

Abstract
Mammalian cells respond to double-stranded RNA (dsRNA) by activating a translation-inhibiting endoribonuclease, RNase L. Consensus in the field indicates that RNase L arrests protein synthesis by degrading ribosomal RNAs (rRNAs) and messenger RNAs (mRNAs). However, here we provide evidence for a different and far more efficient mechanism. By sequencing abundant RNA fragments generated by RNase L in human cells, we identify site-specific cleavage of two groups of noncoding RNAs: Y-RNAs, whose function is poorly understood, and cytosolic tRNAs, which are essential for translation. Quantitative analysis of human RNA cleavage versus nascent protein synthesis in lung carcinoma cells shows that RNase L stops global translation when tRNAs, as well as rRNAs and mRNAs, are still intact. Therefore, RNase L does not have to degrade the translation machinery to stop protein synthesis. Our data point to a rapid mechanism that transforms a subtle RNA cleavage into a cell-wide translation arrest.
Keywords: RNase L, signaling, translation, Y-RNA, tRNA
INTRODUCTION
Protein synthesis is the most energetically expensive cellular process that consumes more than half of a cell's energy (Lane and Martin 2010). This high cost dictates that cells vigorously control translation when they adapt to evolving and often stressful conditions. A particularly dynamic change in protein synthesis takes place in mammalian cells exposed to dsRNA. DsRNA is a potent activator of the innate immune system that triggers type-I interferons (IFNs) and causes biosynthesis of a small molecule intracellular inhibitor of translation, 2-5A (Hovanessian and Kerr 1978).
The 2-5A binds with low-nanomolar affinity to the ankyrin-repeat domain of an 83.5 kDa receptor enzyme, RNase L (Dong and Silverman 1995; Han et al. 2012). The binding of 2-5A drives RNase L self-assembly into catalytically active homodimers and high-order oligomers that cleave single-stranded RNA (ssRNA), but not dsRNA molecules inside cells, and exhibit sequence specificity for UN^N sites (^ marks the cleavage site; N = A, G, U or C [Han et al. 2012, 2014; Huang et al. 2014]). As an arm in the IFN response, RNase L mediates innate immune defense against viruses (Ireland et al. 2009; Goldstein et al. 2016) and bacteria (Li et al. 2008). The functions of RNase L span beyond infection control. It has been shown that RNase L regulates terminal adipocyte differentiation (Fabre et al. 2012), inhibits cell migration and metastasis in a mouse model (Banerjee et al. 2015), and controls proliferation and adhesion in human and murine cells (Rath et al. 2015).
Upon activation by 2-5A, RNase L strongly inhibits global protein synthesis (Hovanessian and Kerr 1978). This inhibitory effect has been attributed to degradation of important cellular RNAs, particularly 28S rRNA (Iordanov et al. 2000) and mRNAs (Toots et al. 1988; Brennan-Laun et al. 2014). However, here we find a different mechanism that RNase L engages before substantial RNA decay takes place. We encountered this phenomenon during an unbiased search for small RNA fragments with 2′,3′-cyclic phosphate, which were predicted to result from RNase L activity (Malathi et al. 2007; Banerjee et al. 2014; Chakrabarti et al. 2015). This transcriptome-wide search revealed that RNase L cleaves highly conserved and essential components of the translation apparatus: tRNAs. This observation raises questions about the precise molecular cause of translation arrest by RNase L and leads us to investigate the effects of this innate immune endoribonuclease on cellular RNAs and protein synthesis.
RESULTS
DsRNA triggers cleavage of small noncoding RNAs at UN^N consensus sites
The dsRNA-activated endoribonuclease RNase L cleaves cellular RNAs via a mechanism that leaves 2′,3′-cyclic phosphate termini. This RNA modification distinguishes RNase L from many enzymes involved in conventional mRNA decay and microRNA processing and allows capture and identification of the RNA products released by RNase L using RNA sequencing (RNA-seq). This technique has been developed recently and used to analyze cleavage of viral and ribosomal RNAs by RNase L (Cooper et al. 2014). Here we developed an alternative approach for 2′,3′-cyclic phosphate RNA-seq analysis based on RtcB ligase (Tanaka et al. 2011), and used it to identify small RNA products of RNase L (≤200 nt; Supplemental Fig. S1A–C).
We began by profiling RNA cleavage in HeLa cells treated with poly(IC), a synthetic dsRNA that triggers the IFN response and 2-5A synthesis (Rath et al. 2015). The poly(IC) treatment induced abundant RNA fragments with UN^N cleavage consensus sites that were absent in naïve cells (Fig. 1A; Supplemental Fig. S1D; Supplemental Dataset S1). The UN^N consensus matches precisely the preference of RNase L (Han et al. 2014). The most highly up-regulated reads map to tRNAs and Y-RNAs (Fig. 1B–D). The dsRNA-activated enzyme attacks preferentially three cytosolic tRNAs: tRNA-His, tRNA-Pro, and tRNA-Gln (Fig. 1E). In contrast, mitochondrial tRNAs are resistant to the effects of dsRNA (Supplemental Fig. S1E).
FIGURE 1.

RtcB RNA-seq analysis of RNA cleavage in poly(IC)-stimulated HeLa cells. (A) Sequence consensus at cleavage sites with 2′,3′-cyclic phosphates before and after poly(IC) treatment. Nontargets (NT) and targets (T) are defined in B. (B) Cleavage site distribution according to fold-induction by poly(IC). Boxed regions mark RNase L-sensitive (T) and resistant (NT) RNAs. (C) Composition of the NT and T groups by main RNA types. The group “other” contains primarily U6 small nucleolar RNA as well as mRNAs, micro-RNAs, and small ncRNAs (Supplemental Dataset S1). (D) Up-regulation of reads for each Y-RNA by poly(IC). Y-RNAs are cleaved at UN^N consensus sites. (E) ASL cleavage sites in observed fragments of cytosolic tRNAs. The stacked bars show basal (gray) and poly(IC)-induced (red) reads for each site.
Whereas tRNAs mediate translation, the precise cellular functions of Y-RNAs are still being investigated. Y-RNAs belong to a class of small noncoding RNAs identified as components of major autoantigens in systemic lupus erythematosus (SLE) and Sjögren's syndrome (Kowalski and Krude 2015), where they are bound to two autoantigen proteins Ro60 and La (van Gelder et al. 1994; Kowalski and Krude 2015). The cleavage of all Y-RNAs in our experiments occurs predominantly in the upper region between nucleotides 24 and 32 at UN^N sites (Fig. 2A,B). Intriguingly, Y-RNAs are cleaved in this region in the serum of cancer patients and during apoptosis (Rutjes et al. 1999; Dhahbi et al. 2014; Kowalski and Krude 2015; Cambier et al. 2017), either due to the action of RNase L or due to sensitivity of these nucleotides to stress endoribonucleases in general. RNY4 stands out in our data set due to its unusual non-UN^N cleavage site CA^G (Fig. 2A). This observation raises the question of whether RNY4 is cleaved by RNase L or by an unknown dsRNA-activated endoribonuclease, which we address experimentally next.
FIGURE 2.

Single-nucleotide resolution profiles of tRNA and Y-RNA cleavage during poly(IC) response. (A) Secondary structure cleavage profiles for Y-RNAs. RNase L targets the upper region at UN^N sites. RNY4 is cleaved predominantly at a single unconventional site CA^G. Stacked bar charts for RNA fragments are colored as in Figure 1E. (B) Graphic model of Y-RNA cleavage by RNase L. The main cleavage sites in all Y-RNAs are located in the region between nucleotides 24 and 32, away from the conserved binding sites for Ro60 and La autoantigens. (C) Secondary structure cleavage profiles for RNase L-sensitive tRNAs. (D) Graphic model for tRNA cleavage by RNase L versus ANG and bacterial endoribonucleases.
The cleavages of tRNA-His, tRNA-Gln, and tRNA-Pro occur at specific sites in the anticodon stem–loop (ASL; Fig. 2C). The action of RNase L therefore gives rise to cleaved tRNAs, a class of signaling tRNA-derived molecules found in a rapidly growing number of processes from regulation of cancer proliferation to epigenetic suppression of retroelements (Ivanov et al. 2011; Dhahbi et al. 2014; Honda et al. 2015; Sharma et al. 2016). Until now, biogenesis of mammalian cleaved tRNAs has been attributed to the action of angiogenin (ANG) (Thompson and Parker 2009; Ivanov et al. 2014; Honda et al. 2015). We show that RNase L is a new and potent source of cleaved tRNAs, activated specifically during dsRNA response. By cleaving tRNAs and by inhibiting cell proliferation, mammalian RNase L resembles endoribonucleases from bacterial toxin–antitoxin systems. Bacteria are known to use tRNA cleavage enzymes colicin E, colicin D, PrrC, zymocin, and PaT to inhibit translation in competing bacteria and to regulate bacteriostasis (Thompson and Parker 2009; Ogawa 2016). The intriguing resemblance between the bacterial enzymes and mammalian RNase L is limited, however. They are phylogenetically unrelated and recognize distinct ASL sites (Fig. 2D).
RNase L is directly responsible for the dsRNA-induced Y-RNA and tRNA cleavage
The UN^N cleavage consensus shown in Figure 1A suggests the action of RNase L. However, dsRNA controls hundreds of genes, and cleavage by multiple directly and indirectly activated endoribonucleases cannot be excluded. This uncertainty applies particularly to RNY4, which has a predominant non-UN^N cleavage site (Fig. 2A). To verify the role of RNase L in RNY4 cleavage, we used two approaches: RNase L overexpression and specific RNase L activation using 2-5A.
Overexpression is sufficient to activate RNase L without additional stimulation due to a spontaneous self-association (Han et al. 2012; Rath et al. 2015). The resulting activity is 20–30-fold weaker than with poly(IC), but sufficient to observe tRNA and Y-RNA cleavage by RtcB RNA-seq. Overexpression of wild-type (WT), but not the inactive RNase L mutant H672N (Rath et al. 2015), induced tRNA cleavage and Y-RNA cleavage, leading to detection of Y-RNA fragments (Y-RFs) with UN^N termini (Supplemental Fig. S2A,B; Supplemental Dataset S1). Enrichment analysis (Rath et al. 2015) shows that overexpression results in globally the same RNA fragments as poly(IC) treatment (Supplemental Fig. S2). RNY4 was cleaved specifically at the non-UN^N position—CA^G— confirming the role of RNase L.
The complementary test using 2-5A was conducted in T47D cells to allow rapid and specific RNase L activation, as described previously (Rath et al. 2015). Although the cell line was different, Y-RNA and tRNA cleavage occurred at the UN^N consensus sites within minutes from 2-5A addition (Supplemental Fig. S3A,B; Supplemental Dataset S1). The identified RNA fragments were globally the same as in HeLa cells treated with poly(IC) (Supplemental Fig. S3C). Cleavage of tRNAs occurred at the expected ASL sites for tRNA-His, tRNA-Pro, and tRNA-Gln (Supplemental Fig. S3D,E). RNY4 was cleaved at the non-UN^N site, CA^G (Supplemental Fig. S3F). Together, the overexpression and the 2-5A treatment experiments confirm the involvement of RNase L in Y-RNA and tRNA cleavage during dsRNA response.
Host proteins and post-transcriptional modifications shape RNase L specificity
The site-specific cleavage of tRNAs and Y-RNAs cannot be explained by recognition of the UN^N sequence alone and predicts involvement of additional factors. In the extreme case of RNY4, recognition of these additional factors overrides the UN^N preference of the active site. To find the molecular origins of RNase L specificity, we tested cleavage of model RNA stem–loops derived from tRNA-His and RNY4. Neither of these minimal substrates was cleaved specifically at the physiologic site (Fig. 3A). Strikingly, the missing specificity was not rescued even with full-length tRNA-His and RNY4 transcribed in vitro (Supplemental Fig. S4). Therefore, cellular components or activities are required for tRNA and Y-RNA recognition by RNase L.
FIGURE 3.

Cleavage of naked human RNA points to distinct specificity determinants for Y-RNAs and tRNAs. (A) Cleavage of model stem–loops from tRNA-His and RNY4 analyzed by polyacrylamide gel electrophoresis (PAGE). Neither model substrate is cleaved preferentially at the physiologic site. (B) RNA chip analysis for cleavage of protein-free total RNA by RNase L. The nonspecific decay observed in this experiment contrasts the site-specific cleavage in cells (Supplemental Fig. S1B). (C) RtcB qPCR analysis to measure cleavage of tRNA-His and RNY4 at physiologic and nonphysiologic sites in naked RNA. Error bars show SE from two qPCR replicates. (D) Cleavage of tRNA-His purified from WT or mutant E.coli lacking a queuosine biosynthesis gene tgt was measured by RtcB qPCR. The queuosine position is shaded gray. Error bars show SE from two biological replicates.
We probed a possible role of cellular proteins by cleaving protein-free (naked) tRNA-His and RNY4 isolated from human cells. To this end, we developed a sensitive method for detection of site-specific RNA cleavage by RNase L, which is suitable for analysis of total RNA extracted from cells: RtcB qPCR (Supplemental Fig. S5; Materials and Methods). We prepared naked RNA by phenol extraction, which effectively disrupts protein–RNA complexes as indicated by changes in the rRNA cleavage pattern (Fig. 3B vs. Supplemental Fig. S1B). RNase L correctly recognized the physiologic cleavage site in the naked cellular tRNA-His (Fig. 3C). However, cellular Y-RNAs were cleaved at nonphysiologic sites (Fig. 3C; Supplemental Fig. S5). These data suggest that RNase L gains site-specificity for Y-RNAs by recognizing cellular protein/Y-RNA complexes. In contrast, recognition of tRNAs, at least in the case of tRNA-His, does not require any helper proteins and likely depends on a post-transcriptional modification.
A bulky queuosine residue at position −2 relative to the RNase L cleavage site stands out in the ASL of tRNA-His. To test whether this queuosine is important for the cleave specificity, we took advantage of E. coli, which also have queuosine in tRNA-His. First, we confirmed that RNase L cleaves E.coli tRNA-His site-specifically at the correct site. Using RtcB qPCR, we demonstrated the same ∼1000-fold preference for site 36 relative to positions 33 and 34, as seen with human tRNA-His (Fig. 3D). This specificity was quantitatively erased in tRNA-His isolated from a genetically engineered strain of E.coli lacking a gene required for queuosine biosynthesis, tgt (Fig. 3D; Noguchi et al. 1982). Therefore, queuosine protects sites 33 and 34 from RNase L, which guides cleavage to site 36 and explains recognition of cellular, but not in vitro transcribed tRNA-His. A post-transcriptional modification thus shapes RNase L specificity for tRNA-His and suggests that RNA modifications are among the key factors that render only certain tRNAs (Fig. 1E) and only certain tRNA nucleotides sensitive to RNase L.
RNase L stops global translation by a signaling-like mechanism engaged ahead of coding and noncoding RNA decay
The cleavage of tRNAs raises an intriguing possibility that tRNA degradation rather than the previously reported cleavage of 28S rRNA promotes the translation arrest by 2-5A. To probe this scenario, we determined the depth of tRNA (and Y-RNA) degradation by RNase L using Northern blots. Activation of RNase L by poly(IC) for 5 h was sufficient for a nearly complete depletion of 28S rRNA (Fig. 4A). Northern blot analysis revealed a similarly strong degradation of all Y-RNAs: RNY1, RNY3, and RNY4 were lost almost completely (Fig. 4B). The extent of tRNA cleavage varied, depending on the tRNA. Full-length tRNA-Pro and tRNA-Glu were not lost at 1 h post poly(IC) treatment and decreased by approximately twofold after 10 h. Histidyl tRNA was down-regulated by 30% 1 h after poly(IC) and more than 10-fold by 5 h (Fig. 4C). These data show that in addition to the ribosomes, RNase L cleaves and depletes tRNA-His, and thus compromises more than one key part of the translation apparatus.
FIGURE 4.
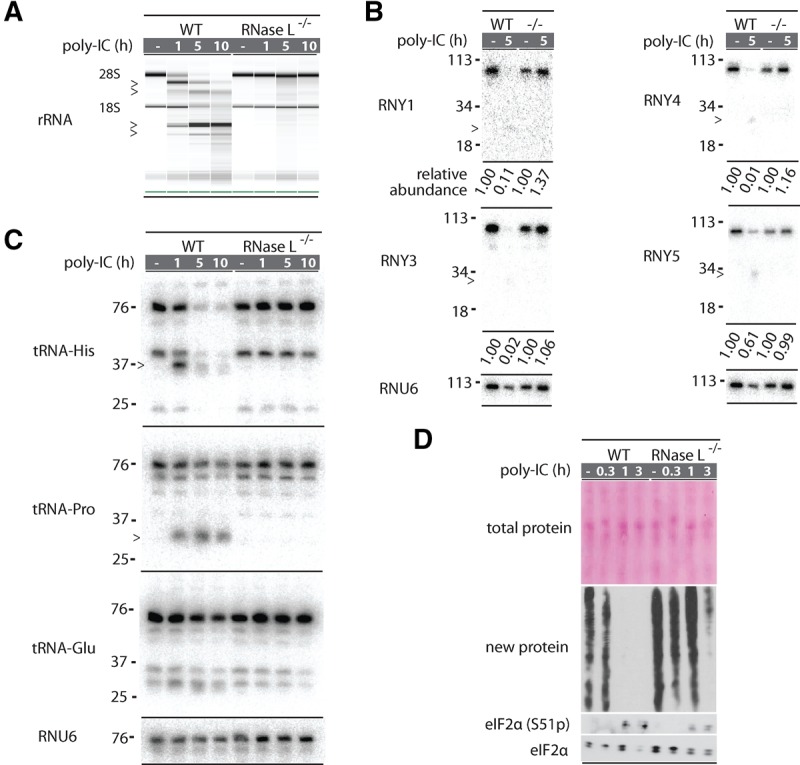
Northern blot analysis of Y-RNA and tRNA cleavage during dsRNA response. (A) RNA chip analysis for cleavage of total RNA in A549 cells treated with poly(IC). Markers show fragments produced by RNase L. RNA is intact in RNase L−/− cells. (B) Northern blot analysis for each Y-RNA after 5 h of poly(IC) treatment. RNA fragments are less abundant than the starting Y-RNAs, but detectable, indicating that they survive for some time in the cells. Relative abundance is normalized to RNU6. (C) Northern blot analysis of tRNA cleavage during poly(IC) treatment. Cleavage of tRNA-His and tRNA-Pro are readily detected but only tRNA-His is depleted upon prolonged treatment. (D) Western blot analysis to detect inhibition of new protein synthesis during poly(IC) treatment. Poly(IC) does not change the abundance of preexisting total protein, but blocks translation of new proteins. RNase L is important for early and strong translation arrest. Western blot for eIF2α phosphorylation status reflects PKR activation.
The exact contribution of RNase L to translation arrest during dsRNA response is obscured by a parallel dsRNA-sensing pathway mediated by the protein kinase PKR that phosphorylates translation initiation factor eIF2α (Smith et al. 2005). To isolate a specific role of RNase L, we compared nascent protein synthesis in WT and RNase L−/− cells using puromycin pulse-labeling (Aulas et al. 2017). Treatment with poly(IC) for as little as 1 h was sufficient to block all detectable translation in WT A549 cells (Fig. 4D). In contrast, translation in RNase L−/− cells was not affected at 1 h and inhibited only partially after 3 h of treatment. Western blotting showed the appearance of phospho-eIF2α in WT and RNase L−/− cells, indicating PKR activation after ∼1 h in both genetic backgrounds. However, PKR alone was insufficient for a rapid translation arrest, revealing a critical role of RNase L in shutting off protein synthesis.
To resolve the mechanism of translation arrest by RNase L in more detail, we compared the temporal dynamics and strengths of RNA cleavage versus translation in cells treated with the specific activator of RNase L, 2-5A. As expected, activity of PKR or other eIF2α kinases was not observed in these experiments, allowing us to focus on translation control by RNase L (Fig. 5A). RNA chip analysis showed that 28S rRNA was predominantly intact after 1 h of 2-5A treatment and that ∼50% still remained intact after 2.5 h (Fig. 5B). However, inhibition of protein synthesis was disproportionately strong. We observed an ∼10-fold decrease in global translation at 1 h and shutdown of translation at 2.5 h (Fig. 5C).
FIGURE 5.

RNA degradation versus translation control by RNase L. (A) Western blot for eIF2α phosphorylation shows absence of strong PKR activation during 2-5A treatment. (B) RNA chip analysis of cellular RNA cleavage during 2-5A treatment. (C) Western blot analysis to detect protein synthesis inhibition by 2-5A. Note that 2-5A does not change the abundance of preexisting proteins, but arrests new protein synthesis. Translation arrest requires RNase L. (D) Northern blot analysis for tRNA cleavage. The expected tRNA halves are observed for tRNA-His and tRNA-Pro, which are sensitive to RNase L. (E) Cellular levels of abundant mRNAs (ACTB1, GAPDH) and an RNase L-sensitive mRNA (CTNND1) (Rath et al. 2015) during 2-5A treatment analyzed by qPCR. Graph shows cycles to threshold (Ct) and SD for two qPCR replicates. (F) RNA-seq quantification of top most abundant mRNAs before and after 2-5A treatment. (G) Transfection of WT, but not the RNase-inactive H672N RNase L (3 h) induces 2-5A-dependent rRNA cleavage and translation block. (H) Proposed model for RNA and translation regulation by RNase L.
The loss of translation by largely intact ribosomes suggests that the inhibition could arise from a loss of tRNA-His or mRNAs. Northern blot analysis of tRNA-His showed a rapid accumulation of cleaved fragments, which reach maximum after approximately 30 min of 2-5A transfection. Nevertheless, the levels of full-length tRNA-His did not decrease, suggesting that tRNA depletion is not the cause of the translation arrest (Fig. 5D; Y-RNAs are intact as well, Supplemental Fig. S6A). The levels of abundant mRNAs that give rise to the influx of new housekeeping proteins detected by puromycin-tagged Western blotting also remained unchanged, as determined by qPCR (Fig. 5E) and poly(A)+ RNA-seq (Fig. 5F). Our data eliminate loss of rRNAs, tRNAs, or mRNAs as the cause of translation arrest by RNase L and suggest that RNase L may activate a signaling mechanism.
In the simplest scenario, signaling could be mediated by the abundant tRNA/Y-RNA cleavage products detected by RtcB RNA-seq (Fig. 1B,C; Supplemental Dataset S1), or by less abundant fragments derived from rRNAs, mRNAs, or other RNAs. Inhibition of translation by fragments derived from specific tRNAs has been suggested for the mammalian stress endoribonuclease ANG (Ivanov et al. 2011) and bacterial toxin–antitoxin endoribonucleases (for review, see Thompson and Parker 2009). A notable common feature of these tRNA-cleaving enzymes is insignificant depletion of full-length tRNAs coupled with pronounced translation arrest, which was attributed to signaling via cleaved tRNAs (Thompson and Parker 2009). The active tRNA fragments generated by angiogenin have a 5′-terminal oligoguanylate motif (TOG) essential for repressing translation initiation by disrupting the eIF4F (Ivanov et al. 2011). The tRNAs cleaved by RNase L lack the TOG motif (Fig. 1E; Supplemental Fig. S6B; Supplemental Dataset S1), indicating that the precise mechanism of ANG is not utilized.
To probe the role of cleaved RNAs, we tested transfection of the most abundant RNA cleavage product derived from tRNA-His (Fig. 1C; Supplemental Fig. S7A,B), and transfections of total cleaved RNAs, as well as small cleaved RNAs extracted from 2-5A-treated human cells under nondenaturing conditions (Supplemental Fig. S7C–E). These transfections did not block protein synthesis (Supplemental Fig. S7BD,E). Quantitative assessments using RtcB qPCR indicate that the levels of the fragments increased by ∼200-fold relative to naïve cells, which is strong, but below the ∼2000-fold increase observed in wild-type A549 cells treated with 2-5A. Therefore, the absence of translational arrest in Supplemental Figure S7 could be due to insufficient levels of cleaved RNAs attainable by transfection. It is also possible that the signaling RNA species function as structured RNAs or as ribonucleoprotein complexes (RNPs) that do not survive extraction from cells. Such denaturant-sensitive translation repressors have been observed among small ncRNAs in plant phloem (Zhang et al. 2009). Although RNase L blocks translation without depleting known RNAs, formally it remains possible as well that signaling is mediated by depletion of an unknown RNA.
All mechanisms that we described above rely on RNA cleavage. To exclude cleavage-independent mechanisms, such as an unexpected kinase output from the pseudokinase domain (Han et al. 2014) or signaling via physical interactions of the high-order RNase L complex that assembles upon binding of 2-5A (Han et al. 2012), we transfected WT RNase L and a catalytically inactive point mutant H672N (Han et al. 2014) into RNase L−/− A549 cells. Protein synthesis was arrested only by WT RNase L, and only in the presence of 2-5A (Fig. 5G). Regulated RNA cleavage therefore provides the trigger for translation arrest by RNase L.
DISCUSSION
We determined that the endoribonuclease activity of RNase L is central to the shut-off of protein synthesis in human A549 cells during dsRNA response. In matching RNase L−/− cells, which have to rely on the dsRNA-sensing kinase PKR, protein synthesis is inhibited only at late time points, and incompletely. By comparing RNA degradation with inhibition of protein synthesis, we show that RNase L blocks translation long before it depletes the RNA components of the translation machinery. Formally, this outcome could be explained by two largely different models: polysome traffic jam and signaling. In the traffic jam model, a few damaged ribosomes would stall many undamaged ribosomes on the polysomes. The model of the traffic jam has limitations due to the absence of experimentally observed robust polysomal stalls (Subramaniam et al. 2014), and recently recognized ribosome rescue and recycling mechanisms (for review, see Brandman and Hegde 2016). Moreover, mRNAs are often translated by monosomes (Heyer and Moore 2016) not subject to traffic jams. Our observations that RNase L can stop all visible translation therefore support the signaling model, whereas experiments with the H672N RNase L show that the signaling-like activity is supplied by the endoribonuclease domain (Fig. 5H).
RNase L is one of three related endoribonucleases in mammalian cells. The other two enzymes are isoforms α and β of the kinase-endoribonuclease, Ire1. Ire1 monitors the quality of protein folding inside the endoplasmic reticulum and carries out splicing of XBP1 mRNA to activate the unfolded protein response (Walter and Ron 2011). Using RtcB RNA-seq we found that Ire1 does not cleave any Y-RNAs or tRNAs under conditions of complete XBP1 mRNA splicing (Supplemental Fig. S8A,B). Cleavage of small noncoding RNAs is therefore not a general property of the kinase-linked endoribonucleases, but a distinct feature of RNase L.
In conclusion, rapid arrest of translation and site-specific cleavage of tRNAs and Y-RNAs emerge as key effects of RNase L that take place during dsRNA response in human cells. The RtcB qPCR assay for RNase L activity that we developed here provides technology for detection of the resulting RNA fragments. We estimate that RtcB qPCR can quantify femtomoles of specific RNA fragments in samples of total RNA. This sensitivity enables conceptually new experimental designs for probing the RNase L pathway, allows detection of trace quantities of immunogenic dsRNAs in mammalian cells and tissues, and provides a platform for developing new diagnostics with which to evaluate activation of the innate immune system in diseases.
MATERIALS AND METHODS
RtcB RNA-seq library preparation
Small miRvana (Life Technologies) purified RNAs from T47D cells (500 ng) or HeLa cells (1 µg) were ligated to 10 µM adaptor, 5′-OH-GAUCGUCGGACTGTAGAACTCTGAAC-3′-desthiobiotin (underlined bases are RNA). The reactions were conducted using 30 µM RtcB, 20 mM HEPES pH 7.5, 110 mM NaCl, 2 mM MnCl2, 100 µM GTP, 40U RiboLock RNase inhibitor (Fermentas), 4 mM DTT, and 0.05% Triton X-100 for 1 h at 37°C. The mixture was quenched by 1 volume of stop buffer (8 M urea, 1 mM EDTA, 0.1% SDS, 0.02% bromophenol blue, and 0.02% xylene cyanol) and fractioned by 10% PAGE 29:1 with 8 M urea. Gels were stained with SYBR-safe, visualized, and RNA larger than free adaptor was excised from the gel. RNA was eluted from gel slices overnight at 4°C with gentle mixing in 20 mM HEPES pH 7.5, 100 mM NaCl, 0.05% Triton X-100. Eluted RNA was recovered by ethanol-precipitation with 25 µg glycogen as a carrier.
RNA was reverse-transcribed using MultiScribe Reverse transcriptase (RT) and 2 pmol of primer complimentary to the ligation adaptor. RNA, RT primer, and dNTPs were incubated for 5 min at 65°C and snap-cooled on ice. A 2× mastermix containing RT buffer, RT, and 40U Ribolock was added to snap-cooled samples for a final volume of 20 µL. Reactions were incubated at 25°C for 10 min, then at 37°C for 1 h. RNA/cDNA hybrids were pulled down with hydrophilic magnetic streptavidin beads (NEB) and washed three times with 1 mL of 20 mM HEPES pH 7.5, 300 mM NaCl, 0.1% Triton X100, 10 min per wash. This step was followed by two 1 mL washes with 20 mM HEPES pH 7.5, 100 mM NaCl. The final wash buffer was removed and RNA/cDNA hybrids were eluted with 10 µL 10 mM biotin. The eluted cDNAs were then added to an equal volume of 20 mM HEPES pH 7.5, 0.1% Triton X100.
An adaptor, 5′-P-TGGAATTCTCGGGTGCCAAGG-3′-amino, was ligated to the 3′ ends of cDNAs using 1 U/µL CircLigase (Epicentre) and 1 µM adaptor. CircLigase reactions were incubated for 1 h at 65°C and quenched by adding EDTA to a final concentration of 8 mM. Quenched CircLigase reactions were PCR amplified for 16 cycles with Phusion DNA polymerase and forward primer with the sequence 5′-AATGATACGGCGACCACCGAGATCTACA CGTTCAGAGTTCTACAGTCCGA-3′ plus NEXTFLEX small RNA barcode primers (BIOO Scientific). Libraries were analyzed by Agilent BioAnalyzer high sensitivity DNA 1000 chip. Equimolar amounts were pooled, gel purified, and sequenced on an Illumina HiSeq 2500, such that the sequencing read is the reverse complement of the captured RNA and the first base of the read represents the base that had 2′,3′-cyclic phosphate after trimming the adapter sequence.
RNA-seq computational analysis
After barcode splitting, sequencing reads with quality scores ≥30 were trimmed of adaptor sequences and converted into reverse complements. The obtained reads were mapped to the human transcriptome using the FASTA algorithm with ≤1 mismatch, which accounts for possible polymorphisms. Sequences of tRNAs were modified to include CCA 3′-ends and G at the −1 position in tRNA-His. All steps past barcode splitting were conducted using software written in-house. Source code and Microsoft Windows binaries are available upon request.
Tissue culture
All cell lines were maintained at 37°C in a 5% CO2 humidified atmosphere. HeLa cells were grown in MEM + 10% FBS. A549 WT and RNase L CRISPR KO cells (Li et al. 2016) and T47D cells were grown in RPMI + 10% FBS. HeLa and T47D cells were a gift from the laboratory of Professor Yibin Kang (Princeton University), and A549 cells were a gift from the laboratory of Professor Susan Weiss (University of Pennsylvania).
Cell treatments for RNA-seq
For poly(IC) treatment, HeLa cells at ∼80%–90% confluence were transfected with 1 µg/mL poly(IC) or mock transfected (PBS) using Lipofectamine 2000 for 8 h. Cells were trypsinized, washed with cold PBS and small RNAs were purified using the miRvana kit. For RNase L overexpression, HeLa cells at ∼80%–90% confluence were transfected with 10 µg pcDNA4/TO encoding WT or H672N RNase L (Han et al. 2014) using Lipofectamine 2000. Cells were harvested as above at 24 h post-transfection and RNA was purified using the miRvana kit. For 2-5A treatment via semi-permeabilization, T47D cells were trypsinized at ∼80% confluence and washed with cold PBS. Cells were pelleted by centrifugation at 1000g for 5 min and at 4°C, and resuspended in 1 mL PBS. Resuspended cells were divided evenly and pelleted again for digitonin semi-permeabilization in the absence or presence of p2-5A3 in phosphate buffer, as described previously (Rath et al. 2015). One-half of each sample was added to 300 µL miRvana lysis buffer at 1 min and 3 min time points. Small RNA was purified according to the miRvana protocol. For Ire1 activation, A549 cells were treated with 2 mM DTT for 3 h and RNA was purified by miRvana. Ire1 activation was confirmed by RT-PCR for XBP1 using primers indicated in Supplemental Table S1.
Transfections
For all experiments with A549 cells, cells were seeded in 12-well dishes to achieve ∼70%–80% confluence. Transfections were carried out using Lipofectamine 2000. Cells were transfected with 1 µM ppp2-5An (mixed 2-5As with n ≥ 3) or 1 µg/mL poly(IC), synthetic RNA, or the indicated amounts of purified native cellular RNA for the durations stated in the figures. For transfection–translation studies, A549 WT cells were transfected with 300 nM synthetic fragments for 3 h. For introducing naturally generated fragments, A549 WT cells were transfected with or without 2-5A for 2 h and total or ≤200 nt RNAs were phenol extracted. The native RNAs were transfected (2.4 µg total or 8 µg for ≤200 nt) into A549 RNase L −/− cells for 2 h. For Figure 5G, the indicated amounts of RNase L were transfected with or without 1 µM 2-5A for 3 h.
RNA purification from human cells
Small RNAs for RtcB RNA sequencing were purified with the miRvana kit (Life Technologies) according to the manufacturer's instructions. RNAs for qPCR were purified using TRIzol (Life Technologies) and resuspended in water. RNAs for Northern blot were purified by TRIzol (Fig. 4A; Supplemental Fig. S6) or a modified TRIzol protocol (Fig. 4B) to purify small RNAs. Briefly, the aqueous phase was transferred to a clean tube, supplemented with one-third volume 100% ethanol and passed over an RNeasy (Qiagen) spin column. Small RNAs in flowthrough were precipitated by adding one-tenth volume of sodium acetate pH 5.2, 20 mg glycogen, and two-thirds volume of 100% ethanol. After washing with 75% ethanol, small RNAs were resuspended in water. Native RNA was purified from mock or 2-5A transfected WT A549 cells in 10 cm dishes by scraping cells in native RNA buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 1 mM MgCl2) followed by extraction with 1 volume of acid phenol:chloroform (5:1), and ethanol precipitation. RNAs were washed twice with 75% ethanol and resuspended in native RNA buffer. To obtain native small RNAs, one-third volume 100% ethanol was added to total native RNA and large RNAs were pelleted by centrifugation at 5000g, 5 min, 4°C. The supernatant containing small RNAs was transferred to a new tube and supplemented with two-thirds volume 100% ethanol. Small RNAs were pelleted by centrifugation at 21,000g for 10 min and at 4°C, washed with 75% ethanol, and resuspended in native RNA buffer.
E. coli RNA purification
WT (BW25113) and tgt::kan E.coli from the Keio knockout collection were generously provided by Dr. Marcin Grabowicz and Dr. Thomas Silhavy (Princeton University). After verifying the indicated genotypes by PCR amplifying the tgt locus and Sanger sequencing, E. coli were grown overnight at 37°C in LB or LB + 50 µg/mL kanamycin. Bacteria were pelleted, resuspended in TRIzol, and vortexed with glass beads. RNA was then purified according to the manufacturer's protocol and resuspended in water. Purified RNA was analyzed by 8 M urea 15% PAGE to verify RNA integrity and subsequently refolded and diluted to a final concentration of 1 mg/mL in 20 mM HEPES pH 7.5, 100 mM NaCl and 1 mM MgCl2.
RtcB enzyme preparation
RtcB was cloned from E. coli into pGEX-6P and expressed with an N-terminal GST-tag. Protein isolation and purification were performed as previously described for human OAS1 (Donovan et al. 2013), but in the absence of divalent metal cations.
Synthesis of model RNAs with 2′,3′-cyclic phosphate
Model RNAs were synthesized by Dharmacon and deprotected according to the manufacturer's instructions. Deprotected RNAs were ethanol-precipitated, resuspended in 20 mM HEPES pH 7.5, and quantified by UV spectrophotometry. Phosphorothioate linkages were converted to 2′,3′-cyclic phosphate in 80%–90% formamide by adding one-tenth volume of I2 (1 mg/mL) in ethanol. The reactions were incubated at 37°C for 10 min. During large-scale preparations, the iodine was fully consumed after 10 min, based on loss of color. Reactions were supplemented with 1/10 volume of 10 mg/mL I2 and incubated for another 10 min. There was no loss of color after the second I2 treatment. RNAs were ethanol-precipitated, resuspended in 20 mM HEPES, pH 7.5, and quantified by UV spectrophotometry. For analysis by PAGE, small amounts of RNA were dephosphorylated with Antarctic phosphatase (NEB) and 5′-end labeled with PNK and [γ-32P]ATP. Model RNA sequences are listed in Supplemental Table S1.
Quantitative PCR (qPCR)
TRIzol purified total RNA was digested with DNase I (Life Technologies), reextracted with acid phenol:chloroform (5:1; Life Technologies) and ethanol-precipitated. RNA was reverse-transcribed with Multiscribe reverse transcriptase and random hexamer priming. Analysis by qPCR was done using primers listed in Supplemental Table S1 and SYBR-green based detection over 40 cycles, with annealing/extension temperature of 60°C and duration of 1 min. GAPDH was used for normalization.
RtcB qPCR: streamlined detection of RNA fragments generated by RNase L
To analyze tRNA and Y-RNA cleavage by RNase L without using RNA-seq, we ligated RNA with 2′,3′-cyclic phosphate to an adaptor with RtcB as described above for RtcB RNA-seq library preparation, but modified to use less RiboLock (10 U) (Supplemental Fig. S5). Reactions were stopped by adding EDTA to a final concentration of 3 mM in 11 µL volume and incubated for ∼10 min at room temperature. EDTA-quenched ligation reaction (1 µL) was used as a template for reverse transcription with Multiscribe RT. A primer with a 3′-end complimentary to the adaptor and a 5′-overhang that serves as a universal priming site (5′-TCCCTATCAGTGATAGAGAGTTC AGAGTTCTACAGTCCG-3′) was used during the qPCR (Supplemental Fig. S5A). Reverse transcription was carried out as described for library preparation, except that 0.4 U Ribolock per reaction and 10 pmol RT primer were used. Reactions were terminated by heating to 95°C for 5 min. SYBR-green based qPCR was conducted using a universal reverse primer that binds to the cDNA overhang (underlined) and cleavage site-specific forward primers designed for each RNA target (Supplemental Table S1). qPCR was carried out for 50 cycles using 62°C annealing/extension for 1 min. U6, which has a naturally occurring 2′,3′-cyclic phosphate and an RNase L independent cleavage site in tRNA-His (position 18, transcript numbering; Supplemental Dataset S1) were used for normalization. The in vitro digest of naked RNA was normalized using only the internal tRNA-His site 18. Cleavage data for in vitro transcribed RNY4 and tRNA-His were normalized using the 3′-end of RNY4 and tRNA-His because the HDV ribozyme generates a 2′,3′-cyclic phosphate directly suitable for RtcB ligation.
Ribozyme constructs for RNA transcription in vitro
T7 RNA polymerase transcription constructs of RNY4 and tRNA-His flanked on the 5′ side by the hammerhead ribozyme and on the 3′ side by the HDV ribozyme were synthesized and cloned into pUC57 by GeneWiz. Sequences are as follows. The T7 promoter is in bold and the RNY4 and tRNA-His sequences are underlined: RNY4: 5′- GAATTCTAATACGACTCACTATAGGGAGAACCATC GGACCAGCCCTGATGAGTCCGTGAGGACGAAACGGTACCCGGTACCGTCGGCTGGTCCGATGGTAGTGGGTTATCAGAACTTATTAACATTAGTGTCACTAAAGTTGGTATACAACCCCCCACTGCTAAATTTGACTGGCTTTTTGGGCGGCATGGTCCCAGCCTCCTCGCTGGCGCCGCCTGGGCAACATGCTTCGGCATGGC GAATGGGACCGGATCC-3′; tRNA-His: 5′- GAATTCTAATACGACTCACTATAGGGAGATATACGATCACGGCCCTGATGAGTCCGTGAGGACGAAACGGTACCCGGTACCGTCGGCCGTGATCGTATAGTGGTTAGTACTCTGCGTTGTGGCCGCAGCAACCTCGGTTCGAATCCGAGTCACGGCACCAGGGCGGCATGGTCCCA GCCTCCTCGCTGGCGCCGCCTGGGCAACATGCTTCGGCAT GGCGAATGGGACCAATACAATAATAAGCATAATAACCAAGG ATCC-3′.
RNA transcription in vitro
Plasmids encoding ribozyme-flanked RNY4 and tRNA-His were linearized with Bam HI (NEB) and transcribed with the MEGAshortscript T7 Transcription Kit (Thermo Fisher). Transcription reactions were treated with DNase I for 15 min at 37°C followed by incubation at 65°C for 15 min and slow cooling for 20 min at room temperature. One volume of 8 M urea gel loading buffer was added to each reaction and samples were briefly denatured at 95°C, resolved by 8 M urea 10% PAGE, and visualized by UV-shadowing. Bands were excised, eluted in TE buffer pH 7.5, and precipitated with 3 volumes of 100% ethanol, one-tenth volume 3 M sodium acetate pH 5.2, and 5 µg glycogen. Recovered RNAs were resuspended in H2O and quantified by UV spectrophotometry. RNY4 and tRNA-His sizes were verified after 5′-end labeling by 8 M urea 10% PAGE with in vitro transcribed radiolabeled size markers.
Cleavage of RNA by recombinant RNase L
RNA from HeLa cytosolic S10 extracts was purified by acid phenol:chloroform extraction and ethanol precipitation. 2-5A and ATP were incubated for 5 min at 20°C in the absence or presence of recombinant human RNase L. Reactions were started by adding RNA (11 µg). Aliquots (5 µL) were quenched by TRIzol at 10, 60, and 300 sec, and RNA was purified. The reactions were conducted in 20 µL volumes using 12 mM sodium phosphate pH 7.0, 140 mM NaCl, 5 mM MgCl2, 1.25 mM β-mercaptoethanol, 0.1% Triton X-100, 1 mM ATP, 5 µM p2-5A3, and 100 nM RNase L. Synthetic model RNAs were synthesized by Dharmacon and 5′-end labeled with PNK and [γ-32P]ATP. The model RNAs were digested in the buffer described above with 100 nM RNase L using 1 µM p2-5A3 in 10 µL volume. Aliquots of each reaction were added to formamide gel loading buffer (90% formamide, 5% glycerol, 0.02% bromophenol blue) at time points 15, 120, and 600 sec. Samples were fractionated on 20% 8 M urea PAGE (29:1).
Transcribed RNAs were incubated at 80°C for 3 min, supplemented with 10 mM NaCl and 2 mM MgCl2, and cooled at room temperature for 20 min. Full-length 32P-5′-end-labeled transcribed RNY4 and tRNA-His were digested for 10 min and reactions quenched by adding 2 volumes of formamide loading buffer. The RNA samples were analyzed by 15% PAGE with 8 M urea. To assess purified cleaved RNA by RtcB qPCR, unlabeled full-length transcribed RNY4 and tRNA-His (2 µg) were 5′-phosphorylated with cold ATP and PNK, extracted with one volume of acid phenol:chloroform (5:1) (Thermo Fisher), and precipitated with ethanol. Recovered RNA was resuspended in 10 µL H2O. RNA (200 ng) was incubated with or without RNase L as above and aliquots at time points 20 sec, 3 min, and 10 min were quenched in TRIzol. RNA was purified accordingly and resuspended in 10 µL of H2O for analysis by RtcB qPCR.
E. coli RNA (2 µg) was digested with RNase L as described for purified human RNA and aliquots of the reactions were added to TRIzol at 10, 60, and 300 sec. Cleaved RNAs were purified and resuspended in H2O. RNA concentrations were determined by UV spectrophotometry. RNase L-mediated cleavage of E. coli tRNA-His was determined by using 50 ng RNA and RtcB qPCR as described above. Site-specific cleavage primer are listed in Supplemental Table S1.
Ribopuromycilation assay and Western blot
Cells were treated with 10 µg/mL puromycin (Invitrogen) in growth medium for 5 min, allowing puromycin to incorporate into the de novo synthesized proteome. Cells were then trypsinized to harvest and cell pellets were split to be lysed in TRIzol (Invitrogen) for RNA purification and in NuPAGE LDS sample buffer for Northern blot and Western blot analyses, respectively. For Western blotting, proteins were separated on 10% BisTris PAGE (NuPAGE), and transferred to PVDF membranes (Life Technologies). The membrane was stained with Ponceau to normalize for sample loading, then washed and blocked with 5% nonfat dry milk in TBST. The membranes were probed with 1:1000 of the following primary antibodies: (i) mouse anti-human puromycin antibody (EMD Millipore) to detect de novo protein synthesis, (ii) mouse anti-human total eIF2α (Cell Signaling Technology), and (iii) rabbit anti-human eIF2α-phosphoS51 (Abcam) followed by horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (1:10,000, Jackson ImmunoResearch).
Northern blot
TRIzol purified RNA was resolved on Novex TBE-urea 15% polyacrylamide gels (Life Technologies) followed by transfer and UV-crosslinking to Brightstar-Plus positively charged nylon membranes (Ambion). Blots were prehybridized in Ultrahyb-Oligo (Ambion) followed by hybridization of 5′-32P-labeled DNA oligonucleotide probes (Supplemental Table S1). Membranes were then washed twice with 2× SSC (300 mM NaCl, 30 mM sodium citrate pH 7.0, 0.5% SDS) and exposed to phosphor-storage screens. Prior to re-probing, membranes were stripped with 2×10 min washes in near-boiling H2O/0.5% SDS.
Error analysis
For experiments that required repeated measurements, such as qPCR analyses, standard errors (SE) were obtained using biological or technical replicates, as stated in figure legends.
DATA DEPOSITION
RtcB RNA-seq data have been deposited to the NCBI GEO server under accession code GSE100520.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Professor Susan Weiss and Yize (Henry) Li (University of Pennsylvania School of Medicine) for the gift of A546 WT and RNase L−/− cells (Li et al. 2016). We thank Dr. Marcin Grabowicz and Professor Thomas Silhavy (Princeton University) for the gift of E. coli strains, and Professor Elizabeth Gavis for sharing instrumentation. We thank Professor Andrei Korostelev for help in designing ribozyme constructs and comments on the manuscript, and Professor Pavel Ivanov (Harvard Medical School) for important discussions about tRNA cleavage. We are grateful to Dr. Wei Wang and the staff of Princeton University's sequencing core facility for help with RNA sequencing. We thank members of the Korennykh laboratory for reading the manuscript and providing valuable comments. We are grateful to Professor Roy Parker for making valuable suggestions during review. This study was funded by Princeton University, National Institutes of Health grants 5T32GM007388 and F99 CA212468-01 (to S.R.), National Institutes of Health grant 1R01GM110161-01 (to A.K.), Sidney Kimmel Foundation grant AWD1004002 (to A.K.), and Burroughs Wellcome Foundation Grant 1013579 (to A.K.).
Author contributions: J.D. designed RtcB RNA-seq and RtcB qPCR studies and prepared RtcB RNA-seq reagents and libraries. J.D. conducted biochemical assays. J.D. and S.R. carried out cell biology work and RtcB qPCR analyses. S.R. conducted studies of translation. D.K.M. generated in vitro transcribed ncRNAs. A.K. processed RtcB RNA-seq data. J.D., S.R., and A.K. wrote the manuscript, and A.K. supervised the work.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062000.117.
REFERENCES
- Aulas A, Fay MM, Lyons SM, Achorn CA, Kedersha N, Anderson P, Ivanov P. 2017. Stress-specific differences in assembly and composition of stress granules and related foci. J Cell Sci 130: 927–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Chakrabarti A, Jha BK, Weiss SR, Silverman RH. 2014. Cell-type-specific effects of RNase L on viral induction of beta interferon. mBio 5: e00856-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Li G, Li Y, Gaughan C, Baskar D, Parker Y, Lindner DJ, Weiss SR, Silverman RH. 2015. RNase L is a negative regulator of cell migration. Oncotarget 6: 44360–44372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Hegde RS. 2016. Ribosome-associated protein quality control. Nat Struct Mol Biol 23: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan-Laun SE, Ezelle HJ, Li XL, Hassel BA. 2014. RNase-L control of cellular mRNAs: roles in biologic functions and mechanisms of substrate targeting. J Interferon Cytokine Res 34: 275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambier L, de Couto G, Ibrahim A, Echavez AK, Valle J, Liu W, Kreke M, Smith RR, Marbán L, Marbán E. 2017. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol Med 9: 337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti A, Banerjee S, Franchi L, Loo YM, Gale M Jr, Núñez G, Silverman RH. 2015. RNase L activates the NLRP3 inflammasome during viral infections. Cell Host Microbe 17: 466–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DA, Banerjee S, Chakrabarti A, Garcia-Sastre A, Hesselberth JR, Silverman RH, Barton DJ. 2014. Ribonuclease L targets distinct sites in influenza A virus RNAs. J Virol 89: 2764–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhahbi JM, Spindler SR, Atamna H, Boffelli D, Martin DI. 2014. Deep sequencing of serum small RNAs identifies patterns of 5′ tRNA half and YRNA fragment expression associated with breast cancer. Biomark Cancer 6: 37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Silverman RH. 1995. 2-5A-dependent RNase molecules dimerize during activation by 2-5A. J Biol Chem 270: 4133–4137. [DOI] [PubMed] [Google Scholar]
- Donovan J, Dufner M, Korennykh A. 2013. Structural basis for cytosolic double-stranded RNA surveillance by human oligoadenylate synthetase 1. Proc Natl Acad Sci 110: 1652–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre O, Salehzada T, Lambert K, Boo Seok Y, Zhou A, Mercier J, Bisbal C. 2012. RNase L controls terminal adipocyte differentiation, lipids storage and insulin sensitivity via CHOP10 mRNA regulation. Cell Death Differ 19: 1470–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SA, Thornbrough JM, Zhang R, Jha BK, Li Y, Elliott R, Quiroz-Figueroa K, Chen AI, Silverman RH, Weiss SR. 2016. Lineage A Betacoronavirus NS2 proteins and the homologous Torovirus Berne pp1a carboxy-terminal domain are phosphodiesterases that antagonize activation of RNase L. J Virol 91: e02201–e02216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Whitney G, Donovan J, Korennykh A. 2012. Innate immune messenger 2-5A tethers human RNase L into active high-order complexes. Cell Rep 2: 902–913. [DOI] [PubMed] [Google Scholar]
- Han Y, Donovan J, Rath S, Whitney G, Chitrakar A, Korennykh A. 2014. Structure of human RNase L reveals the basis for regulated RNA decay in the IFN response. Science 343: 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer EE, Moore MJ. 2016. Redefining the translational status of 80S monosomes. Cell 164: 757–769. [DOI] [PubMed] [Google Scholar]
- Honda S, Loher P, Shigematsu M, Palazzo JP, Suzuki R, Imoto I, Rigoutsos I, Kirino Y. 2015. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc Natl Acad Sci 112: E3816–E3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanessian AG, Kerr IM. 1978. Synthesis of an oligonucleotide inhibitor of protein synthesis in rabbit reticulocyte lysates analogous to that formed in extracts from interferon-treated cells. Eur J Biochem 84: 149–159. [DOI] [PubMed] [Google Scholar]
- Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, Thevakumaran N, Talukdar M, Pillon MC, Ceccarelli DF, et al. 2014. Dimeric structure of pseudokinase RNase L bound to 2-5A reveals a basis for interferon-induced antiviral activity. Mol Cell 53: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iordanov MS, Paranjape JM, Zhou A, Wong J, Williams BR, Meurs EF, Silverman RH, Magun BE. 2000. Activation of p38 mitogen-activated protein kinase and c-Jun NH2-terminal kinase by double-stranded RNA and encephalomyocarditis virus: involvement of RNase L, protein kinase R, and alternative pathways. Mol Cell Biol 20: 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland DD, Stohlman SA, Hinton DR, Kapil P, Silverman RH, Atkinson RA, Bergmann CC. 2009. RNase L mediated protection from virus induced demyelination. PLoS Pathog 5: e1000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, Emara MM, Villen J, Gygi SP, Anderson P. 2011. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol Cell 43: 613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov P, O'Day E, Emara MM, Wagner G, Lieberman J, Anderson P. 2014. G-quadruplex structures contribute to the neuroprotective effects of angiogenin-induced tRNA fragments. Proc Natl Acad Sci 111: 18201–18206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski MP, Krude T. 2015. Functional roles of non-coding Y RNAs. Int J Biochem Cell Biol 66: 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane N, Martin W. 2010. The energetics of genome complexity. Nature 467: 929–934. [DOI] [PubMed] [Google Scholar]
- Li XL, Ezelle HJ, Kang TJ, Zhang L, Shirey KA, Harro J, Hasday JD, Mohapatra SK, Crasta OR, Vogel SN, et al. 2008. An essential role for the antiviral endoribonuclease, RNase-L, in antibacterial immunity. Proc Natl Acad Sci 105: 20816–20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Wang Y, Goldstein SA, Dong B, Gaughan C, Silverman RH, Weiss SR. 2016. Activation of RNase L is dependent on OAS3 expression during infection with diverse human viruses. Proc Natl Acad Sci 113: 2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M Jr, Silverman RH. 2007. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448: 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi S, Nishimura Y, Hirota Y, Nishimura S. 1982. Isolation and characterization of an Escherichia coli mutant lacking tRNA-guanine transglycosylase. Function and biosynthesis of queuosine in tRNA. J Biol Chem 257: 6544–6550. [PubMed] [Google Scholar]
- Ogawa T. 2016. tRNA-targeting ribonucleases: molecular mechanisms and insights into their physiological roles. Biosci Biotechnol Biochem 80: 1037–1045. [DOI] [PubMed] [Google Scholar]
- Rath S, Donovan J, Whitney G, Chitrakar A, Wang W, Korennykh A. 2015. Human RNase L tunes gene expression by selectively destabilizing the microRNA-regulated transcriptome. Proc Natl Acad Sci 112: 15916–15921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutjes SA, van der Heijden A, Utz PJ, van Venrooij WJ, Pruijn GJ. 1999. Rapid nucleolytic degradation of the small cytoplasmic Y RNAs during apoptosis. J Biol Chem 274: 24799–24807. [DOI] [PubMed] [Google Scholar]
- Sharma U, Conine CC, Shea JM, Boskovic A, Derr AG, Bing XY, Belleannee C, Kucukural A, Serra RW, Sun F, et al. 2016. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 351: 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Schmechel SC, Williams BR, Silverman RH, Schiff LA. 2005. Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J Virol 79: 2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam AR, Zid BM, O'Shea EK. 2014. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell 159: 1200–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Chakravarty AK, Maughan B, Shuman S. 2011. Novel mechanism of RNA repair by RtcB via sequential 2′,3′-cyclic phosphodiesterase and 3′-phosphate/5′-hydroxyl ligation reactions. J Biol Chem 286: 43134–43143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DM, Parker R. 2009. Stressing out over tRNA cleavage. Cell 138: 215–219. [DOI] [PubMed] [Google Scholar]
- Toots UE, Kel've MB, Saarma M. 1988. [Degradation of messenger RNA, ribosomal RNA and 2-5A-dependent inhibition of protein biosynthesis]. Mol Biol (Mosk) 22: 1473–1481. [PubMed] [Google Scholar]
- van Gelder CW, Thijssen JP, Klaassen EC, Sturchler C, Krol A, van Venrooij WJ, Pruijn GJ. 1994. Common structural features of the Ro RNP associated hY1 and hY5 RNAs. Nucleic Acids Res 22: 2498–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086. [DOI] [PubMed] [Google Scholar]
- Zhang S, Sun L, Kragler F. 2009. The phloem-delivered RNA pool contains small noncoding RNAs and interferes with translation. Plant Physiol 150: 378–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.