

Abstract
Uridine insertion and deletion RNA editing generates functional mitochondrial mRNAs in Trypanosoma brucei, and several transcripts are differentially edited in bloodstream (BF) and procyclic form (PF) cells correlating with changes in mitochondrial function. Editing is catalyzed by three ∼20S editosomes that have a common set of 12 proteins, but are typified by mutually exclusive RNase III KREN1, N2, and N3 endonucleases with distinct cleavage specificities. KREPB4 is a common editosome protein that has a degenerate RNase III domain lacking conserved catalytic residues, in addition to zinc-finger and Pumilio/fem-3 mRNA binding factor (PUF) motifs. Here we show that KREPB4 is essential for BF and PF growth, in vivo RNA editing, and editosome integrity, but that loss of KREPB4 has differential effects on editosome components and complexes between BF and PF cells. We used targeted mutagenesis to investigate the functions of the conserved PUF and RNase III domains in both life-cycle stages and show that the PUF motif is not essential for function in BF or PF. In contrast, specific mutations in the RNase III domain severely inhibit BF and PF growth and editing, and disrupt ∼20S editosomes, while others indicate that the RNase III domain is noncatalytic. We further show that KREPB4, specifically the noncatalytic RNase III domain, is required for the association of KREN1, N2, and N3 with PF editosomes. These results, combined with previous studies, support a model in which KREPB4 acts as a pseudoenzyme to form the noncatalytic half of an RNase III heterodimer with the editing endonucleases.
Keywords: RNA editing, Trypanosoma brucei, editosome, endonuclease, RNase III
INTRODUCTION
Trypanosoma brucei is a kinetoplastid protozoan parasite that causes human African trypanosomiasis (HAT) in humans and nagana in animals. It is transmitted by the tsetse fly and is a health and economic threat to millions of people and animals in sub-Saharan Africa. Kinetoplastids are named for their mitochondrial “kinetoplast” DNA network, which is composed of interlocked maxicircle and minicircle DNA molecules (Stuart and Feagin 1992; Shapiro and Englund 1995). Several identical maxicircles encode two ribosomal RNAs (rRNAs) and messenger RNAs (mRNAs) for 18 mitochondrial proteins. Twelve of these mRNAs undergo post-transcriptional RNA editing, a phenomenon that was first described in kinetoplastids (Benne et al. 1986; Feagin et al. 1987; Shaw et al. 1988; Bhat et al. 1990; Koslowsky et al. 1990; Souza et al. 1992, 1993). Kinetoplastid RNA editing is essential (Schnaufer et al. 2001) and involves the precise insertion and deletion of uridylylates (Us) at hundreds and tens of editing sites (ESs), respectively, to generate translatable mitochondrial transcripts. ESs and edited sequences are specified by guide RNAs (gRNAs) that are encoded in thousands of heterogeneous minicircles (Blum and Simpson 1990; Blum et al. 1990; Pollard et al. 1990; Sturm and Simpson 1990; Pollard and Hajduk 1991; Stuart et al. 2005; Aphasizhev and Aphasizheva 2011). Each gRNA typically contains information for multiple ESs, and editing of most mRNAs requires several gRNAs. Editing occurs by rounds of coordinated catalytic steps: mRNA cleavage by endonucleases, U addition by terminal uridylyl-transferase (TUTase), U removal by U-specific exoribonuclease (exoUase), and RNA rejoining by ligases. The enzymes required for editing, in addition to proteins that have no known catalytic functions, form multiprotein ∼20S complexes called editosomes or RNA editing core complexes (RECCs) (Panigrahi et al. 2001a,b, 2003a,b, 2006; Ernst et al. 2003; Carnes et al. 2005, 2008, 2011; Stuart et al. 2005; Trotter et al. 2005; Lerch et al. 2012).
There are three similar, but compositionally and functionally distinct versions of these ∼20S editosomes. In addition to a common set of 12 proteins, each contains a different endonuclease along with a uniquely associated specific partner protein and has different ES cleavage specificity (Carnes et al. 2005, 2008, 2011; Trotter et al. 2005; Panigrahi et al. 2006; Guo et al. 2012). One complex contains the KREN1/KREPB8 protein pair plus the KREX1 exonuclease and cleaves deletion ESs. The other two complexes contain the KREN2/KREPB7 or KREN3/KREPB6 protein pairs and cleave insertion sites, albeit with different preferences (Carnes et al. 2005, 2008, 2011, 2017; Trotter et al. 2005; Panigrahi et al. 2006; Guo et al. 2012). KREN1, KREN2, and KREN3 each have a single RNase III domain that contains conserved catalytic residues found in all characterized RNase III endonucleases. Loss of any one of these endonucleases, or mutation of these residues eliminates in vivo editing and in vitro cleavage of editing sites (Carnes et al. 2005, 2008; Trotter et al. 2005; Panigrahi et al. 2006).
The common set of proteins contains two related proteins, KREPB4 and KREPB5, that each have a degenerate RNase III domain that lacks universally conserved catalytic residues, in addition to a U1-like zinc-finger motif, and a Pumilio/fem-3 mRNA binding factor (PUF) motif (Worthey et al. 2003; Carnes et al. 2012; McDermott et al. 2015b, 2016). Because all characterized RNase III endonucleases function as dimers to cleave double-stranded RNA (dsRNA) (MacRae and Doudna 2007; Nicholson 2014), and because the endonucleases are each present as a single copy per editosome (Carnes et al. 2011), we have hypothesized that the RNase III domain of KREPB4 and/or KREPB5 forms a heterodimeric RNase III active site with the editing endonucleases (Carnes et al. 2012; Nicholson 2014; McDermott et al. 2015a,b). Recent cross-linking and mass spectrometry (CXMS) analyses of editosomes revealed proximity between KREPB4 and all three endonucleases and the endonuclease partner proteins KREPB6 and KREPB7 (Supplemental Fig. S1; McDermott et al. 2016), providing evidence that KREPB4 is a major interaction partner of the editing endonucleases.
RNAi knockdown has previously shown that KREPB4 is essential for growth of procyclic form (PF) cells where it disrupts the structural integrity of ∼20S editosomes, leading to accumulation of ∼5–10S subcomplexes and loss of endonuclease activity in vitro (Babbarwal et al. 2007). Previous studies also showed that expression of the T. cruzi ortholog following RNAi silencing can complement KREPB4 knockdown, but that point mutations in the zinc-finger motif prevent complementation and incorporation into ∼20S editosomes (Carnes et al. 2012). The KREPB4 RNase III domain has not yet been subject to mutational analysis, and its function is yet to be studied (Carnes et al. 2012). The role of KREPB4 in bloodstream form (BF) cells is also unknown. We hypothesize that the function of KREPB4 in BF differs from that in PF, as a growing body of evidence shows that elimination or mutation of specific editosome proteins, including KREPB5, differentially affects cell viability, RNA editing and editosomes in BF and PF, and similar results have been also observed with other mitochondrial RNA processing protein complexes (Aphasizheva et al. 2015; McDermott et al. 2015a,b). Editosome complexes and components, including the related KREPB5, are therefore implicated in the processes that control differential editing of several mitochondrial transcripts between BF and PF cells, that in turn reflect changes in the mitochondrion and energy generation between the different life-cycle stages (Feagin et al. 1987; Feagin and Stuart 1988; Bhat et al. 1990; Koslowsky et al. 1990, 1992; Souza et al. 1992, 1993; Riley et al. 1995).
In this work, we generated BF and PF KREPB4 conditional null (CN) cell lines to fully analyze the function of this protein in both BF and PF life-cycle stages. We demonstrate that KREPB4 is essential in PF and BF for growth, in vivo RNA editing, and for ∼20S editosome, but not GAP2-containing mitochondrial RNA binding 1 (MRB1) complex integrity. Furthermore, we show that loss of KREPB4 has differential effects on the editosome component and complex integrity between BF and PF cells. We investigated the functions of specific KREPB4 domains in both life-cycle stages by exclusive expression of mutant alleles, and showed that the conserved PUF motif is not essential for function in BF or PF. Similarly, mutation of a potentially conserved catalytic residue in the KREPB4 RNase III domain produced no defect in function, providing experimental evidence that KREPB4 is noncatalytic. In contrast, substitution of a highly conserved glycine in the RNase III domain severely inhibits growth, in vivo editing and disrupts ∼20S editosomes to the same extent as loss of KREPB4 in both BF and PF. We finally show that KREPB4, and specifically the RNase III domain, is required for the association of the editing endonucleases KREN1, KREN2, and KREN3 with PF editosome complexes, validating protein proximities and thus interactions from cross-linking and mass spectrometry analyses. Together these results are fully consistent with the hypothesis that KREPB4 forms the noncatalytic half of an RNase III enzyme–pseudoenzyme heterodimer with the editing endonucleases.
RESULTS
KREPB4 is essential and is required for RNA editing in both BF and PF T. brucei
To examine KREPB4 function in both BF and PF life-cycle stages, we prepared BF and PF KREPB4 CN cells. These are cells in which both endogenous KREPB4 alleles were deleted, and in which a tetracycline (tet)-regulatable wild-type (WT) KREPB4 allele was inserted into the rRNA locus. Depletion of KREPB4 by tet withdrawal resulted in severe growth inhibition in both BF and PF CN cells (Table 1; Fig. 1A). RT-qPCR analysis showed that mRNA coding for KREPB4 was reduced by >98% upon withdrawal of tet from these CN cells (Fig. 1B). The knockdown in both BF and PF also resulted in a reduction in the relative levels of multiple edited mRNAs, but had little effect on the never-edited mRNAs COI and ND4. All edited mRNAs that were assayed in BF were reduced by 98% or more, and the levels of edited CYb and COIII were below detection. In PF, edited mRNAs were reduced by between 66% (COII) and 90% (CYb). As is commonly observed upon depletion of ∼20S editosome components (Carnes et al. 2005, 2008, 2017; Trotter et al. 2005; Salavati et al. 2006; Guo et al. 2008, 2012; Ernst et al. 2009; McDermott et al. 2015b), the levels of pre-edited mRNAs were generally increased by various amounts or were unchanged (Fig. 1B) in both BF and PF.
TABLE 1.
BF and PF KREPB4 CN cell doubling times (in hours) were monitored in vitro over 48 (BF) or 96 (PF) h in the presence (tet-regulatable copy expressed; E) or absence (tet-regulatable copy repressed; R) of tet

The table reports mean values ± SD from three independent experiments, and the calculated numbers of cell generations within the time-frames are given in parentheses.
FIGURE 1.
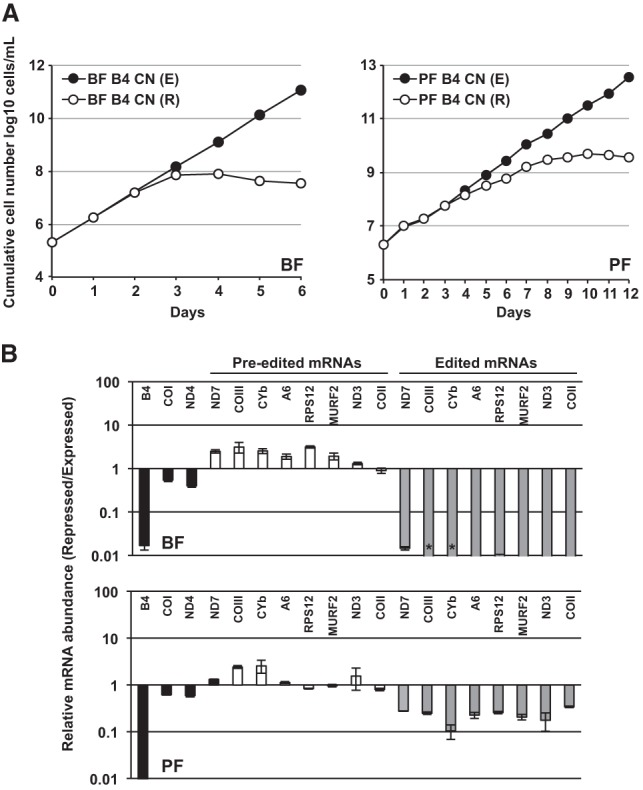
(A) Cumulative growth of BF and PF CN cells in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R). (B) RT-qPCR analysis of BF and PF CN cells in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R) for 2 and 4 d, respectively. The abundances of KREPB4, and never-edited (COI and ND4), pre-edited, and edited mitochondrial mRNAs in repressed cells were calculated relative to cells expressing WT KREPB4, and plotted on a log scale. For each target amplicon, relative abundance was determined using TERT as an internal control. Data represent mean of three experiments, and error bars indicate mean ± SEM. Asterisks indicate that the target amplicon was not detected in the repressed cells.
Loss of KREPB4 has different effects on BF and PF editosomes
Knockdown of KREPB4 had differential effects on editosome component and complex integrity between BF and PF cells. Western analysis was performed on total BF CN cell lysates that were grown in the presence or absence of tet for 2 and 4 d, and on PF CN cell lysates that were grown in the presence or absence of tet for 4 and 8 d. These time-points were chosen to allow comparison of BF and PF cells at approximately equivalent generation times following tet withdrawal (Table 1). Blots were probed using a mixture of monoclonal antibodies for four ∼20S editosome proteins (KREPA1, KREPA2, KREL1, and KREPA3) in addition to GAP2 (Acestor et al. 2009), which is a component of the Core (also known as the gRNA-binding, or GRBC) subcomplex of MRB1 (which is also known as the RNA editing substrate binding complex, RESC) (Hashimi et al. 2008; Panigrahi et al. 2008; Weng et al. 2008; Ammerman et al. 2012; Aphasizheva et al. 2014; Read et al. 2016), and mitochondrial Hsp70 (mtHsp70), which is not involved in editing. Repression of KREPB4 expression resulted in a loss of ∼20S editosome components, particularly KREPA1, KREPA2, and KREL1, after 2 d in BF (Fig. 2A). GAP2 and mtHsp70 were unchanged in the absence of tet, indicating that protein loss at this stage is specific to editosome proteins. After 4 d of tet withdrawal in BF, editosome proteins were undetectable, while levels of GAP2 and mtHsp70 remained unchanged (Fig. 2A). In contrast, editosome components were retained in PF cells following 4 and 8 d of tet withdrawal (Fig. 2A). Western analysis of glycerol-gradient fractions from the same BF lysates revealed a dramatic loss of ∼20S editosome complexes that mirrored the loss of editosome components following KREPB4 depletion (Fig. 2B). Complexes were retained in PF, even after 8 d of tet withdrawal. However, complex integrity was disrupted as KREPA1, KREPA2, KREL1, and KREPA3 all shifted to smaller S values (Fig. 2B). KREPB4 mRNA was dramatically depleted in both PF and BF cells grown in the absence of tet (99.5% in PF, 98.3% in BF), indicating that the greater retention of editosome components and complexes in PF was not due to a greater KREPB4 knockdown in BF than in PF. The differential effects of KREPB4 loss on BF and PF editosome complexes and components have previously been observed upon loss of KREPB5 (McDermott et al. 2015b), and provide further evidence for intrinsic differences between BF and PF editosomes. Consistent with previous analyses (Babbarwal et al. 2007; Acestor et al. 2009), this work additionally shows that loss or disruption of editosomes, with consequent depletion of edited mRNAs, does not affect sedimentation of the MRB1 core complex component GAP2.
FIGURE 2.
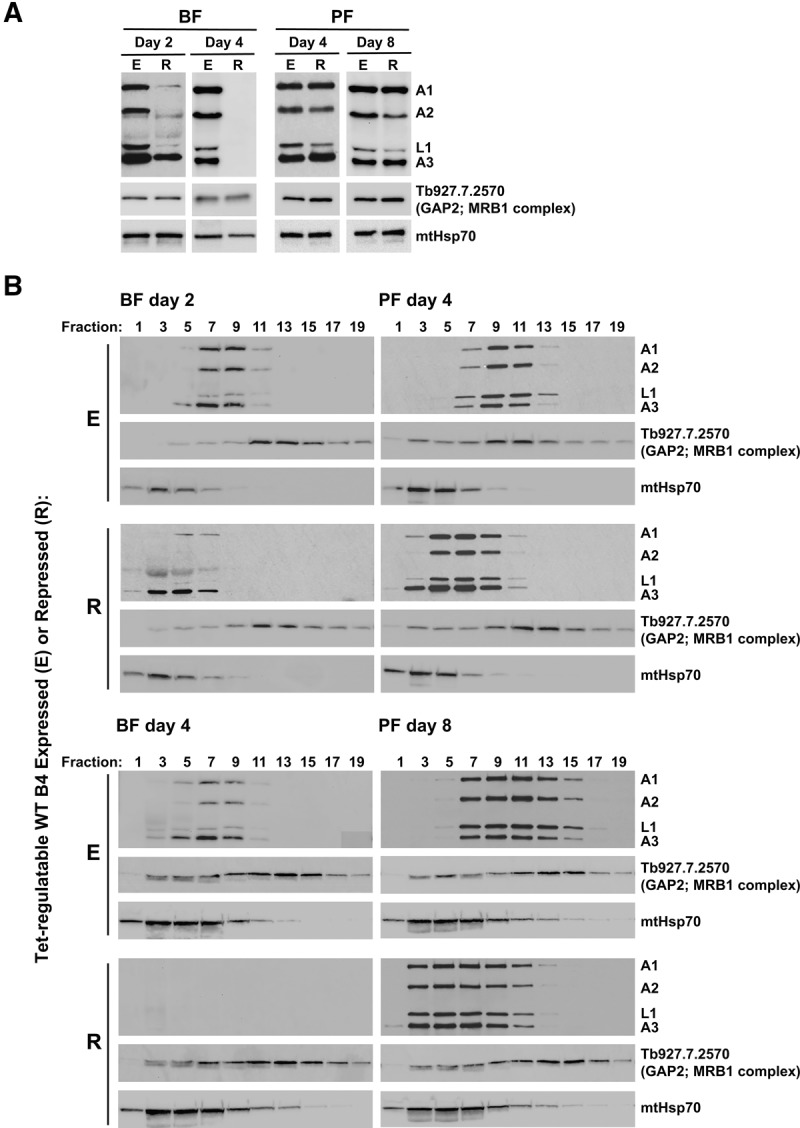
(A) Western analysis of total lysates (equivalent of 1 × 107 BF cells/lane, and 5 × 106 PF cells/lane) from BF and PF CN cells in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R) for 2 and 4 d (BF), and 4 and 8 d (PF). The Western blots were probed with monoclonal antibodies against editosome proteins KREPA1, KREPA2, KREL1, and KREPA3, the MRB1 complex component GAP2, and mitochondrial Hsp70. (B) Western analysis of 5 h 10%–30% glycerol-gradient fractions of lysates from BF and PF CN cells in which KREPB4 was expressed or repressed as in A.
The evidence presented here therefore shows that loss of KREPB4 generally has similar effects on editing and on editosome components and complexes in each life-cycle stage as the loss of KREPB5 (McDermott et al. 2015b). We also previously showed that alanine substitution of the conserved histidine H233, or a “PUF-triple” substitution of C232, H233, and E236, within the KREPB5 PUF motif differentially affects editing and editosomes in PF but not BF (McDermott et al. 2015b). Based on alignment with PUF motif sequences from a diverse range of organisms, these residues were predicted to make specific contacts with RNA via stacking interactions or hydrogen bonds and van der Waals contacts (McDermott et al. 2015b). Thus, we sought to test whether equivalent substitutions within the KREPB4 PUF motif also have differential effects on editing between life-cycle stages. We individually substituted H281, T280, and E284 with alanine, and also prepared a combined “PUF-triple” T280A/H281A/E284A mutant (McDermott et al. 2015b). WT or mutant KREPB4 alleles were tagged with a V5-epitope tag and constitutively expressed from the β-tubulin locus of BF and PF KREPB4 CN cells (Fig. 3A). Western blotting confirmed that the V5-tagged alleles were expressed in the presence and absence of tet (Supplemental Fig. S2A). Exclusive expression of the V5-tagged WT allele in the absence of tet permitted normal BF and PF growth, as did expression of the PUF motif mutant alleles (Fig. 3B). Parallel to the effects on cell growth, exclusive expression of V5-tagged WT KREPB4 rescued in vivo editing defects observed in the parental BF and PF CN cells as did exclusive expression of the PUF-triple mutant allele (Supplemental Fig. S2B,C). These results show that PUF motif substitutions do not affect KREPB4 function in either BF or PF, and indicate that the PUF motifs in KREPB4 and KREPB5 do not have equivalent functions.
FIGURE 3.

(A) Schematic showing tet-regulated and constitutive expression of WT and mutant KREPB4 alleles in BF and PF CN cells. Withdrawal of tet results in the exclusive expression of V5-tagged WT or mutant KREPB4. (B) Cumulative growth of BF and PF CN cells constitutively expressing V5-tagged WT or PUF motif mutant versions of KREPB4, and in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R). Exclusive expression of V5-tagged WT and all mutant KREPB4 proteins (each indicated by R) permitted normal growth in BF and PF.
The KREPB4 RNase III domain is essential for function in BF and PF
BF loss and PF disruption of editosomes upon repression of KREPB4 indicate an important structural role within ∼20S complexes in both life-cycle stages. This is consistent with cross-linking and mass spectrometry analyses of PF editosomes that revealed extensive cross-linking between KREPB4 and other editosome proteins (Supplemental Fig. S1; McDermott et al. 2016). Notably, KREPB4 was in proximity to all three editing endonucleases and the endonuclease partner proteins KREPB6 and KREPB7. This was intriguing given the presence of the degenerate RNase III domain in KREPB4 which lacks a conserved catalytic center, but contains a highly conserved glycine residue, G163, previously shown to be essential for KREPB5 function in both BF and PF (Fig. 4A,B; Carnes et al. 2012; McDermott et al. 2015a,b, 2016). To assess the role of the KREPB4 RNase III domain, we substituted G163 with either arginine, or more conservatively, valine. We also generated an alanine substitution at glutamic acid E164, which corresponds to a conserved aspartic acid residue in catalytic RNase III endonucleases (Fig. 4A). RNase III mutant KREPB4 alleles were tagged with a V5-epitope tag and inserted into the β-tubulin locus of BF and PF KREPB4 CN cells. Western blotting confirmed that the V5-tagged alleles were expressed in the presence and absence of tet (Fig. 4C). Exclusive expression of G163R or G163V mutant alleles resulted in severe growth inhibition of both BF and PF cells (Fig. 4D). In contrast, exclusive expression of the E164A mutant allele did not affect BF or PF growth (Fig. 4E). Together, these results show that the conserved glycine in the KREPB4 RNase III domain is essential for function in both BF and PF. However, E164, which aligns with a conserved catalytic residue, is not required for function, providing experimental evidence that the degenerate RNase III domain lacks direct catalytic activity.
FIGURE 4.
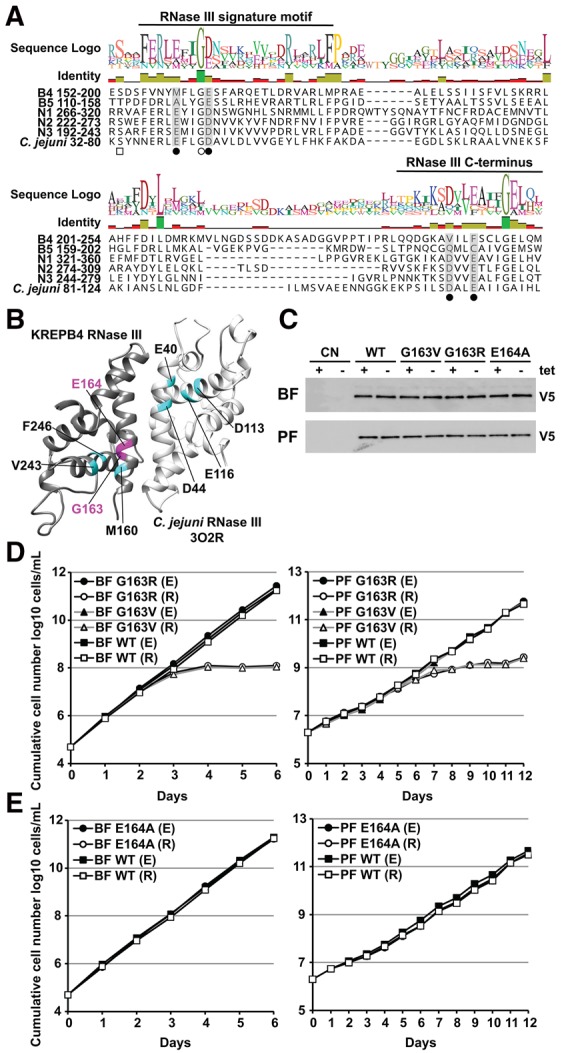
(A) Alignment of RNase III domain sequences from KREPB4-B5, KREN1-N3, and Campylobacter jejuni (accession Q9PM40; PDB ID 3O2R). A total of 172 sequences from 35 kinetoplastid species and strains were aligned using MUSCLE (Edgar 2004), and only T. brucei sequences are shown for clarity. Closed circles denote residues required for metal ion coordination and RNase III catalysis (Nicholson 2014). These residues are conserved in KREN1-N3, but degenerate in KREPB4-B5. Open circle indicates a universally conserved glycine. Substitution of this glycine in KREPB5 prevents function in both BF and PF (Carnes et al. 2012; McDermott et al. 2015b). Phosphorylation of a residue equivalent to C. jejuni S34, denoted by the open square, in E. coli increases RNase III catalytic activity (Gone et al. 2016). (B) Comparative model of the KREPB4 RNase III domain generated using the C. jejuni RNase III nuclease domain (PDB ID 3O2R) as a template (McDermott et al. 2016). Modeled KREPB4 structure is shown in dark gray, while template RNase III domain dimer is shown in light gray. RNase III residues required for metal ion coordination and catalysis, and aligned KREPB4 residues, are shown in cyan. The universally conserved glycine G163, and aspartic acid E164 in the KREPB4 RNase III domain that were subject to mutagenesis are highlighted in magenta. (C) Western analysis with anti-V5 tag monoclonal antibody showing constitutive expression of V5-tagged mutant or WT KREPB4 proteins from the β-tubulin locus in BF and PF CN cells (equivalent of 4 × 106 and 2 × 106 cells/lane, respectively) in the presence and absence of tet. (D,E) Cumulative growth of BF and PF CN cells that constitutively express V5-tagged WT, G163R/V (D), or E164A (E) RNase III mutant versions of KREPB4, and in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R).
In addition, mutations were made at KREPB4 RNase III residues S153, S218, S219, and S224 (Fig. 4A; Supplemental Fig. S3). S153 aligns with an RNase III serine residue whose phosphorylation in E. coli increases catalytic activity (Fig. 4A; Gone et al. 2016), and S218, S219, and S224 were each predicted to be phosphorylated using the PhosTryp method (Supplemental Table S1; Palmeri et al. 2011). To study these putatively modified residues, we substituted them individually, and in combination for S218, S219, and S224, with alanine or glutamic acid, which would be inhibitory or mimetic for phosphorylation, respectively. None of these substitutions had any effect on the growth of BF or PF upon exclusive expression (Supplemental Fig. S3). Thus, the predictions may have been incorrect and these residues are not phosphorylated. Alternatively, they could be phosphorylated in combination with modifications to other editosome proteins, and substitutions to several modified residues in multiple proteins might be required to see functional effects in editing.
The effects of RNase III domain glycine substitution on editing and editosomes in BF and PF were evaluated by RT-qPCR and glycerol-gradient sedimentation of lysates from BF and PF CN cells containing V5-tagged WT, G163R or G163V mutant alleles grown in the presence or absence of tet. These analyses revealed that exclusive expression of G163R and G163V generally had the same effects on in vivo editing and editosomes as the loss of KREPB4 in BF and PF. Edited mRNAs were reduced by 98% or more in BF upon exclusive expression of G163V and G163R alleles, while in PF the reduction in edited mRNAs ranged from 49% (ND7) to 91% (CYb) (Fig. 5). Editosome complexes were essentially undetectable in BFs that exclusively expressed either mutant (Fig. 6A). Upon exclusive expression of the mutant alleles in PFs, complexes containing editosome proteins were present, but shifted to smaller S values (Fig. 6B), as observed in the CN cells upon loss of KREPB4. The V5-tagged G163V and G163R proteins generally cosedimented with editosome proteins in the presence of tet in both BF and PF, but were also shifted to smaller S values in the absence of tet (Fig. 6A,B). Thus, G163V and G163R were fully incorporated into ∼20S complexes only in the presence of tet-regulatable WT KREPB4 in both BF and PF. This dependency of RNase III mutant KREPB4 on WT KREPB4 for incorporation into ∼20S complexes suggests that there may be multiple copies of KREPB4 per editosome. Together these results show that exclusive expression of either G163R or G163V had the same effect on in vivo editing and editosome integrity as the loss of KREPB4 in BF and PF, and that the highly conserved glycine in the KREPB4 RNase III domain is essential for the proper homo- and heteromeric interactions of KREPB4 with other editosome proteins.
FIGURE 5.

(A) RT-qPCR analysis of BF and PF CN cells that constitutively express V5-tagged WT, G163R, or G163V mutant versions of KREPB4, and in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R) for 2 and 4 d, respectively. The abundances of transcripts from KREPB4 alleles (B4 reg = tet-regulatable WT allele; B4 EE = exclusively expressed V5-tagged WT or mutant allele; B4 ORF = B4 open reading frame), and never-edited (COI and ND4), pre-edited, and edited mitochondrial mRNAs in repressed cells were calculated relative to cells expressing tet-regulatable WT KREPB4. For each target amplicon, relative abundance was determined using TERT as an internal control. Heat map shows the log10-transformed relative abundances of RNAs from BF cells (left) and PF cells (right), as indicated by the scale bar (bottom). Data represent mean of three experiments. (B) Data from A, presented as means ± SEM, plotted on a log10 scale. Asterisks indicate that the target amplicon was not detected in the repressed cells.
FIGURE 6.

Western analysis of 5 h 10%–30% glycerol-gradient fractions of lysates from (A) BF and (B) PF CN cells constitutively expressing V5-tagged WT, G163R or G163V KREPB4 alleles, and in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R) for 2 (BF) or 4 (PF) days. Western blots were probed with monoclonal antibodies against editosome proteins KREPA1, KREPA2, KREL1, and KREPA3, and with anti-V5-tag antibody to detect the constitutively expressed V5-tagged WT or mutant KREPB4 proteins.
KREPB4 is required for endonuclease association with editosomes
To assess the effects of KREPB4 loss and substitution on the editing endonucleases, we used in situ tagging to generate PF B4 CN cell lines carrying C-terminally myc-tagged (Oberholzer et al. 2006) endogenous KREN1, KREN2, or KREN3. Lysates were prepared from tagged cells that were grown in the presence or absence of tet for 4 d, and fractionated on glycerol gradients. Western analysis revealed that upon repression in the absence of tet, each of the endonucleases shifted to smaller S values in the gradients, mirroring the shifts in KREPA1, KREPA2, KREL1, and KREPA3 upon loss of KREPB4 (Fig. 7A). Unlike KREPA1, KREPA2, KREL1, and KREPA3, we also observed decreases in the total abundances of each of the endonucleases following KREPB4 repression (Fig. 7A,B). The effect of KREPB4 loss on endonuclease association with editosomes in the gradients was examined further by KREPA2 immunoprecipitation from pooled ∼20S glycerol-gradient fractions 8–11. Input fractions from cells in which KREPB4 was repressed contained reduced amounts of editosome components and complexes compared to those from cells in which KREPB4 was expressed (Fig. 7C). Despite this, we observed some coimmunoprecipitation of KREPA1, KREL1, and KREPA3 with KREPA2 in the absence of KREPB4. In contrast, KREN1, KREN2, and KREN3 did not coimmunoprecipitate following the loss of KREPB4 (Fig. 7C). We cannot discount that lack of endonuclease interactions could be due to general disruption of editosome structure upon loss of KREPB4. However, these complexes were isolated from ∼20S peak glycerol-gradient fractions. Furthermore, the immunoprecipitation of the insertion subcomplex component KREPA1, in addition to KREPA3, by the deletion subcomplex component KREPA2 shows that the complexes do not fully disintegrate in the absence of KREPB4. Thus, these data indicate that KREPB4 is required for the association of the endonucleases with editosome complexes.
FIGURE 7.

(A) Western analysis of 5 h 10%–30% glycerol-gradient fractions 5–13 of lysates from PF CN cells with myc-tagged endogenous KREN1, KREN2, or KREN3, and in which the tet-regulatable WT KREPB4 allele was expressed (E) or repressed (R) for 4 d. Western blots were probed with monoclonal antibodies against the myc-epitiope tag to detect the endonucleases, and against editosome proteins KREPA1, KREPA2, KREL1, and KREPA3. (B) Western analysis of input lysates for glycerol-gradient fractionation described in A. (C) anti-KREPA2 IP of editosomes from pooled ∼20S glycerol-gradient fractions 8–11 shown in A. Immunoprecipitates and input pooled fractions were probed with antibodies as in A. Asterisk indicates the heavy chain of the IP antibody. (D) Anti-KREPA2 IP and (E) anti-V5 IP of editosomes from total lysates of PF CN cells with myc-tagged endogenous KREN1, KREN2, or KREN. Cells were either not expressing KREPB4 (CN), or were exclusively expressing V5-tagged WT, G163V, or G163R KREPB4 alleles. Immunoprecipitates and input lysates were probed with antibodies as in A and with anti-V5-tag antibody to detect the V5-tagged WT or mutant KREPB4 proteins.
To investigate the effect of KREPB4 RNase III mutation on endonuclease association with editosome complexes, we inserted the V5-tagged WT or mutant G163V and G163R KREPB4 mutant alleles into the β-tubulin locus of our CN cell lines that also contained the myc-tagged endonucleases. We then performed immunoprecipitation of KREPA2 and of the V5-epitope tag from total cell lysates. To specifically query the effects of the mutant proteins, we prepared lysates from cells that were grown in the absence of tet for 4 d, and were therefore exclusively expressing V5-tagged WT or mutant alleles. We observed coimmunoprecipitation of KREPB4-V5, KREPA1, KREL1, KREPA3, and each of the endonucleases with KREPA2 from cells that were exclusively expressing WT-KREPB4. In comparison, we observed decreased coimmunoprecipitation of KREPA2, KREPA1, KREL1, KREPA3, and no coimmunoprecipitation of KREN1, KREN2, or KREN3 from cells conditionally lacking KREPB4, or cells that were exclusively expressing KREPB4 G163V or G163R (Fig. 7D). We also observed decreased coimmunoprecipitation of KREPB4-V5 with KREPA2 upon exclusive expression of the mutant alleles. Similarly, upon immunoprecipitation of the V5-tagged KREPB4 G163V or G163R proteins with anti-V5 antibody, we observed a decrease in coimmunoprecipitation of editosome proteins and loss of the endonucleases compared to V5-tagged WT KREPB4 (Fig. 7E). It is interesting to note that while loss of KREPB4 leads to reductions in total abundances of each of the endonucleases (Fig. 7A–E), the G163V and G163R substitutions result in a significant reduction in the abundance of only KREN1 in total cell lysates (Fig. 7D,E), indicating that KREN1 may be particularly sensitive to changes in the function, conformation, or interactions of KREPB4. Together these results are consistent with those of gradient sedimentation displayed in Figure 6 and immunoprecipitation in Figure 7C, indicating that the loss or G163V and G163R substitution of KREPB4 results in loss of the endonucleases from editosome complexes and disruption of editosome integrity. Furthermore, the G163V and G163R substitutions decrease the incorporation of KREPB4 into editosomes, consistent with the requirement of the RNase III domain for the proper interactions of KREPB4 with other editosome proteins.
DISCUSSION
Here we show that KREPB4 is essential for growth, RNA editing, and editosome integrity in both BF and PF cells. The loss of KREPB4 has comparable effects on editosome components and complexes in each life-cycle stage as the loss of KREPB5 (McDermott et al. 2015b), in that we observed a dramatic and specific loss of editosomes and editosome components in BF cells, but retention of components and of complexes with smaller S values in PF cells. Loss of KREPB4 therefore has differential effects on editosome component and complex integrity between life-cycle stages, providing further evidence for intrinsic differences between BF and PF editosomes. These life-cycle stage differences could be based in editosome protein/complex conformation or stability, interactions with other proteins, complexes, or RNAs, and/or post-translational modifications, with consequent effects on assembly and interactions among proteins within the editosomes.
BF loss and PF disruption of editosomes upon knockdown of KREPB4 indicate an important structural role within ∼20S complexes in both life-cycle stages. This is consistent with previous studies that used RNAi to knock down KREPB4 expression in PF (Babbarwal et al. 2007), and with extensive cross-linking between KREPB4 and other core editosome proteins (Supplemental Fig. S1; McDermott et al. 2016) in CXMS. Within the set of proteins common to all types of editosome, KREPB4 cross-linked with both the insertion (KRET2, KREPA1, and KREL2) and deletion (KREPA2) heterotrimeric subcomplexes, and with KREPA4, A5, and A6, that likewise bridged the heterotrimeric subcomplexes. Moreover, KREPB4 also cross-linked to KREPB5, which is similarly essential for the integrity or stability of editosomes. In addition to ∼20S editosomes, we also analyzed the effect of KREPB4 loss on GAP2-containing complexes. This work shows that loss of KREPB4, with subsequent disruption of ∼20S editosomes and of edited mRNAs in BF and PF, does not alter levels or sedimentation of GAP2. GAP2 is essential for RNA editing in BF and PF, and is required for binding and stabilization of guide RNAs within the MRB1 complex, which is thought to functionally cooperate with ∼20S editosomes to achieve fully processive RNA editing. This work is consistent with evidence to date indicating that GAP2 and other components of MRB1 may interact with ∼20S editosomes, but do so in an indirect, e.g., mediated by RNA, or transitory manner, such that the integrities or stabilities of the complexes do not depend upon one another (Panigrahi et al. 2003a; Fisk et al. 2008; Weng et al. 2008; Acestor et al. 2009; Kafková et al. 2012; Aphasizheva et al. 2014).
Previous studies have shown that single amino substitutions in KREPB5 have differential effects on growth, editing, and complexes in BF and PF life-cycle stages (McDermott et al. 2015b). Substitution of key residues predicted to contact RNA within the KREPB5 PUF motif differentially affect editing and editosomes in PF but not BF. The PUF motif and these critical residues are conserved in KREPB4, and we hypothesized that they also have stage-specific functions. However, we show here that their substitution with alanine does not affect growth, editing, or editosomes in either BF or PF. PUF proteins typically bind specific RNA sequences via multiple tandem motifs (Zamore et al. 1997; Wang et al. 2002; Cheong and Hall 2006; Koh et al. 2011). KREPB4 and KREPB5 each have a single PUF motif as do KREN1, KREN2, and KREN3 (Carnes et al. 2012; McDermott et al. 2015b), and we had previously hypothesized that these proteins function together to interact with editing substrates in a fashion that is preferential for specific nucleotide sequences. Our results here indicate that this may not be the case, at least for KREPB4, and that the KREPB5 PUF motif has a specific function, perhaps in the recognition of differentially edited substrates in PF, or in its interactions with other editosome proteins. It is possible that the KREPB4 PUF motif has a specific function in other unassayed life-cycle stages, or a nonspecific or redundant role that can be performed by other PUF-motif containing proteins in the complex. Thus, even though KREPB4 and KREPB5 share equivalent protein domains, the functions of these proteins and their domains are not necessarily equivalent. This is further supported by previous mutational analyses showing that the U1-like zinc finger domain is essential for KREPB4, but not KREPB5 function (Carnes et al. 2012), although further investigation of this domain is required in both BF and PF life-cycle stages. KREPB4 and KREPB5 also have different patterns of cross-linking within PF editosomes in CXMS (Supplemental Fig. S1; McDermott et al. 2016), suggesting that they are in proximity to different proteins and are functionally distinct.
In contrast to the PUF motifs, the degenerate RNase III domains in KREPB4 and KREPB5 are critical for the functions of both proteins in maintaining the structural integrity of editosomes. A highly conserved glycine, G163, in the KREPB4 RNase III domain is essential for growth, editing, association with editosome proteins, and editosome integrity in both BF and PF, but E164, which aligns with a conserved catalytic RNase III residue, is not. Substitutions in the equivalent residues in KREPB5 have similar effects, providing evidence that the RNase III domains in both proteins are noncatalytic but essential for function. Homology modeling of the KREPB4 and B5 RNase III domains places the conserved glycine in the α-helical region important for dimerization, which in turn creates the functional catalytic center in other RNase III endonucleases (MacRae and Doudna 2007; Carnes et al. 2012; Nicholson 2014; McDermott et al. 2015a, 2016). Substitution of this glycine is thus predicted to result in steric interference that prevents normal RNase III dimerization, supporting the hypothesis that noncatalytic RNase III domains in KREPB4 and KREPB5 form heterodimeric structures with the catalytic RNase III domain-containing endonucleases. We cannot discount that the glycine substitutions generally disrupt protein folding, although this seems unlikely given the more conservative nature of G163V. We have also previously identified several editosome proteins, in addition to KREPB4 and KREPB5, with RNase III domains that lack key conserved catalytic residues (McDermott et al. 2016). These include the uniquely associated endonuclease partner proteins KREPB6, KREPB7, and KREPB8 (Carnes et al. 2011; Guo et al. 2012), as well as KREPB9 and KREPB10 (Lerch et al. 2012). It is possible that all different RNase III domain-containing proteins within editosomes, potentially including multiple copies of the same protein as suggested here for KREPB4, form different heterodimers at different times during cycles of editing, or in different life-cycle stages. This organization would be unprecedented among known RNase III proteins, and we hypothesize that it would allow for the recognition of a range of RNA substrates, and for the specific cleavage of numerous different insertion and deletion sites by different editosomes.
This work with previous knockdown and CXMS analyses together indicate that KREPB4 particularly interacts with the catalytic endonucleases. Here we show for the first time that KREPB4, specifically its noncatalytic RNase III domain, is required for the association of the editing endonucleases with ∼20S editosomes, and loss of KREPB4 reduces the steady-state abundances of the endonucleases without significantly affecting the levels of other editosome proteins. In CXMS, the C-terminal domain, and a region flanking the RNase III domain of KREPB4 were in proximity to the RNase III domains of each of the endonucleases, and to the C-terminal domains of KREPB6 and KREPB7 (Supplemental Fig. S1E). Altogether, these data suggest that the heteromeric interactions of the catalytically active RNase III endonucleases with KREPB4 are examples of enzyme–pseudoenzyme interactions. Inactive pseudoenzymes in complex with active enymes have previously been observed in trypanosomes where they influence catalysis of polyamine synthesis (Willert et al. 2007; Nguyen et al. 2013) and protein arginine methylation (Kafková et al. 2017). Such RNase III heterodimers in editosomes could cleave mRNA, and leave gRNA intact, and an analogous RNase III mechanism for nicking one strand of dsRNA has been described, albeit in an artificial system (Conrad et al. 2002; Meng and Nicholson 2008). Detailed biochemical and structural analysis is now required to confirm the nature of heteromeric RNase III interactions within editosomes. The enzymatic cleavage of RNA by RNase III is key to eukaryotic, bacterial, and viral gene expression and regulation (Nicholson 2014). Thus, understanding the different paradigms for how RNA is processed by RNase III enzymes, and how these enzymes can be regulated, including within editosomes, is of fundamental importance.
MATERIALS AND METHODS
Growth of cells in vitro
BF cells were grown in HMI-9 (Hirumi and Hirumi 1989) with 10% FBS at 37°C, 5% CO2. PF cells were grown in SDM-79 (Brun and Schönenberger 1979) with 10% FBS at 27°C. For growth curve analysis, cell density was measured using a Coulter Counter. BF were reseeded at 2 × 105 cells/mL in 10 mL every day, while PF were reseeded at 2 × 106 cells/mL in 10 mL every 2 d. Transfections of BF cell lines with the Amaxa Nucleofector (Lonza), and of PF cell lines with the BTX transfection device (Harvard Apparatus, Inc.), were carried out as previously described (Merritt and Stuart 2013). Concentrations of drugs used for selection and tet-regulated expression of transgenes are as follows. For BF: 2.5 µg/mL G418, 5 µg/mL hygromycin, 2.5 µg/mL phleomycin, 0.5 µg/mL tet, 0.1 µg/mL puromycin. For PF: 15 µg/mL G418, 25 µg/mL hygromycin, 2.5 µg/mL phleomycin, 0.5 µg/mL tet, 1 µg/mL puromycin, 10 µg/mL blasticidin, 25 µg/mL ganciclovir.
Generation of conditional null cell lines
WT KREPB4, including the stop codon and flanked in frame with attB Gateway recombination sites, was PCR amplified using primers described in Supplemental Table S2. BP Clonase II (Thermo Fisher Scientific) was used to transfer the PCR product into the Gateway entry vector pDONR221. LR Clonase II (Thermo Fisher Scientific) was then used to transfer the KREPB4 sequence into the Gateway destination vector pLEW100v5(BLE)GW (Merritt and Stuart 2013), which allows for tet-regulatable expression of WT KREPB4 from the rRNA locus. The resulting pLEW100v5(BLE)-KREPB4 plasmid was linearized with NotI, transfected into PF 29.13, or BF SM427 cells, and transgenic lines were selected by phleomycin resistance. Preparation of DNA constructs and transfections for endogenous KREPB4 allele knockouts in the regulatable PF cell line were generated and carried out as previously described for the generation of the PF KREPB5 CN cell line (McDermott et al. 2015b), using primers described in Supplemental Table S2. The drug selection cassettes in the resulting PF CN cell line were excised using transient expression of Cre recombinase and selection with ganciclovir (Invivogen) as previously described (McDermott et al. 2015b). DNA constructs for KREPB4 allele knockouts in the regulatable BF cell line were generated by PCR amplification of hygromycin and blasticidin drug cassettes from pyrFEKO-HYG (Addgene plasmid #24020; George Cross) and SM06 (Merritt and Stuart 2013). Targeting sequences were also amplified using sequences described in Supplemental Table S2, and combined with drug cassettes in fusion PCR reactions (Merritt and Stuart 2013). The resulting constructs were transfected stepwise into the regulatable BF cell line, transgenic lines selected by hygromycin and blasticidin resistance, and correct insertion of knockout cassettes assessed by PCR.
Epitope tagging
PF KREPB4 CN cell lines carrying myc-tagged versions of either KREN1, KREN2, or KREN3, were generated by in situ tagging with a 3x c-myc epitope tag. Constructs for transfection were generated by a single round of PCR as previously described (Oberholzer et al. 2006), using the pMOTag43M-crelox-PUR plasmid (Addgene plasmid #24032; George Cross), a forward primer containing sequence preceding the stop codon of the target endonuclease, and a reverse primer containing the reverse complement of the 3′UTR of the target endonuclease. Transgenic lines were selected by puromycin resistance. Primers are described in Supplemental Table S2. Transient expression of pLEW100Cre_del_tetO (Addgene plasmid #24019; George Cross) and ganciclovir (Invivogen) selection were used as previously described (McDermott et al. 2015b) to excise and allow reuse of the puromycin drug resistance cassette.
Generation of exclusive expression cell lines
WT KREPB4, lacking the stop codon and flanked in frame with attB Gateway recombination sites, was PCR amplified using primers described in Supplemental Table S2 and transferred into pDONR221. pENTR-KREPB4 (-stop) was used in a LR reaction with the destination vector pHD1344tub(PAC)GW-Cterm3V5, which allows for constitutive expression of C-terminal 3xV5 tagged KREPB4 in the β-tubulin locus. The resulting pHD1344tub(PAC)-KREPB4-Cterm3V5 plasmid was used as a template for site-directed mutagenesis (QuikChange II kit; Agilent) using forward and reverse primers listed in Supplemental Table S2. NotI digested plasmids were transfected into the BF and PF KREPB4 CN cell lines. Transgenic lines were selected by puromycin resistance and constitutive expression of KREPB4-3xV5 was confirmed by Western blot. BF and PF B5 cell lines exclusively expressing mutant alleles were prepared as previously described (McDermott et al. 2015b) using primers in Supplemental Table S2.
Fractionation on glycerol gradients
Glycerol-gradient fractionation was carried out on total cell lysates from 2 × 109 PF or BF cells in the presence or absence of 0.5 µg/mL tet. Cell cultures were counted using a haemocytometer to ensure that only living cells were harvested for fractionation. Following lysis in 650 µL lysis buffer (10 mM Tris–HCl pH 7.2, 10 mM MgCl2, 100 mM KCl, 1% Triton X-100) and centrifugation (12000 rpm, 10 min, 4°C), cleared lysates were loaded onto 11 mL 10%–30% glycerol gradients and centrifuged at 38,000 rpm for 5 h at 4°C in a SW40Ti rotor (Beckman). Twenty-four fractions of 500 µL were collected from top to bottom, flash frozen in liquid nitrogen and stored at −80°C.
Immunoprecipitation
Cleared lysate was prepared by lysis of 2 × 108 cells in 1 mL IPP150 (10 mM Tris–HCl pH 8.0, 150 mM NaCl, 0.1% Nonidet P-40, Roche Complete protease inhibitors) with 1% Triton X-100, followed by centrifugation at 12,000 rpm at 4°C. For each immunoprecipitation, 0.5 mL cleared lysate (1 × 108 cells) was incubated overnight with 1 μL of rabbit antibody (Rockland Immunochemicals; Supplemental Table S3) specific for the V5 epitope tag. Of note, 12.5 μL of Protein G Mag Sepharose Xtra (GE Healthcare) were washed twice with 1 mL of 1× PBS, 0.1% BSA and once with 1 mL IPP150. Beads were then incubated for 4 h rotating at 4°C with cleared lysate/antibody. Anti-KREPA2 immunoprecipitations were alternatively carried out by incubation of 0.5 mL cleared lysate (1 × 108 cells) with goat anti-mouse IgG dynabeads (Therno Fisher Scientific) that had been coated in mAb P1H3-D7 (Supplemental Table S3; Panigrahi et al. 2001a) as previously described (Panigrahi et al. 2001a,b; Ernst et al. 2003). After incubation, the supernatant was removed, and beads were washed four times with 1 mL IPP150. Complexes bound to beads were eluted with 100 μL of 2 × SDS sample buffer, heated for 5 min at 95°C. Immunoprecipitations from pooled ∼20S peak glycerol-gradient fractions were carried out as above, except inputs were 100 μL each of fractions 8–11 combined and brought to a total volume of 500 μL with IPP150 plus protease inhibitors.
SDS-PAGE and Western blotting
SDS-PAGE loading buffer was added to cleared whole cell lysates, or to samples containing purified protein complexes and resolved on 10% SDS-PAGE gels (Criterion Tris–HCl, Bio-Rad). For Western analysis, resolved proteins were transferred to Immobilon-P PVDF membranes (Millipore) and probed using antibodies described in Supplemental Table S3. Blots were developed with an enhanced chemiluminescence kit (Thermo Scientific) according to the manufacturer's instructions, and imaged using the FluorChem E system (ProteinSimple) or X-ray film (Kodak).
RNA isolation and RT-qPCR analysis
Total RNA was harvested using TRIzol and treated with TURBO DNase (Life Technologies) according to manufacturer's instructions. RNA integrity was confirmed using a RNA nanochip on a BioAnalyzer (Agilent Technologies). Two micrograms of total RNA was reverse-transcribed using TaqMan Reverse Transcription Reagents and MultiScribe Reverse Transcriptase (Life Technologies), preamplified in multiplex specific target amplification (STA) reactions using TaqMan PreAmp Master Mix (Life Technologies), and treated with Exonuclease I (NEB). The abundance of reference, never-edited, pre-edited, and edited transcript cDNAs were then analyzed by high-throughput real-time PCR on the BioMark HD system as previously described (McDermott et al. 2015b). Primers are described in Supplemental Table S2. Calculations of RNA levels in samples following tet withdrawal, relative to the presence of tet, were done using the 2 [−ΔΔC(T)] method (Livak and Schmittgen 2001) using TERT as an internal reference. Technical duplicates of each cDNA sample were assayed for each target and internal reference per experiment and C(T) data averaged before performing the 2 [−ΔΔC(T)] calculation. Experiments were repeated using two or three biological replicates.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Jason Carnes for comments on the manuscript. This work was supported by National Institutes of Health grant R01AI014102 to K.S.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062786.117.
REFERENCES
- Acestor N, Panigrahi AK, Carnes J, Ziková A, Stuart KD. 2009. The MRB1 complex functions in kinetoplastid RNA processing. RNA 15: 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammerman ML, Downey KM, Hashimi H, Fisk JC, Tomasello DL, Faktorová D, Kafková L, King T, Lukes J, Read LK. 2012. Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res 40: 5637–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizhev R, Aphasizheva I. 2011. Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip Rev RNA 2: 669–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Zhang L, Wang X, Kaake RM, Huang L, Monti S, Aphasizhev R. 2014. RNA binding and core complexes constitute the U-insertion/deletion editosome. Mol Cell Biol 34: 4329–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Maslov D, Qian Y, Huang L, Wnag Q, Costello C, Aphasizhev R. 2015. Ribosome-associated PPR proteins function as translational activators in mitochondria of trypanosomes. Mol Microbiol 99: 1043–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbarwal VK, Fleck M, Ernst NL, Schnaufer A, Stuart K. 2007. An essential role of KREPB4 in RNA editing and structural integrity of the editosome in Trypanosoma brucei. RNA 13: 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benne R, Van den Burg J, Brakenhoff JP, Sloof P, Van Boom JH, Tromp MC. 1986. Major transcript of the frameshifted coxII gene from trypanosome mitochondria contains four nucleotides that are not encoded in the DNA. Cell 46: 819–826. [DOI] [PubMed] [Google Scholar]
- Bhat GJ, Koslowsky DJ, Feagin JE, Smiley BL, Stuart K. 1990. An extensively edited mitochondrial transcript in kinetoplastids encodes a protein homologous to ATPase subunit 6. Cell 61: 885–894. [DOI] [PubMed] [Google Scholar]
- Blum B, Simpson L. 1990. Guide RNAs in kinetoplastid mitochondria have a nonencoded 3′ oligo(U) tail involved in recognition of the preedited region. Cell 62: 391–397. [DOI] [PubMed] [Google Scholar]
- Blum B, Bakalara N, Simpson L. 1990. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 60: 189–198. [DOI] [PubMed] [Google Scholar]
- Brun R, Schönenberger. 1979. Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta Trop 36: 289–292. [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K. 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci 102: 16614–16619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol 28: 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Soares CZ, Wickham C, Stuart K. 2011. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J Biol Chem 286: 19320–19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Schnaufer A, McDermott SM, Domingo G, Proff R, Steinberg AG, Kurtz I, Stuart K. 2012. Mutational analysis of Trypanosoma brucei editosome proteins KREPB4 and KREPB5 reveals domains critical for function. RNA 18: 1897–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, McDermott S, Anupama A, Oliver BG, Sather DN, Stuart K. 2017. In vivo cleavage specificity of Trypanosoma brucei editosome endonucleases. Nucleic Acids Res 45: 4667–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong CG, Hall TM. 2006. Engineering RNA sequence specificity of Pumilio repeats. Proc Natl Acad Sci 103: 13635–13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Schmitt JG, Evguenieva-Hackenberg E, Klug G. 2002. One functional subunit is sufficient for catalytic activity and substrate specificity of Escherichia coli endoribonuclease III artificial heterodimers. FEBS Lett 518: 93–96. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst NL, Panicucci B, Igo RP Jr, Panigrahi AK, Salavati R, Stuart K. 2003. TbMP57 is a 3′ terminal uridylyl transferase (TUTase) of the Trypanosoma brucei editosome. Mol Cell 11: 1525–1536. [DOI] [PubMed] [Google Scholar]
- Ernst NL, Panicucci B, Carnes J, Stuart K. 2009. Differential functions of two editosome exoUases in Trypanosoma brucei. RNA 15: 947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagin JE, Stuart K. 1988. Developmental aspects of uridine addition within mitochondrial transcripts of Trypanosoma brucei. Mol Cell Biol 8: 1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feagin JE, Jasmer DP, Stuart K. 1987. Developmentally regulated addition of nucleotides within apocytochrome b transcripts in Trypanosoma brucei. Cell 49: 337–345. [DOI] [PubMed] [Google Scholar]
- Fisk JC, Ammerman ML, Presnyak V, Read LK. 2008. TbRGG2, an essential RNA editing accessory factor in two Trypanosoma brucei life cycle stages. J Biol Chem 283: 23016–23025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gone S, Alfonso-Prieto M, Paudyal S, Nicholson AW. 2016. Mechanism of ribonuclease III catalytic regulation by serine phosphorylation. Sci Rep 6: 25448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Ernst NL, Stuart KD. 2008. The KREPA3 zinc finger motifs and OB-fold domain are essential for RNA editing and survival of Trypanosoma brucei. Mol Cell Biol 28: 6939–6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Carnes J, Ernst NL, Winkler M, Stuart K. 2012. KREPB6, KREPB7, and KREPB8 are important for editing endonuclease function in Trypanosoma brucei. RNA 18: 308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimi H, Ziková A, Panigrahi AK, Stuart KD, Lukes J. 2008. TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14: 970–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirumi H, Hirumi K. 1989. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol 75: 985–989. [PubMed] [Google Scholar]
- Kafková L, Ammerman ML, Faktorová D, Fisk JC, Zimmer SL, Sobotka R, Read LK, Lukes J, Hashimi H. 2012. Functional characterization of two paralogs that are novel RNA binding proteins influencing mitochondrial transcripts of Trypanosoma brucei. RNA 18: 1846–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafková L, Debler EW, Fisk JC, Jain K, Clarke SG, Read LK. 2017. The major protein arginine methyltransferase in Trypanosoma brucei functions as an enzyme-prozyme complex. J Biol Chem 292: 2089–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YY, Wang Y, Qiu C, Opperman L, Gross L, Tanaka Hall TM, Wickens M. 2011. Stacking interactions in PUF-RNA complexes. RNA 17: 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowsky DJ, Bhat GJ, Perrollaz AL, Feagin JE, Stuart K. 1990. The MURF3 gene of T. brucei contains multiple domains of extensive editing and is homologous to a subunit of NADH dehydrogenase. Cell 62: 901–911. [DOI] [PubMed] [Google Scholar]
- Koslowsky DJ, Riley GR, Feagin JE, Stuart K. 1992. Guide RNAs for transcripts with developmentally regulated RNA editing are present in both life cycle stages of Trypanosoma brucei. Mol Cell Biol 12: 2043–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerch M, Carnes J, Acestor N, Guo X, Schnaufer A, Stuart K. 2012. Editosome accessory factors KREPB9 and KREPB10 in Trypanosoma brucei. Eukaryot Cell 11: 832–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- MacRae IJ, Doudna JA. 2007. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol 17: 138–145. [DOI] [PubMed] [Google Scholar]
- McDermott SM, Carnes J, Stuart K. 2015a. Identification by random mutagenesis of functional domains in KREPB5 that differentially affect RNA editing between life cycle stages of Trypanosoma brucei. Mol Cell Biol 35: 3945–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott SM, Guo X, Carnes J, Stuart K. 2015b. Differential editosome protein function between life cycle stages of Trypanosoma brucei. J Biol Chem 290: 24914–24931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott SM, Luo J, Carnes J, Ranish JA, Stuart K. 2016. The architecture of Trypanosoma brucei editosomes. Proc Natl Acad Sci 113: E6476–E6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng W, Nicholson AW. 2008. Heterodimer-based analysis of subunit and domain contributions to double-stranded RNA processing by Escherichia coli RNase III in vitro. Biochem J 410: 39–48. [DOI] [PubMed] [Google Scholar]
- Merritt C, Stuart K. 2013. Identification of essential and non-essential protein kinases by a fusion PCR method for efficient production of transgenic Trypanosoma brucei. Mol Biochem Parasitol 190: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen S, Jones DC, Wyllie S, Fairlamb AH, Phillips MA. 2013. Allosteric activation of trypanosomatid deoxyhypusine synthase by a catalytically dead paralog. J Biol Chem 288: 15256–15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AW. 2014. Ribonuclease III mechanisms of double-stranded RNA cleavage. Wiley Interdiscipl Rev RNA 5: 31–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberholzer M, Morand S, Kunz S, Seebeck T. 2006. A vector series for rapid PCR-mediated C-terminal in situ tagging of Trypanosoma brucei genes. Mol Biochem Parasitol 145: 117–120. [DOI] [PubMed] [Google Scholar]
- Palmeri A, Gherardini PF, Tsigankov P, Ausiello G, Späth GF, Zilberstein D, Helmer-Citterich M. 2011. PhosTryp: a phosphorylation site predictor specific for parasitic protozoa of the family trypanosomatidae. BMC Genomics 12: 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Gygi SP, Ernst NL, Igo RP Jr, Palazzo SS, Schnaufer A, Weston DS, Carmean N, Salavati R, Aebersold R, et al. 2001a. Association of two novel proteins, TbMP52 and TbMP48, with the Trypanosoma brucei RNA editing complex. Mol Cell Biol 21: 380–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Carmean N, Igo RP Jr, Gygi SP, Ernst NL, Palazzo SS, Weston DS, Aebersold R, Salavati R, et al. 2001b. Four related proteins of the Trypanosoma brucei RNA editing complex. Mol Cell Biol 21: 6833–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Allen TE, Stuart K, Haynes PA, Gygi SP. 2003a. Mass spectrometric analysis of the editosome and other multiprotein complexes in Trypanosoma brucei. J Am Soc Mass Spectrom 14: 728–735. [DOI] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Ernst NL, Wang B, Carmean N, Salavati R, Stuart K. 2003b. Identification of novel components of Trypanosoma brucei editosomes. RNA 9: 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ernst NL, Domingo GJ, Fleck M, Salavati R, Stuart KD. 2006. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 12: 1038–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Zikova A, Dalley RA, Acestor N, Ogata Y, Anupama A, Myler PJ, Stuart KD. 2008. Mitochondrial complexes in Trypanosoma brucei: a novel complex and a unique oxidoreductase complex. Mol Cell Proteomics 7: 534–545. [DOI] [PubMed] [Google Scholar]
- Pollard VW, Hajduk SL. 1991. Trypanosoma equiperdum minicircles encode three distinct primary transcripts which exhibit guide RNA characteristics. Mol Cell Biol 11: 1668–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard VW, Rohrer SP, Michelotti EF, Hancock K, Hajduk SL. 1990. Organization of minicircle genes for guide RNAs in Trypanosoma brucei. Cell 63: 783–790. [DOI] [PubMed] [Google Scholar]
- Read LK, Lukes J, Hashimi H. 2016. Trypanosome RNA editing: the complexity of getting U in and taking U out. Wiley interdiscip Rev RNA 7: 33–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley GR, Myler PJ, Stuart K. 1995. Quantitation of RNA editing substrates, products and potential intermediates: implications for developmental regulation. Nucleic Acids Res 23: 708–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salavati R, Ernst NL, O'Rear J, Gilliam T, Tarun S Jr, Stuart K. 2006. KREPA4, an RNA binding protein essential for editosome integrity and survival of Trypanosoma brucei. RNA 12: 819–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnaufer A, Panigrahi AK, Panicucci B, Igo RP Jr, Wirtz E, Salavati R, Stuart K. 2001. An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science 291: 2159–2162. [DOI] [PubMed] [Google Scholar]
- Shapiro TA, Englund PT. 1995. The structure and replication of kinetoplast DNA. Annu Rev Microbiol 49: 117–143. [DOI] [PubMed] [Google Scholar]
- Shaw JM, Feagin JE, Stuart K, Simpson L. 1988. Editing of kinetoplastid mitochondrial mRNAs by uridine addition and deletion generates conserved amino acid sequences and AUG initiation codons. Cell 53: 401–411. [DOI] [PubMed] [Google Scholar]
- Souza AE, Myler PJ, Stuart K. 1992. Maxicircle CR1 transcripts of Trypanosoma brucei are edited and developmentally regulated and encode a putative iron-sulfur protein homologous to an NADH dehydrogenase subunit. Mol Cell Biol 12: 2100–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza AE, Shu HH, Read LK, Myler PJ, Stuart KD. 1993. Extensive editing of CR2 maxicircle transcripts of Trypanosoma brucei predicts a protein with homology to a subunit of NADH dehydrogenase. Mol Cell Biol 13: 6832–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K, Feagin JE. 1992. Mitochondrial DNA of kinetoplastids. Int Rev Cytol 141: 65–88. [DOI] [PubMed] [Google Scholar]
- Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. 2005. Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30: 97–105. [DOI] [PubMed] [Google Scholar]
- Sturm NR, Simpson L. 1990. Kinetoplast DNA minicircles encode guide RNAs for editing of cytochrome oxidase subunit III mRNA. Cell 61: 879–884. [DOI] [PubMed] [Google Scholar]
- Trotter JR, Ernst NL, Carnes J, Panicucci B, Stuart K. 2005. A deletion site editing endonuclease in Trypanosoma brucei. Mol Cell 20: 403–412. [DOI] [PubMed] [Google Scholar]
- Wang X, McLachlan J, Zamore PD, Hall TM. 2002. Modular recognition of RNA by a human pumilio-homology domain. Cell 110: 501–512. [DOI] [PubMed] [Google Scholar]
- Weng J, Aphasizheva I, Etheridge RD, Huang L, Wang X, Falick AM, Aphasizhev R. 2008. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol Cell 32: 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert EK, Fitzpatrick R, Phillips MA. 2007. Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc Natl Acad Sci 104: 8275–8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthey EA, Schnaufer A, Mian IS, Stuart K, Salavati R. 2003. Comparative analysis of editosome proteins in trypanosomatids. Nucleic Acids Res 31: 6392–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamore PD, Williamson JR, Lehmann R. 1997. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 3: 1421–1433. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.