPS cells. This observation holds despite the fact that the majority of sites bound by the endogenously expressed OCT4 and SOX2 (in iPS cells) are different from those bound in day 5 cells. We imagine that this “repositioning” of OCT4 and SOX2 (day 5 vs. iPS) is accounted for by changes in DNA binding partners, and the following observations are consistent with this idea.
We found a large number of locations where OCT4 and SOX2 were bound in the same 2 Kb fragment at day 5 of reprogramming with OSKM, but also many sites at which one or the other was bound singly – OCT4 with no nearby SOX2, and vice versa. For these ‘singly’ bound locations, only a minority was also occupied in iPS cells, a finding also reported by others (Sridharan et al., 2009). We imagine that the changed locations on DNA of these ‘singly’ bound OCT4 and SOX2 are attributable to changes in binding partners. Others have reported (and we have observed – not shown) that many transcription factors expressed in fibroblasts are turned off and other new transcription factors are produced in iPS cells (Yu et al., 2007). In contrast to singly bound sites, most of the locations that were co-bound by OCT4 and SOX2 in day 5 cells, were found co-bound by endogenously encoded OCT4/SOX2 in iPS cells. The well-characterized ability of OCT4/SOX2 to bind DNA cooperatively likely explains why a site bound by an OCT4/SOX2 pair at day 5 tends to be bound by that pair in iPS cells.
Enhancer creation/destruction
We detected about the same total number of putative enhancers (ca. 110,000, defined as 2 Kb fragments bearing strong putative enhancer signals) in the three cell types: parental fibroblasts, day 5 cells, and iPS cells. This constancy in number holds despite widespread putative enhancer creation/destruction. By day 5 of reprogramming, using any of the four mixes, thousands of putative enhancers have been created at locations devoid of signals in parental fibroblasts. Most of these new putative enhancers are not present in iPS cells, and, in turn, many new ones appear in iPS cells at locations different from those found in day 5 cells. Only 10–20% of the newly created putative enhancers in day 5 include bound OCT4 or SOX2, so the changes in the positions of most putative enhancers (day 5 to iPS) cannot be explained by the repositioning of OCT4 or SOX2. Rather, these changes are likely due to changes in the transcription factors other than OCT4 or SOX2. As measured by enhancer/transcription factor locations, day 5 cells apparently represent an ‘intermediate’ along the pathway from fibroblasts to iPS cells. Put another way, consistent with suggestions of others (Golipour et al., 2012; Zunder et al., 2015), reprogramming is not simply the turning off of parental fibroblast enhancers and the creation of new ones in iPS cells, but rather involves a transient state of gene expression in day 5 cells.
In addition to transient changes, certain changes occurring by day 5 are permanent. For example most of putative enhancers present in parental fibroblasts that had been extinguished by day 5 remained extinguished in iPS cells. The DNA segments of these extinguished putative enhancers bore little or no OCT4 or SOX2. We suggest that it is unlikely that phenomena detected by ChIP-Seq (e.g. enhancer formation) when detected at day 5 is only observed in a sub-population of cells but rather, occurs in a majority of the cells. This is because the measured strengths of putative enhancers at Day 5 were quite similar to those in iPS cells or fibroblasts. This would not be the case were the Day 5 enhancers present in only a subpopulation of cells.
EXPERIMENTAL PROCEDURES
Lentiviral constructs
Constructs listed in Table S1 have been deposited in Addgene (IDs 100103 to 100125). OCT4 fusions were derived from the pLM-vexGFP-OCT4 lentiviral construct (Addgene ID 22240), SOX2 fusions were derived from the pLM-mCitrine-SOX2 lentiviral construct (Addgene ID 23242), KLF4 fusions were derived from the pLM-mCherry-KLF4 lentiviral construct (Addgene ID 23243), and cMYC fusions were derived from the pLM-mCerulean-cMYC lentiviral construct (Addgene ID 23244).
Cell culture and human iPSC derivation
Human iPS derivation from donor fibroblasts was reviewed and authorized by the MSKCC TRI-SCI ESCRO committee (Protocol # 2010-05-001). Information on donor fibroblasts used in this study with respect to their age, gender, race and source is provided in Supplementary Table S2. Human iPSC reprogramming was performed as previously described (Hammachi et al., 2012) with minor modifications and is detailed in the supplementary materials and methods section. Other methods including ChIP-Seq and data analysis are also presented in detail in the Supplementary materials and methods section.
Statistical analysis
Reprogramming efficiency data plotted in Figure 1 are expressed as mean +/− SD with n = at least 6 (fetal fibroblasts) and n = 2 for older donor fibroblasts. Nuclear staining of age-associated markers (Supplementary Figure S1) were analyzed in Excel by comparing mean nuclear staining in each cell type with that of fetal fibroblasts (n = at least 100 cells for each cell type per marker) by two sample t-tests. p values of < 0.01 were considered statistically significant. Microarray data in were analyzed using Partek’s Genomics Suite, a p-value <0.01 and a log2 two fold change in differentially expressed genes (Supplementary Figure S2) was considered statistically significant. Additional details on data analysis for all experiments are provided in the Supplementary materials and methods section.
Supplementary Material
Highlights.
SOX2-VP16 improves reprogramming of human fibroblasts to iPSCs.
OCT4, SOX2, and SOX2-VP16 create, or perpetuate, enhancers where they bind.
SOX2-VP16-created enhancers are stronger than SOX2-created enhancers.
Many transcription factors change locations during reprogramming.
Acknowledgments
We thank Lorenz Studer (MSKCC, NY) and members of the Ptashne lab for discussions; Michel Sadelain (MSKCC, NY) and Eirini Papapetrou (MSSM, NY) for wild type O, S, K, M lentiviral constructs and initial involvement in this project; the WiCell Research Institute for the H1 (WA01) and H9 (WA09) human ES cells; Mark Tomishima (funded by NYSTEM C029153) at the SKI Stem Cell Core as well as other members of core facilities at MSKCC: Molecular cytology, Integrated genomics, Molecular cytogenetics and Flow cytometry. This work was funded by NYSTEM (N09G-217 and N11G-262) to MP and the MSK Cancer Center Support Grant/Core Grant (P30 CA008748).
Footnotes
AUTHOR CONTRIBUTIONS
S.N and M.P conceived the project; S.N designed and constructed lentiviral vectors, performed and analyzed reprogramming experiments, FACS analysis, QPCR and microarray experiments; G. Bryant performed and analyzed ChIP-endo-Seq experiments; S.N and S.S made and titered lentiviruses, S.S performed reprogramming and QPCR experiments and harvested cells for ChIP-endo-Seq experiments. G. Berrozpe helped write and improve the manuscript. S.N, G. Bryant, G. Berrozpe and M.P wrote the manuscript.
ACCESSION NUMBER
The data discussed in this manuscript have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE81900 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81900)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adewumi O, Aflatoonian B, Ahrlund-Richter L, Amit M, Andrews PW, Beighton G, Bello PA, Benvenisty N, Berry LS, Bevan S, et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat Biotechnol. 2007;25:803–816. doi: 10.1038/nbt1318. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Benoist C, Chambon P. In vivo sequence requirements of the SV40 early promotor region. Nature. 1981;290:304–310. doi: 10.1038/290304a0. [DOI] [PubMed] [Google Scholar]
- Berrozpe G, Agosti V, Tucker C, Blanpain C, Manova K, Besmer P. A distant upstream locus control region is critical for expression of the Kit receptor gene in mast cells. Mol Cell Biol. 2006;26:5850–5860. doi: 10.1128/MCB.01854-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrozpe G, Bryant GO, Warpinski K, Ptashne M. Regulation of a mammalian gene bearing a CpG island promoter and a distal enhancer. Cell Rep. 2013;4:445–453. doi: 10.1016/j.celrep.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, Donaghey J, Smith ZD, Ratanasirintrawoot S, Zhang X, et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell. 2015;162:412–424. doi: 10.1016/j.cell.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Tomlinson SR. The transcriptional foundation of pluripotency. Development. 2009;136:2311–2322. doi: 10.1242/dev.024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chew JL, Loh YH, Zhang W, Chen X, Tam WL, Yeap LS, Li P, Ang YS, Lim B, Robson P, et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol Cell Biol. 2005;25:6031–6046. doi: 10.1128/MCB.25.14.6031-6046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K. Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell. 2017;168:442–459. e420. doi: 10.1016/j.cell.2016.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do JT, Scholer HR. Regulatory circuits underlying pluripotency and reprogramming. Trends Pharmacol Sci. 2009;30:296–302. doi: 10.1016/j.tips.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert SF, Barressi MJF. Developmental Biology. 11. Sinauer Associates; 2016. [Google Scholar]
- Golipour A, David L, Liu Y, Jayakumaran G, Hirsch CL, Trcka D, Wrana JL. A late transition in somatic cell reprogramming requires regulators distinct from the pluripotency network. Cell Stem Cell. 2012;11:769–782. doi: 10.1016/j.stem.2012.11.008. [DOI] [PubMed] [Google Scholar]
- Hammachi F, Morrison GM, Sharov AA, Livigni A, Narayan S, Papapetrou EP, O’Malley J, Kaji K, Ko MS, Ptashne M, et al. Transcriptional activation by Oct4 is sufficient for the maintenance and induction of pluripotency. Cell Rep. 2012;1:99–109. doi: 10.1016/j.celrep.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hirai H, Tani T, Katoku-Kikyo N, Kellner S, Karian P, Firpo M, Kikyo N. Radical acceleration of nuclear reprogramming by chromatin remodeling with the transactivation domain of MyoD. Stem Cells. 2011;29:1349–1361. doi: 10.1002/stem.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue F, Kircher M, Martin B, Cooper GM, Witten DM, McManus MT, Ahituv N, Shendure J. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 2017;27:38–52. doi: 10.1101/gr.212092.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582. doi: 10.1016/j.cell.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Maherali N, Ahfeldt T, Rigamonti A, Utikal J, Cowan C, Hochedlinger K. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3:340–345. doi: 10.1016/j.stem.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martello G, Smith A. The nature of embryonic stem cells. Annu Rev Cell Dev Biol. 2014;30:647–675. doi: 10.1146/annurev-cellbio-100913-013116. [DOI] [PubMed] [Google Scholar]
- McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HP, Schaffner W. Transcriptional enhancers can act in trans. Trends Genet. 1990;6:300–304. doi: 10.1016/0168-9525(90)90236-y. [DOI] [PubMed] [Google Scholar]
- Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106. doi: 10.1038/nbt1374. [DOI] [PubMed] [Google Scholar]
- Papapetrou EP, Tomishima MJ, Chambers SM, Mica Y, Reed E, Menon J, Tabar V, Mo Q, Studer L, Sadelain M. Stoichiometric and temporal requirements of Oct4, Sox2, Klf4, and c-Myc expression for efficient human iPSC induction and differentiation. Proc Natl Acad Sci U S A. 2009;106:12759–12764. doi: 10.1073/pnas.0904825106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146. doi: 10.1038/nature06534. [DOI] [PubMed] [Google Scholar]
- Paull D, Sevilla A, Zhou H, Hahn AK, Kim H, Napolitano C, Tsankov A, Shang L, Krumholz K, Jagadeesan P, et al. Automated, high-throughput derivation, characterization and differentiation of induced pluripotent stem cells. Nat Methods. 2015;12:885–892. doi: 10.1038/nmeth.3507. [DOI] [PubMed] [Google Scholar]
- Perry MW, Boettiger AN, Levine M. Multiple enhancers ensure precision of gap gene-expression patterns in the Drosophila embryo. Proc Natl Acad Sci U S A. 2011;108:13570–13575. doi: 10.1073/pnas.1109873108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M, Gann A. Genes and Signals. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2002. [Google Scholar]
- Rodda DJ, Chew JL, Lim LH, Loh YH, Wang B, Ng HH, Robson P. Transcriptional regulation of nanog by OCT4 and SOX2. J Biol Chem. 2005;280:24731–24737. doi: 10.1074/jbc.M502573200. [DOI] [PubMed] [Google Scholar]
- Rohani L, Johnson AA, Arnold A, Stolzing A. The aging signature: a hallmark of induced pluripotent stem cells? Aging Cell. 2014;13:2–7. doi: 10.1111/acel.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- Scholer HR, Ruppert S, Suzuki N, Chowdhury K, Gruss P. New type of POU domain in germ line-specific protein Oct-4. Nature. 1990;344:435–439. doi: 10.1038/344435a0. [DOI] [PubMed] [Google Scholar]
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet. 2014;15:272–286. doi: 10.1038/nrg3682. [DOI] [PubMed] [Google Scholar]
- Sridharan R, Tchieu J, Mason MJ, Yachechko R, Kuoy E, Horvath S, Zhou Q, Plath K. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, Vernot B, Cheng JB, Thurman RE, Sandstrom R, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154:888–903. doi: 10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L, Vera E, Cornacchia D. Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell Stem Cell. 2015;16:591–600. doi: 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Theunissen TW, Jaenisch R. Molecular control of induced pluripotency. Cell Stem Cell. 2014;14:720–734. doi: 10.1016/j.stem.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triezenberg SJ, LaMarco KL, McKnight SL. Evidence of DNA: protein interactions that mediate HSV-1 immediate early gene activation by VP16. Genes Dev. 1988;2:730–742. doi: 10.1101/gad.2.6.730. [DOI] [PubMed] [Google Scholar]
- Wang Y, Chen J, Hu JL, Wei XX, Qin D, Gao J, Zhang L, Jiang J, Li JS, Liu J, et al. Reprogramming of mouse and human somatic cells by high-performance engineered factors. EMBO Rep. 2011;12:373–378. doi: 10.1038/embor.2011.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP, Jaenisch R. c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell. 2008;2:10–12. doi: 10.1016/j.stem.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, Yamanaka S. The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell. 2014;14:40–52. doi: 10.1016/j.stem.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Yuan H, Corbi N, Basilico C, Dailey L. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 1995;9:2635–2645. doi: 10.1101/gad.9.21.2635. [DOI] [PubMed] [Google Scholar]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- Zunder ER, Lujan E, Goltsev Y, Wernig M, Nolan GP. A continuous molecular roadmap to iPSC reprogramming through progression analysis of single-cell mass cytometry. Cell Stem Cell. 2015;16:323–337. doi: 10.1016/j.stem.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.