

Abstract
Seryl-tRNA synthetase (SerRS) attaches L-serine to the cognate serine tRNA (tRNASer) and the noncognate selenocysteine tRNA (tRNASec). The latter activity initiates the anabolic cycle of selenocysteine (Sec), proper decoding of an in-frame Sec UGA codon, and synthesis of selenoproteins across all domains of life. While the accuracy of SerRS is important for overall proteome integrity, it is its substrate promiscuity that is vital for the integrity of the selenoproteome. This raises a question as to what elements in the two tRNA species, harboring different anticodon sequences and adopting distinct folds, facilitate aminoacylation by a common aminoacyl-tRNA synthetase. We sought to answer this question by analyzing the ability of human cytosolic SerRS to bind and act on tRNASer, tRNASec, and 10 mutant and chimeric constructs in which elements of tRNASer were transposed onto tRNASec. We show that human SerRS only subtly prefers tRNASer to tRNASec, and that discrimination occurs at the level of the serylation reaction. Surprisingly, the tRNA mutants predicted to adopt either the 7/5 or 8/5 fold are poor SerRS substrates. In contrast, shortening of the acceptor arm of tRNASec by a single base pair yields an improved SerRS substrate that adopts an 8/4 fold. We suggest that an optimal tertiary arrangement of structural elements within tRNASec and tRNASer dictate their utility for serylation. We also speculate that the extended acceptor-TΨC arm of tRNASec evolved as a compromise for productive binding to SerRS while remaining the major recognition element for other enzymes involved in Sec and selenoprotein synthesis.
Keywords: SerRS, selenocysteine, serine, tRNA, aminoacylation, tRNA recognition, serylation
INTRODUCTION
Aminoacyl-tRNA synthetases (AaRSs) are ubiquitously expressed enzymes that play an essential role in the maintenance of the genetic code by coupling proteinogenic amino acids to their cognate tRNA(s) (Ibba and Söll 2000). The accuracy of AaRSs, exemplified by their ability to select correct substrates, is vital for the fidelity of ribosome-catalyzed mRNA translation as the ribosome cannot assess whether the charged tRNA carries its cognate amino acid (Ibba and Söll 1999). While the catalytic and editing sites of AaRSs select cognate amino acids and exclude near- and noncognate residues (Bullwinkle and Ibba 2014), tRNA selection relies on an intricate network of interactions formed at the AaRS–tRNA molecular interface. These interactions may serve to “read” specific sequences such as the anticodon sequence, the discriminator base, and/or a particular base pair(s) (Giegé et al. 1998). Alternatively, they could also mediate recognition of elements that define the local and/or global shape of the tRNA molecule. Typically, the anticodon loop and anticodon sequence are described as the major recognition elements, without which a productive AaRS–tRNA complex cannot form (Giegé et al. 1998).
However, several AaRSs never acquired the ability to “read” the anticodon sequence. Among those, particularly interesting is seryl-tRNA synthetase (SerRS)—the enzyme that couples L-serine to the cognate serine tRNA (tRNASer) and to the noncognate selenocysteine tRNA (tRNASec). While under normal circumstances such misacylation would be detrimental, in this instance the “error” is essential as it initiates tRNA-dependent synthesis of the amino acid selenocysteine (Sec). The final reaction product of Sec synthesis, Sec–tRNASec, facilitates proper recoding of an in-frame opal Stop codon into a signal for Sec incorporation in a relatively small subset of proteins called selenoproteins. Paradoxically, it is the expanded substrate specificity of SerRS, and not its presumed stringent selectivity that is pivotal for synthesis of functional selenoproteins and selenoenzymes. The significance of SerRS substrate promiscuity is further accentuated by the fact that serylation of tRNASec is the only absolutely conserved reaction in the anabolic cycle of Sec across kingdoms (Yuan et al. 2006, 2010; Xu et al. 2007). As mentioned above, selenoproteins usually represent a minor fraction of the cellular proteome (Lobanov et al. 2006, 2009; Mariotti et al. 2012), but nonetheless are vital for basic cellular processes, particularly those relying on redox reactions (Brigelius-Flohé and Flohé 2016; Reich and Hondal 2016; Steinbrenner et al. 2016). Also, mutations in genes encoding selenoproteins, Sec-synthetic enzymes, and RNA and protein factors supporting decoding of the Sec UGA codon cause a variety of pathologies affecting diverse organ systems (Rayman 2012; Schmidt and Simonović 2012; Hatfield et al. 2014). Taken together, these data suggest that proper synthesis and incorporation of Sec into a nascent selenoprotein is a fundamental biological process.
Although tRNASec and tRNASer are considered structural homologs, supposedly because of their extended variable arms, their structures are fundamentally different. tRNASer adopts the canonical 7/5 fold (7 and 5 refer to the number of base pairs in the acceptor and TψC arms, respectively) and harbors a 12 base pair (bp)-long acceptor-TψC “helix.” In contrast, tRNASec assumes either a 9/4 fold in eukaryotes and archaea or an 8/5 fold in bacteria, resulting in a 13-bp-long acceptor-TψC arm (Itoh et al. 2009, 2013a,b; Palioura et al. 2009; Chiba et al. 2010; Sherrer et al. 2011). Previous work established that the discriminator base G73 and the orientation, but not sequence, of the variable arm are the minimal recognition elements required by the SerRS dimer (Sampson and Saks 1993; Wu and Gross 1993; Asahara et al. 1994; Heckl et al. 1998; Mizutani et al. 1998; Gruic-Sovulj et al. 2006; Rokov-Plavec et al. 2013). The studies also concluded that the acceptor arm is of lesser importance, that the T- and D-arms are needed to close the L-shaped structure of the tRNA, and that the anticodon arm is of no importance for serylation. These results are largely in agreement with X-ray crystal structures of the bacterial SerRS–tRNASer (Biou et al. 1994) and human SerRS–tRNASec (Wang et al. 2015) binary complexes, which also showed that the α-helical N-terminal domain (NTD) of SerRS is in close contact with the variable arm. Other biochemical and enzymatic studies identified “cryptic” recognition elements in the acceptor arm of bacterial tRNASer (Saks and Sampson 1996), and elements that were required to switch identity of tRNALeu (Normanly et al. 1986, 1992) and tRNAVal (Achsel and Gross 1993) to tRNASer.
We sought to extend those studies by further analyzing the ability of human cytosolic SerRS to bind and act on tRNASer and tRNASec—very close structural homologs (Fig. 1). Both tRNAs harbor G73 as the discriminator base, a long-variable arm, equivalent sequences in the D-loop and the T stem, and two equivalent G–C pairs in the acceptor arm (G1:C72 in both tRNAs, and G5:C68 and G4:C69 in tRNASec and tRNASer, respectively [Fig. 1]). The significance of these elements for serylation by SerRS is well established in different systems. However, tRNASec and tRNASer are also quite distinct (“we shall not discuss anticodon sequences for they are irrelevant from the SerRS standpoint”). As mentioned, human tRNASec adopts a 9/4 fold, whereas tRNASer folds into a canonical 7/5 structure (Fig. 1). Consequently, the spatial arrangement of stems and loops in the two tRNAs is quite different. For instance, an extra base pair causes all groups in tRNASec to be translated by 3.4 Å and rotated by ∼33° when compared to tRNASer. In addition, tRNASec harbors an enlarged D-arm when compared to tRNASer (6 bp versus 4 bp). Also, instead of the highly conserved U8 present in tRNASer, tRNASec carries A8 that is splayed out toward solution (Itoh et al. 2009; Palioura et al. 2009). This is a notable modification, as U8 stabilizes the tertiary tRNA structure through H-bonding between the D- and T-arms, and a comparable structure-stabilizing interaction is absent from tRNASec (Itoh et al. 2009; Palioura et al. 2009). Lastly, tRNASec carries the G19:U20:C56 base triple that closes the D- and T-loops. The role of this element for tRNASec recognition by Sec-synthetic enzymes, including SerRS, is still unclear (Sherrer et al. 2008; Itoh et al. 2009; Palioura et al. 2009; Chiba et al. 2010; Wang et al. 2015).
FIGURE 1.
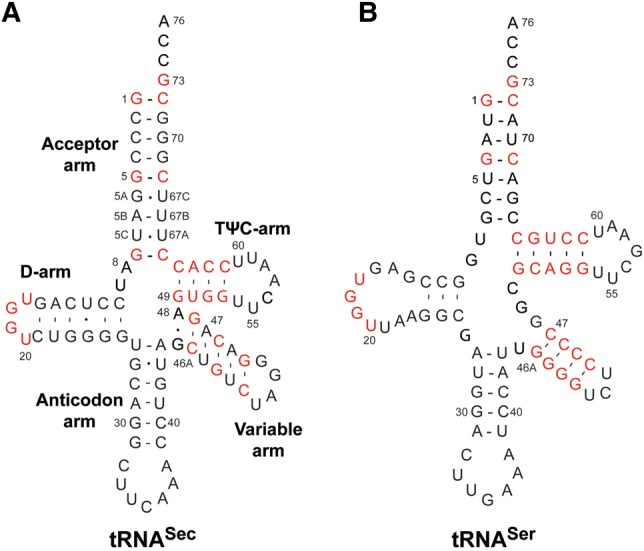
Comparison of human tRNASec and tRNASer. Human tRNASec (A) and tRNASer (B) are shown in a standard cloverleaf representation. tRNASec adopts a distinct 9/4 fold which stands in contrast to the canonical 7/5 fold of tRNASer. Conserved and similar sequences in the acceptor-, D-, variable-, and TψC arms are highlighted in red.
Given the structural divergence between the two tRNA species, we asked if human SerRS prefers one tRNA, and if so, what structural elements determine such preference? We also wondered whether transposition of particular elements from tRNASer into tRNASec alters the utility of engineered chimeric tRNASec/Ser constructs for serylation by SerRS. In this study, we assessed the significance for the initial recognition and utility in the serylation reaction of the global tRNA fold, the length of the acceptor-TψC, identity of the eighth nucleotide, and the length and sequence of the D-arm and D-loop. Our approach consisted of engineering tRNASec/Ser chimeric constructs in which specific elements from tRNASer were transposed into tRNASec. Then, the ability of SerRS to bind and serylate the tRNAs was monitored along with assays that probed the structural integrity of the tRNA constructs. Our results show that human SerRS only subtly prefers tRNASer. The tRNA discrimination occurs at the level of the serylation reaction and not at the level of the initial encounter. While all chimeras and mutants bind to SerRS, only those predicted to adopt a novel 8/4 fold do so in a productive manner. Furthermore, irrespective of the identity of the eighth nucleotide, the 8/4 mutants are better substrates for serylation when compared to tRNASec, but still inferior to tRNASer. Lastly, SerRS does not tolerate the 8/5 fold, the 7/5 fold, and the D-arm and D-loop from tRNASer in the context of human tRNASec. We speculate that the overall shape of tRNASec and tRNASer and spatial arrangement of variable and/or acceptor arms dictate the efficiency of the serylation reaction. We also suggest that SerRS tolerates the 13-bp-long acceptor-TψC arm within the tRNASec frame but with certain restrictions, which could be kingdom-specific. This extended element probably evolved as a compromise for productive binding to SerRS while remaining the major recognition motif for other enzymes in the Sec cycle.
RESULTS
Design of tRNASec/Ser mutants and chimeras
We began our study with the premise that tRNASer, being a cognate tRNA, is a preferred SerRS substrate. Our desire was to utilize some of the tRNASer-specific elements to engineer a better tRNASec-based substrate for SerRS. As a collateral, we could also investigate how certain structural motifs operate within the context of the body or frame of the “other” tRNA (Table 1; Fig. 2). First, the U6:U67 base pair was removed from tRNASec because such a pair is not present in tRNASer. This gave rise to Mutant 1, which is predicted to adopt an 8/4 fold (Fig. 2). The only difference between Mutant 1 and 2 is in the A8→U mutation (Table 1; Fig. 2). The concern was that the absence of U8 (or presence of A8) could compromise the ability of the tRNA to properly fold. To assess the impact of this change on serylation, this mutation was introduced in Mutant 2. This approach was kept throughout our study and thus half of the constructs harbor the A8→U mutation (Table 1; Fig. 2). In either case, Mutants 1 and 2 served to probe whether a shorter acceptor arm and the identity of the eighth nucleotide are critical for serylation. Further, insertion of the G51:C63 pair into the TψC arm of Mutants 1 and 2 gave rise to Mutants 3 and 4, respectively, both of which are predicted to assume an 8/5 fold (Fig. 2). These particular constructs helped assess if human SerRS tolerates the bacterial-like fold of tRNASec. Next, removing the G5a:U67b pair from the acceptor arm generated Mutants 5 and 6, which more closely resemble tRNASer (Fig. 2). These mutants served to test if human SerRS prefers the canonical 7/5 fold even when in the context of a tRNASec body. In addition, Mutants 5 and 6 helped addressing the ongoing question of whether the global fold (i.e. the length of the acceptor-TψC arm) or specific motifs in tRNASec (i.e., the G19:U20:C56 base triple or G73) are the most critical recognition motifs for Sec-synthetic enzymes (Chiba et al. 2010; Itoh et al. 2013a; Wang et al. 2015). Lastly, the entire D-arm and/or D-loop from tRNASer were transplanted into tRNASec and this yielded Chimeras 7 and 9, respectively (Fig. 2). Introduction of U8 into both mutants gave rise to Chimeras 8 and 10 (Fig. 2). In these chimeras, which were predicted to adopt a 9/4 structure, the length of the D-stem was kept at 3 bp as in WT tRNASer, but the exact sequence was varied. The D-stem chimeras permitted evaluation of the significance of the size and/or sequence of the D-arm of tRNASer in the context of a tRNASec frame for serylation.
TABLE 1.
tRNASec mutants and tRNASec/Ser chimeras used in the study
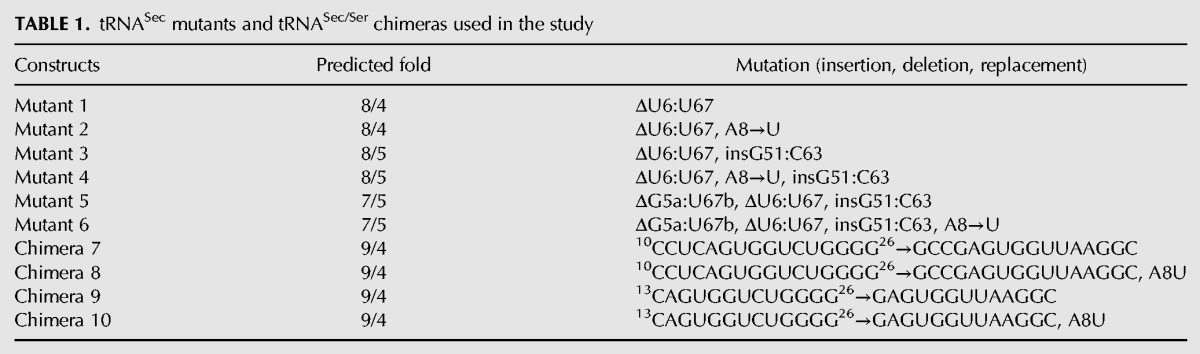
FIGURE 2.

tRNASec mutants and tRNASec/Ser chimeras used in the study. (A) Secondary structure diagram of human tRNASec. Nucleotides to be mutated or replaced with elements from tRNASer are demarcated with boxes, while an arrow points to a position where the G51:C63 base pair is to be inserted in the TψC arm. (B–E) Secondary structure diagrams showing only elements that are altered in particular mutants or chimeric constructs. The remainder of each construct is the same as WT tRNASec. Sequences derived from human tRNASer are in bold red and the G–C insertion in the TψC arm is in bold light blue. Diagrams above each tRNA construct designate the predicted fold with the number of base pairs in the acceptor- (pink), D- (green), and TψC (cyan) arms shown. The anticodon and variable arms are gray and yellow, respectively; the CCA end is not shown for brevity. The tRNA fold is determined by the base pairs in the acceptor-TψC “helix.” Hence, WT tRNASec (A) and Chimeras 7–10 (E) adopt the 9/4-fold, Mutants 1 and 2 fold into the 8/4 structure (B), Mutants 3 and 4 adopt the bacterial-like 8/5 fold (C), and Mutants 5 and 6 are representative of the canonical 7/5 fold (D).
Mutations do not disrupt the structure of the tRNASec/Ser mutants and chimeras
Although our tRNAs were purified under nondenaturing conditions, we performed several experiments to ensure the mutant and chimeric tRNASer/Sec constructs adopted the predicted structures (folds). We decided to probe the structural integrity of each construct by monitoring thermal unfolding and the ability of each tRNA to bind to SerRS in solution.
To assess the effect of mutations on tRNA folding, we performed a melt curve analysis based on the intercalation of the SYBR Green I fluorophore. SYBR Green I fluoresces only when bound to double-stranded nucleic acids. Upon thermal denaturation of the nucleic acid, the fluorophore is freed and the fluorescence signal decreases sharply at the melting temperature (Tm) of the nucleic acid. Plotting the negative derivative of such decrease against the temperature yields the melting or unfolding curve for a given nucleic acid. Any significant changes in folding and stability of tRNASec/Ser constructs due to the introduced mutations would likely alter the corresponding melting curve profiles. The melting curve of WT tRNASec features two major peaks that presumably correspond to distinct unfolding events occurring at Tm1 of +57°C and Tm2 of +64°C (Table 2; Fig. 3A). This is quite different from a curve obtained from the RNase A-treated tRNASec that contains a single Tm value of +58°C (Table 2; Fig. 3), suggesting that the fold contributing to this event is resistant to RNase activity. Melting curves of the other tRNASec/Ser mutants and chimeras feature two maxima whose position, but not the size, is consistent with the WT curve.
TABLE 2.
Melting temperatures (Tm) of human tRNASec/Ser mutants and chimeras used in the study
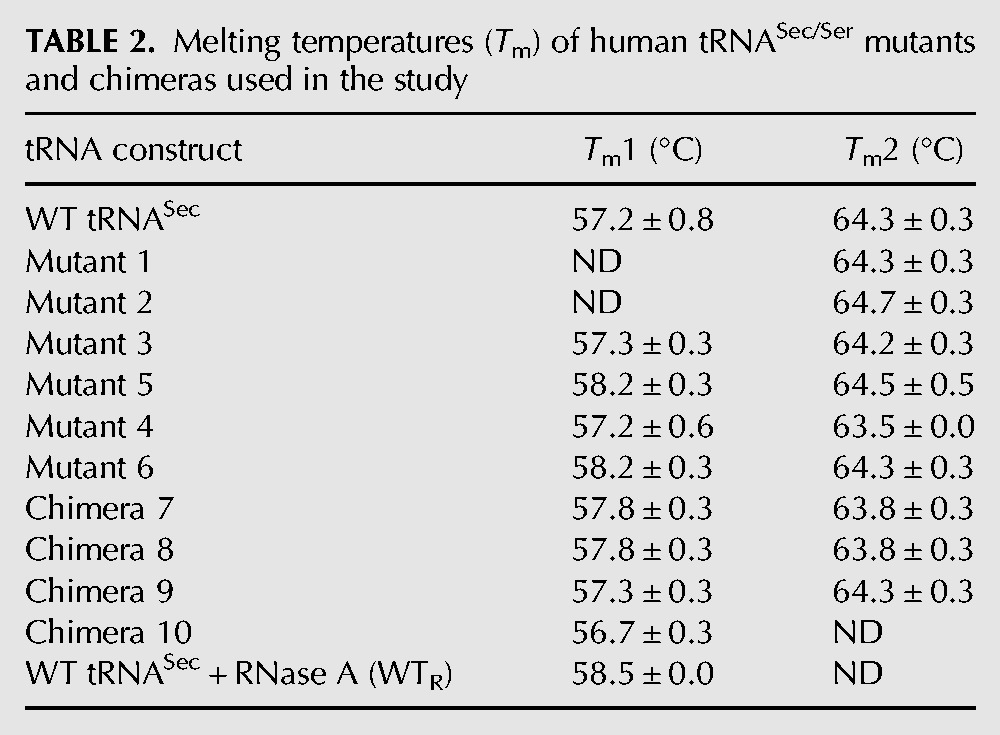
FIGURE 3.

Thermal unfolding shows that tRNASec/Ser mutants and chimeras exhibit a similar unfolding profile as the WT tRNASec. (A–C) A negative first derivative of melting curves for the acceptor- (A) and acceptor-TΨC arm mutants (B), and the D-arm chimeras (C). WT tRNASec (blue) exhibits a melting curve with two peaks at +57 (Tm1) and +64.5°C (Tm2). The RNase A-treated WT tRNASec has a different unfolding curve with a single Tm at +58.5°C (gray). While Mutants 1–6 (A,B) and Chimeras 7 and 8 (C) display similar unfolding profiles as the WT tRNA, Chimeras 9 and 10 (C) differ in that Tm1 is the major peak and Tm2 represents a shoulder in the curve. This suggests that either the stability of these constructs is decreased or that simply their folding pathway is altered. Each melting curve is the average of three independent measurements. (D) The TBU gel analysis revealed that WT and tRNASec/Ser mutants/chimeras used in thermal folding experiments consist of a single species. No band was observed in the RNase A-treated WT tRNASec sample (WTR).
To ensure that the two peaks represent two unfolding events as suggested, we analyzed tRNAs on a denaturing gel. We showed that each tRNA is pure and devoid of degraded or contaminating fragments (Fig. 3D). In spite of the overall similarity, there are specific differences among individual melting curves. The Tm for the first unfolding event could not be determined with high confidence for Mutants 1 and 2 (Fig. 3A). Conversely, Chimeras 9 and 10 did not appear to even undergo the second event (Fig. 3C), arguing that the D-arm modifications destabilize these constructs relative to the WT tRNASec. Also, based on the unfolding trajectories, Mutants 5 and 6 represent intermediate cases between these extremes (Fig. 3B), while the remainder (e.g., Mutants 3 and 4, and Chimeras 7 and 8) closely resembles WT tRNASec (Fig. 3B,C). In conclusion, the melt curve analysis indicates that tRNAs under study fold in a manner similar to WT tRNASec, as illustrated by similar Tm values. Nonetheless, changes in the D- (Chimeras 9, 10) and the acceptor arms (Mutants 1, 2) exerted distinct effects on the tRNASec folding trajectory. Also, with the exception of Chimeras 9 and 10, the second unfolding event occurring at +64°C seems to be the dominant one.
Besides unfolding, it is plausible that mutations caused misfolding of the mutant tRNAs. To rule out this proposition, we tested the ability of tRNAs to form a stable complex with SerRS in solution by a electrophoretic mobility shift assay (EMSA). We showed that addition of SerRS to any tRNA alters tRNA mobility, resulting in formation of a new, single band running close to the top of the gel (Fig. 4A,B). Subsequent protein staining confirmed that the upper band contains SerRS (Fig. 4C,D). Because of its slower electrophoretic mobility compared to SerRS alone (Fig. 4C,D), we conclude that the upper band represents a stable SerRS–tRNA binary complex. Moreover, the size and relative intensity of the shifted band appears similar irrespective of the tRNA used, suggesting a consistent nature of the binary complex. Taken together, these data strongly argue that tRNASec/Ser mutants and chimeras adopted predicted structures in solution, thus providing further credence for our binding and activity assay results.
FIGURE 4.

EMSA reveals that all tRNASec/Ser mutants and chimeras form a single, discrete complex with SerRS. Free tRNA and complexed SerRS-tRNA were resolved on a polyacrylamide gel. (A,B) Gel staining with ethidium bromide showed that SerRS binds to tRNASer, tRNASec, and all of the mutant/chimeric constructs, which resulted in the appearance of a single band near the top of the gel. This band shift, when compared to free tRNA, indicates that a stable binary SerRS–tRNA complex is formed in each case. (C,D) Coomassie staining of the same gels further confirmed this proposition as it established that the shifted band contains SerRS. In all instances, a single, distinct complex, similar in size to SerRS-tRNASec and SerRS-tRNASer, is formed. Moreover, migration of binary complexes is slower when compared to SerRS alone, thus indicating significant changes in molecular size and charge.
SerRS cannot discriminate between tRNASer, tRNASec, and tRNASec/Ser constructs during the initial encounter
The binding affinities to SerRS of WT tRNASer, WT tRNASec, and tRNASec/Ser mutants/chimeras were measured with an in vitro SPR-based assay. Human SerRS was expressed with a C-terminal Avi-tag that served as an attachment point for a biotin residue. The biotinylated SerRS was purified and then immobilized onto a streptavidin (SA) chip (Fig. 5). Various tRNA constructs were then run over the chip and binding events were recorded as sensorgrams (Fig. 5). Our results show that SerRS binds WT tRNASer and tRNASec, and all tRNASec/Ser constructs used in our study with high affinity, suggesting that the enzyme cannot effectively discriminate the cognate tRNASer from the noncognate tRNASec during formation of the binary complex.
FIGURE 5.

Diagram of the SPR-based assay used to study interactions between SerRS and tRNA constructs. (1) Human SerRS (blue), expressed and purified as the C-terminal Avi-tagged (green) fusion construct, was immobilized to the streptavidin chip (yellow rectangle on black bar) through interactions between biotin (dark orange sphere) and streptavidin. (2) Subsequently, tRNA constructs (red) were run over the chip and association and dissociation phases were recorded. Analyses of sensorgrams yielded kinetic parameters describing the initial encounter. The SerRS dimer most likely binds one tRNASec molecule, but it is plausible that the enzyme would interact with two tRNAs (red, highlighted with dashed line).
tRNASer and tRNASec bind strongly to SerRS with similar affinities of 3.3 and 5.9 nM, respectively (Table 3; Fig. 6). Intriguingly, tRNASec/Ser mutants and chimeras retain the ability to interact with the enzyme. Although the binding affinities are weaker, they remained in the nanomolar range (Table 3; Fig. 6; Supplemental Fig. S1), and are still reflective of strong interactions. The change in binding affinities ranged from an ∼26-fold decrease for Mutant 3 to an ∼224-fold decrease for Chimera 8. It is important to note that our SPR binding data were explained best with a two-state binding model and by separately analyzing kinetics of the association and dissociation phases. In other words, our binding curves could not be interpreted using a simple binding model. In the two-state binding model, molecules A (analyte; tRNA) and B (ligand; SerRS) combine into an initial complex AB, which then undergoes a conformational change and adopts a stable AB* complex. The process is mathematically described by the following equation:
| (1) |
where KA = (ka1/kd1)(1 + ka2/kd2) and KD = 1/KA. In all instances, the first binding event is characterized by fast on- (ka1) and slower off-rates (kd1), while the rates of forward and reversed conformational change range from 0.003–0.087 s−1 characterize the second event (Table 3; Fig. 6; Supplemental Fig. S1). Given that the SerRS dimer typically binds one tRNA at a time and the observed low frequency of the event, we suggest that the second binding step is indicative of a slow and large-scale structural adjustment that takes place after the initial encounter between the enzyme and tRNA. We cannot, however, unambiguously determine with the SPR assay if one or two tRNAs concurrently bind to the immobilized SerRS. Yet, our results do show that tRNASec carrying either 12- or 13-bp-long acceptor-TψC arm, A8 or U8, and the D-arm/loop from tRNASer bind equally well to SerRS. The enzyme is apparently oblivious and tolerant, at least at the level of the initial encounter, to relatively minor changes in the tRNA fold and even to larger substitutions in the D-arm and D-loop.
TABLE 3.
Binding affinities to human SerRS of WT and mutant/chimeric tRNA constructs determined by SPR
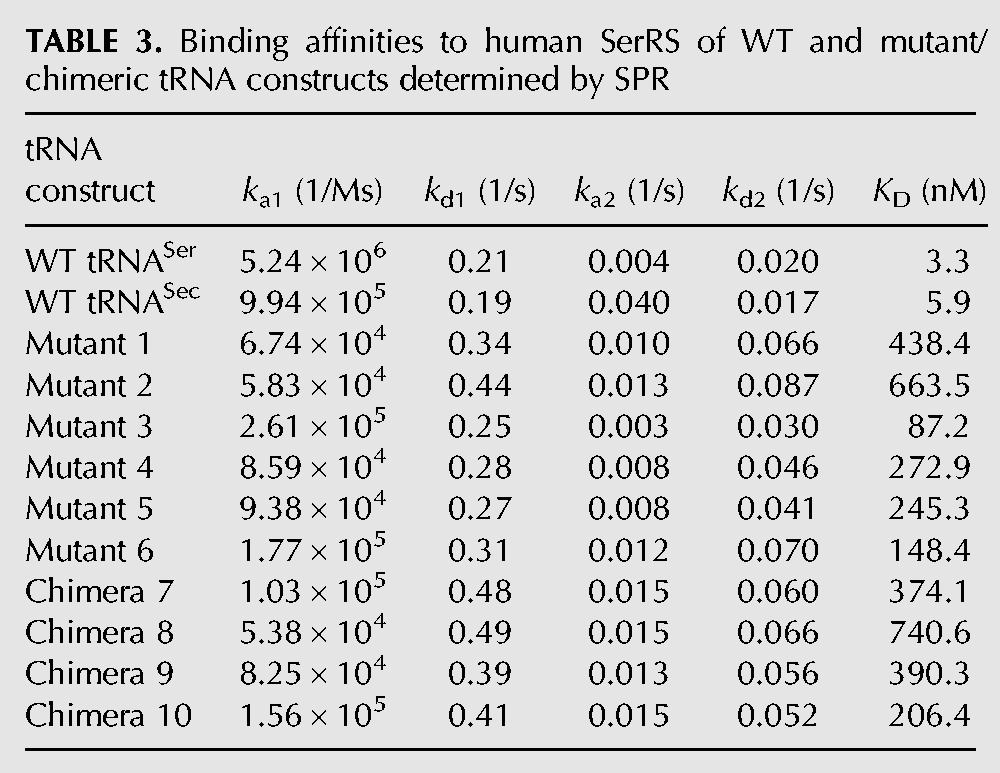
FIGURE 6.

Binding of WT tRNASec, WT tRNASer, and mutant/chimeric tRNA constructs to human cytosolic SerRS is a high-affinity, two-state process. Binding of WT tRNASer (A), WT tRNASec (B), and Mutants 1 (C), and 2 (D) to SerRS was monitored on the Biacore T200 instrument. Serial dilutions of tRNA samples were run over the immobilized enzyme (see Fig. 5). Sensorgrams are colored according to tRNA concentrations used in a particular series. The baseline is shown as solid black line and increasing tRNA concentrations are colored red, green, and purple in each series. In the case of WT tRNASer and tRNASec, the concentration range was 12–111 nM, whereas in the case of Mutants 1 and 2, the range was 37–333 nM. Theoretical curves (black) were fitted with a two-state reaction model using the Biacore T200 evaluation software v.3.0. The goodness-of-fit is represented with fitting residuals, which are displayed below each panel. Kinetics of association and dissociation was analyzed separately while optimizing the RUmax values locally.
Serylation activity of SerRS depends on the length of the acceptor-TψC arm of tRNA
We measured the activity of SerRS in the presence of tRNA constructs to test whether the binding of a given tRNA to SerRS led to formation of a productive complex or if it were simply representative of nonproductive (i.e., “false positive”) interactions with the enzyme. First, we asked if human SerRS indeed prefers tRNASer to tRNASec. Our kinetic assays demonstrate that this is indeed the case. However, the aminoacylation assays demonstrate the extent to which SerRS prefers the tRNASer substrate is quite subtle. Under the experimental conditions used, we determined that KM for tRNASer and tRNASec is 7 and 31 µM, respectively. Likewise, SerRS acts more readily on tRNASer as evidenced by the kcat values (Table 4; Fig. 7). The relative ratio of kcat/KM for each of the substrates provides a good measure of the enzyme's ability to act on those substrates. This analysis shows that human SerRS prefers the cognate tRNASer 6.3-fold over tRNASec (Table 4). While different from the bacterial system where SerRS exhibits ∼100-fold preference for tRNASer over tRNASec (Baron and Böck 1991), our findings are in good agreement with the earlier study on the human system (Heckl et al. 1998). It is important to note that relationships between kcat/KM values were preserved when only linear parts of slopes at low tRNA concentrations were analyzed (data not shown). Taken together, the tRNA fold significantly impacts the overall serylation activity, despite having only modest impacts on the initial binding to SerRS.
TABLE 4.
Kinetic parameters of serylation of WT and mutants/chimeric tRNA constructs by human SerRS
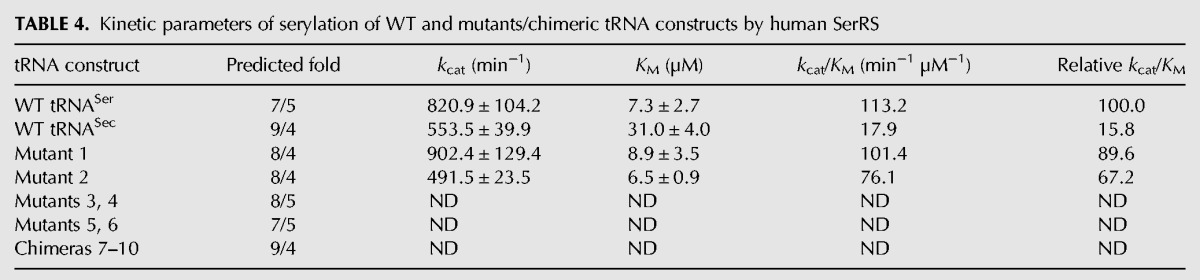
FIGURE 7.

Human SerRS prefers the cognate tRNASer, and Mutants 1 and 2 to the noncognate WT tRNASec. (A) Representative time-course curves of the serylation reaction of various tRNA constructs (10 µM) by human SerRS. The left panel shows the entire time course, whereas the right panel magnifies the initial stage of the reaction during which there is a linear dependence of the product formation and time. The curves clearly demonstrate that SerRS prefers tRNASer (red spheres) to tRNASec (blue triangles). Also, Mutants 1 (solid diamond) and 2 (open square), predicted to adopt the 8/4 fold, recapitulate ∼70% of the activity toward the cognate substrate and are better substrates than tRNASec. Data set labels are shown below the left panel. (B) Michaelis–Menten curves derived from serylation reactions of tRNASer (blue), tRNASec (red), and Mutants 1 (gold) and 2 (green) by SerRS are shown. Kinetic parameters were extracted from these curves using nonlinear curve fitting (Table 4).
Next, we analyzed serylation levels of chimeric tRNASer/Sec constructs. Intriguingly, Mutants 1 and 2, which presumably adopt a novel 8/4 fold, are better SerRS substrates than the WT tRNASec and just slightly worse than the WT tRNASer (Table 4; Fig. 7). In fact, these two mutants recovered ∼90% (Mutant 1) and 67% (Mutant 2) of the serylation activity of the cognate substrate (Table 4; Fig. 7B). This suggests that human SerRS prefers the 12-bp-long acceptor-TψC “helix” and that it is indifferent about identity of the eighth nucleotide.
Given the slight preference for 12 bp in the acceptor-TψC “helix,” we wondered whether human SerRS would prefer human tRNASec that folds into a bacterial-like 8/5 structure or one resembling more closely the cognate tRNASer. Quite unexpectedly, human SerRS failed to charge these mutants (Table 4; Fig. 7), thus arguing that perhaps the frame of human tRNASec is not suitable to support either the canonical 7/5 or bacterial 8/5 folds.
Lastly, we analyzed the impact of the D-arm and D-loop sequences on the serylation reaction. By using Chimeras 7–10, we tested whether the D-arm and D-loop of tRNASer in the context of the 9/4 structure of tRNASec could bolster serylation levels. Our results unequivocally demonstrate that SerRS cannot serylate such constructs (Table 4; Fig. 7), implying that the D-arm and D-loop, though not directly interacting with SerRS (Biou et al. 1994; Wang et al. 2015), play important roles in stabilizing the tertiary structure of the given tRNA. It is reasonable to postulate that the D-stem coevolved with other structural and functional elements (i.e., acceptor-TψC and extra arms), and that it cannot be simply transplanted with the aim to recapitulate the functionality of another tRNA species.
In summary, our data show that removing the nonconserved base pair from the acceptor arm generates an improved tRNASec-based substrate for human SerRS. Also, we demonstrate that incorporation into human tRNASec of the canonical 7/5 fold, the bacterial-like 8/5 fold, or elements of the D-arm from human tRNASer diminish serylation levels.
DISCUSSION
Stringent substrate specificities, often combined with the ability to edit reaction products, make AaRSs into potent protectors of the genetic code. Given that elongation factors and ribosome particles cannot monitor and/or establish identity of the amino acid that is attached to any given tRNA, the significance of AaRSs for fidelity of gene translation cannot be overstated. Only in rare instances do AaRSs broaden their scope (i.e., indirect aminoacylation reactions). One such extraordinary example is SerRS, the enzyme that couples Ser to cognate tRNASer and noncognate tRNASec. Far removed from being an extravagant functional property, this capacity of SerRS to err is vital as it initiates synthesis of the amino acid Sec and all selenoproteins. This reaction also represents the only completely conserved step in the anabolic cycle of Sec across all domains of life (Yuan et al. 2006; Xu et al. 2007). A number of studies have been conducted to dissect various aspects of the tRNA “promiscuity” of SerRS and they all converged to a set of conclusions. It is now well established that the discriminator base G73, the spatial orientation but not the sequence of the variable arms, and several elements in the acceptor arm in tRNASer represent recognition motifs for SerRS (Sampson and Saks 1993; Wu and Gross 1993; Asahara et al. 1994; Heckl et al. 1998; Mizutani et al. 1998; Gruic-Sovulj et al. 2006; Jaric et al. 2009; Rokov-Plavec et al. 2013). Also, meticulous and elegant biochemical studies revealed minimal sets of identity elements in tRNASer required for altering the tRNA identity (Normanly et al. 1986, 1992; Achsel and Gross 1993). However, no further discussion of the fundamental problem of the imperfection of the tRNA-selection mechanism by SerRS is available. Here, we wished to build on previous efforts and posed a set of important questions concerning the tRNA-recognition and serylation mechanisms of human SerRS. Firstly, given that human tRNASec and tRNASer adopt fundamentally distinct structures, we sought to determine whether the tRNA fold is the major determinant for substrate recognition and serylation. Secondly, we wondered if the absence of the conserved U8 position in tRNASec is of any consequence for binding to SerRS and the efficiency of serylation. Lastly, we wished to ascertain if the D-arm and D-loop sequences from the cognate tRNASer support binding to SerRS and subsequent serylation when placed in the context of the noncognate tRNASec. We used a two-pronged approach in which WT tRNASer, WT tRNASec, and 10 tRNA mutant constructs (Table 1; Figs. 1, 2) were analyzed by in vitro binding and aminoacylation assays. The binding assays probed if substrate selection and discrimination is dictated at the level of the initial recognition/encounter of the tRNA body, while the aminoacylation assays assessed whether SerRS could then charge these tRNAs.
As one might expect, WT tRNASer and WT tRNASec bind to SerRS quite strongly and their affinities are almost indistinguishable (3.3 versus 5.9 nM). This suggests that SerRS registers, at least at the level of the first encounter, both tRNAs as cognate species. We recorded stronger binding affinities between SerRS and tRNAs than in previous reports (Jaric et al. 2009; Xu et al. 2013). These discrepancies are most likely due to either significantly different assays used (Xu et al. 2013) or species-dependent peculiarities (Jaric et al. 2009). Somewhat surprising was the observation that all mutant and chimeric tRNA constructs used in our study retained the ability to bind to SerRS with high, albeit decreased, affinity (Table 3; Fig. 6; Supplemental Fig. S1). In spite of the drop in the affinity, the dissociation constants (KD) stayed in the nanomolar range. Deleting 1 bp from the acceptor stem of tRNASec attenuated the strength of the interaction of Mutants 1 and 2 by ∼130- and 200-fold, respectively (Table 3; Fig. 6). Changing the overall fold of tRNASec into 8/5 or 7/5 had a relatively mild effect on KD values (i.e., decrease from ∼26- to 80-fold). Likewise, the effect of replacing the D-loop and/or D-arm in tRNASec with analogous elements from tRNASer varied from an ∼62-fold decrease for Chimera 10 to >200-fold drop for Chimera 8 (Table 3; Supplemental Fig. S1). Intriguingly, detailed analysis of SPR curves suggests the binding of tRNAs to SerRS is a two-step event. While fast association and slow dissociation rates govern the initial encounter or the first step, low frequency processes characterize the second step (Table 3; Equation 1). Because SerRS typically binds and acts on one tRNA at a time and given that low-frequency events were observed, we suggest that the second event is reflective of a relatively large-scale conformational adjustment(s) in the initial SerRS–tRNA complex and not of the binding of the second tRNA equivalent. This interpretation of our results is supported by observed structural dynamics in SerRS and tRNASec in crystal structures of human SerRS (Xu et al. 2013), human SerRS–tRNASec (Wang et al. 2015), human SepSecS–tRNASec (Palioura et al. 2009), and archaeal PSTK–tRNASec (Chiba et al. 2010; Sherrer et al. 2011), as well as in the CryoEM structure of the bacterial SelB:Sec–tRNASec complex bound to the 70S ribosome (Fischer et al. 2016). Also, this is in agreement with suggestions by us and others that the ability of murine and human tRNASec to alter and adjust its conformation may be of functional significance (cf. Fig. 8 with Fig. 2 in Yuan et al. 2010 and Fig. 5D,E in Ganichkin et al. 2011). Moreover, a similar two-state binding was observed in a study on tRNA recognition by SerRS from methanogenic archaeon Methanosarcina barkeri (Jaric et al. 2009). Finally, it is well established that AaRSs read the tRNA identity elements at the transition state (Ibba et al. 1999). Taken together, we argue that SerRS developed a tRNA-recognition mechanism that compensates for structural differences between tRNASer and tRNASec, and that such mechanism cannot discern between the cognate tRNASer and noncognate tRNASec at the level of the initial encounter. Because SerRS exhibits tolerance toward a variety of tRNASec mutants and chimeras, we suggest that SerRS recognizes, at first, a particular shape of the tRNA. This fast binding event presumably requires interactions between NTD of SerRS and the long-variable arm of tRNA. Subsequent formation of sequence-specific interactions leads to structural adjustments in NTD of SerRS and variable and acceptor arms of tRNA that allow positioning of the CCA-end within the catalytic groove. This second event is significantly slower than the first one and it dictates the binding affinity. It is important to mention that our interpretation and speculation is consistent with the proposal put forward by Weygand-Djurasevic and colleagues (Jaric et al. 2009), thus implying that SerRS orthologs of the archaeal/eukaryal subtype could behave in a similar manner where tRNA recognition is concerned. One of the shortcomings of our study is that we did not test whether SerRS would bind with the same high affinity a tRNASer/Sec mutant devoid of the variable arm or some other, structurally divergent, canonical tRNA that does not contain the extended variable arm. Although the significance of the variable arm for recognition and function of tRNASec and tRNASer was previously established (Sampson and Saks 1993; Wu and Gross 1993; Asahara et al. 1994; Heckl et al. 1998; Mizutani et al. 1998; Gruic-Sovulj et al. 2006; Rokov-Plavec et al. 2013), revisiting this particular aspect in the context of chimeric constructs is warranted.
FIGURE 8.

tRNASec is flexible and undergoes a conformational change upon interaction with enzymes. Superimpositioning of crystal structures of human tRNASec in isolation (beige; PDBID: 3A3A), human tRNASec bound to SepSecS (red; PDBID: 3HL2), and archaeal tRNASec in complex with PSTK (blue; PDBID: 3ADD) reveals that the acceptor-, TψC- and variable arms adjust their orientation upon binary complex formation. The acceptor arm swings toward the anticodon stem–loop, while the variable arm can pivot either to or from the anticodon stem. Two views related by ∼90o rotation around the vertical axis are displayed. For more detailed analysis, compare these panels with Figure 2 in Yuan et al. (2010) and Figures 5E and D in Ganichkin et al. (2011).
Concurrently with the SPR studies, aminoacylation assays were used to assess which of the tRNA mutants could be used in serylation reaction. In other words, the aim was to discern tRNAs that formed a productive complex with SerRS from those that interacted with the enzyme in a nonproductive manner. Early in our study we posited that human SerRS, analogous to the bacterial enzyme, prefers tRNASer to tRNASec. Our kinetic measurements showed that this preference is subtle and when translated into the realm of numbers it amounts to ∼6.3-fold difference (Table 4; Fig. 7). Although we did not use post-transcriptionally modified tRNAs, our data on tRNASer are in strong agreement with the previously published results on bovine liver SerRS (Mizutani et al. 1984) and human system (Heckl et al. 1998). This is not entirely surprising as tRNASec carries only three or four modifications, one of which is in the anticodon loop. Interestingly, when the nonconserved U6:U67 was removed from the acceptor arm of tRNASec, a significantly improved substrate was obtained (Table 4; Fig. 7). In fact, Mutants 1 and 2 recapitulated ∼67%–90% of the substrate prowess of the WT tRNASer, irrespective of the A8→U mutation (Table 4). Interestingly, the U6:U67 base pair is in the vicinity of the proposed “cryptic” recognition elements (i.e., base pairs at positions 1–72 and 5–68) for the bacterial SerRS (Saks and Sampson 1996), one of which is an antideterminant (e.g., A5:U68) for the archaeal PSTK (Sherrer et al. 2008). Given its minute, if any, effects on serylation in our study, we conclude that identity of the eighth nucleotide is not relevant for serylation. This is somewhat surprising as it was suggested that it is the absence of interactions that U8 forms with the core of the tRNA that may be responsible for structural plasticity of tRNASec (Palioura et al. 2009; Yuan et al. 2010; Ganichkin et al. 2011). On the other hand, A8 is not conserved in tRNASec across domains, suggesting that any base in this position would adopt a similar orientation as A8 in human tRNASec.
While our findings might suggest that 12 bp in the acceptor-TψC “helix” represent the major differentiating element between tRNASer and tRNASec, the case is not that simple. This was particularly evident when we observed that SerRS could not serylate the tRNASec/Ser mutants that were predicted to adopt either the 8/5 structure of the bacterial tRNASec (Mutants 3 and 4) or the canonical 7/5 fold of tRNASer (Mutants 5 and 6) (Table 4; Fig. 7). Intriguingly, the deletion of specific nonconserved base pair(s) from the acceptor arm of tRNASec had the same effect on the aminoacylation activity as the replacement of the entire arm with that of tRNASer (Wu and Gross 1993). It is thus reasonable to argue that the length and even sequence of the acceptor-TψC arm, or any other structural element for that matter, must only be considered within the context of the given tRNA. Another explanation is that the inserted G51:C63 pair within the TψC stem is a strong antideterminant for human SerRS and that its effect is dominant in Mutants 3-6. Addition of this base pair could misposition bases in the T-loop, thus causing the tRNA to misfold. Because the sequence of the TψC stem is not particularly conserved in tRNASer and tRNASec across species, our initial goal was to enable formation of particular folds and not to probe effect(s) of the exact sequence. Additional studies are needed to establish if incorporation of the C50:G64 pair, as found in human tRNASer, would restore or hinder the serylation activity. In either case, it seems that the overall fold of tRNASec is important for serylation. This postulate is corroborated by analyses of serylation activities of Chimeras 7–10. In these particular constructs, the D-arm or the D-loop of tRNASec was replaced with the analogous elements from tRNASer, while retaining the 9/4 fold of tRNASec and the 3-bp-long D-stem of tRNASer (Fig. 2). It is important to emphasize that Chimeras 7–10 are different from the D-arm mutants used before (Wu and Gross 1993). In that study, either one nucleotide or the entire D-arm was substituted with the sequence from tRNASer and a drastic drop in the aminoacylation activity was observed. In the latter construct, a linker between the acceptor- and D-arms was not preserved, which could have had damaging effects on the overall tRNA structure. In our chimeras, the linker is preserved and the length of the D-arm is kept constant. Our approach basically provided a way of probing whether the sequence or length of the D-arm is an important determinant for serylation. Our results show that the ability of SerRS to serylate Chimeras 7–10 is greatly diminished (Table 3; Fig. 6), thus implying that simple transposition of an element from one tRNA to another, however similar the two may be, is not sufficient to elicit a new functionality. Also, while the sequence of the D-stem seems unimportant for serylation, the length may be significant within the context of the particular tRNA frame. The structural comparison of tRNASer and tRNASec reveals that changes in the D-stem, D-loop, and TψC arm may impact the positioning of the D- and T-loop nucleotides needed for closing the L-shaped molecule. It is thus reasonable to propose that human SerRS recognizes the molecular surface and shape of substrate tRNAs, which is analogous to bacterial (Asahara et al. 1994) and fungal orthologs (Gruic-Sovulj et al. 2006).
It is important to mention that we ruled out the possibility that unfolding and/or misfolding of tRNASec/Ser mutants is the cause for the observed loss in serylation activity by providing several lines of evidence: (i) tRNAs were prepared and purified under nondenaturing conditions, (ii) tRNAs exhibited unfolding trajectories similar to that of WT tRNASec and different from the RNase A-treated tRNA (Table 2; Fig. 3), and (iii) all tRNA constructs bound to SerRS in solution and the binary complexes were of consistent size and stoichiometry (Fig. 4). Furthermore, tRNAs were devoid of impurities and degradation fragments (Fig. 3). The differences in electrophoretic mobility are most likely due to residual secondary structure elements in certain, perhaps more stable, constructs. This is a reasonable explanation since a combination of high temperature with 8 M urea is often needed to break the most stable secondary structure elements (Hegg and Thurlow 1990). Because our gels were run at room temperature, the stable structures might have survived the urea treatment and consequently appeared on the gel as bands with altered mobility. Our data, on the other hand, suggested that certain changes in the D- and acceptor arm might impact tRNASec in a distinct manner; the former, as one would expect, decreases its stability, while the latter increases it. Regardless of these local and perhaps subtle effects, we have provided a solid body of evidence that supports our binding and activity assay results.
One of the drawbacks of our study is that we could not directly address the significance for serylation of the distinct G19:U20:C56 base triple in the D-loop of tRNASec. It has been suggested that U20, which is the only solvent-exposed base in the D-loop, is critical for the structural integrity of tRNASec (Itoh et al. 2009) and for binding to the Sec-synthetic enzymes including SerRS (Chiba et al. 2010; Wang et al. 2015). However, other studies remarked that the base of U20 does not interact with human SepSecS (Palioura et al. 2009) and archaeal PSTK (Sherrer et al. 2008, 2011). As we have seen, insertion of the G51:C63 pair in the TψC arm diminishes serylation. This could be due to mispositioning of the T-loop in relation to the D-loop and the inability of the tRNA to fold. Alternatively, the tRNA chimera could assume the predicted structure, but without the essential base triple formed. In either instance, we cannot unequivocally establish the significance of the base triple for serylation and further analyses of this motif are needed.
In summary, we have shown here that human cytosolic SerRS only subtly prefers the cognate tRNASer to tRNASec and that such discrimination occurs not during the initial encounter but during the serylation step. Further, we have demonstrated that human SerRS tolerates only minimal changes in the acceptor arm of the substrate tRNA that presumably do not disturb the spatial arrangement of the TψC- and variable arms. The removal of the nonconserved U6:U67 couple from the acceptor arm yielded an 8/4 tRNA that was an improved tRNASec substrate for serylation. Other changes in the acceptor-, TψC-, and D-arms diminished the utility of tRNASec for serylation reaction. However, we are aware that the limited set of mutations used in this study may not have been sufficient to fully delineate the importance of all structural elements in tRNASec and tRNASer for serylation. Although our work extends previous findings, additional studies on 8/4 fold mutants and mutants that carry different sequences in the acceptor-TψC or variable arms are warranted to completely understand tRNA-recognition and discrimination mechanisms used by human SerRS. Equally interesting would be to analyze if the principles defined for the human system hold in other species. Lastly, these efforts could lead to engineering of even more superior serylation substrates that could find use in various synthetic biology initiatives.
MATERIALS AND METHODS
Expression and purification of human cytosolic SerRS
The human cytosolic SARS gene was cloned into a pQE-80 expression vector (Qiagen) with an N-terminal His-tag, followed by a Tev protease site. An Avi-tag sequence was added to the C terminus. His-Tev-SerRS-Avi was overexpressed for 18 h at +15°C in the BL21 (DE3) (Stratagene) E. coli expression strain following the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) to the culture. The cells were lysed in 20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10 mM MgCl2, 40 mM imidazole, 10% (v/v) glycerol, 3 mM β-ME; a protease inhibitor tablet (Roche) was added to the lysate. The recombinant protein was captured on a Ni2+-affinity column (GE Healthcare). The nonspecifically bound protein was removed by rinsing the column with lysis buffer. His-Tev-SerRS-Avi was eluted with 250 mM imidazole. The eluate was diluted threefold with 20 mM Tris–HCl pH 8.0, loaded onto a Resource-Q column (GE Healthcare) and purified using a linear gradient of NaCl (0.1–1 M) in 20 mM Tris–HCl, pH 8.0 and 0.5 mM TCEP. The protein peak was collected and diluted to 0.5 mg/mL in the Tev protease reaction buffer (50 mM Tris–HCl, pH 8.0, 0.1 mM EDTA and 1 mM DTT). Tev protease was added to a molar ratio of 1:50 (excess of His-Tev-SerRS-Avi) and the reaction was incubated at +22°C for 12–16 h. Subsequently, 0.5 mM MgCl2 was added to the sample before another round of purification over the Ni2+ column. SerRS-Avi, found in the flow-through fraction, was subjected to a HiLoad 26/600 Superdex 200pg size-exclusion column (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5% glycerol (v/v) and 0.5 mM TCEP. The enzyme was concentrated, flash-frozen in liquid nitrogen and stored at −80°C.
Expression and purification of Tev protease
Tev protease, which contains an N-terminal His-tag, was overexpressed for 18 h at +15°C in the BL21 (DE3) (Stratagene) E. coli strain following the addition of 0.5 mM IPTG. The cells were lysed in 50 mM Tris–HCl, pH 8.0, 200 mM NaCl, 25 mM imidazole, 10% (v/v) glycerol, and 10 mM β-ME, and the protein was captured from the cell lysate using a Ni2+-affinity column and eluted with 300 mM imidazole. The sample was then subjected to a HiLoad 16/600 Superdex 75pg size-exclusion column (GE Healthcare) equilibrated with 20mM Tris–HCl, pH 8.0, 100 mM NaCl, and 5% (v/v) glycerol. The purified protein was concentrated, and 2 mM EDTA, 5 mM DTT, and 50% (v/v) glycerol were added to the sample. The enzyme was stored at −20°C.
Expression and purification of T7 RNA polymerase
T7 RNA polymerase, cloned into a pQE8 expression vector with an N-terminal His-tag, was overexpressed for 3 h at +37°C in BL21 (DE3) using 0.5 mM IPTG. The cells were lysed in 20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10 mM imidazole, 10% (v/v) glycerol, 5 mM β-ME. After purification on a Ni2+-affinity column, the enzyme sample was dialyzed twice against 20 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10% (v/v) glycerol, and 1 mM DTT at +4°C. The sample was cleared by centrifugation, glycerol was added to 50% (v/v), and the sample was stored at −20°C.
Cloning of tRNASec/Ser mutants and chimeras
The pUC19 plasmid containing the human tRNASec gene (Palioura et al. 2009) was mutated with the QuikChange Lightning kit (Agilent Technologies). Standard protocol was followed using the following primers (only “forward” primers in 5′→3′ direction are shown for brevity):
| Mutant 1 | CTCACTATAGCCCGGAGATCCTCAGTGGGGTTC AATTCCACCTTCGGGCGCCAGGA |
| Mutant 2 | CACTATAGCCCGGAGTTCCTCAGTGGTCT |
| Mutants 3,4 | GTAGCTGTCTAGGGACAGAGTGGGTTCAGTGG GTTCAATTCCCACCTTCGGGCGCC |
| Mutant 5 | GACTCACTATAGCCCGAGATCCTCAGTGGTCT CCTGTAGCTGTCTAGCGACAGAGTGGGTTCA GTTCAATTCCCACCTCGGGCGCCAGGAA |
| Mutant 6 | GACTCACTATAGCCCGAGTTCCTCAGTGGTCT |
| Chimera 7 | TACGACTCACTATAGCCCGGATGATGCTGAGTG GTTAAGGCTGCAGGCTTCAAAC TAGCCCGGATGATGCCGAGTGGTTAAGGCTG |
| Chimera 8 | CACTATAGCCCGGATGTTGCCGAGTGGTTAAGG |
| Chimera 9 | GACTCACTATAGCCCGGATGATCCTGAGTGGTT AAGGCTGCAGGCTTCAAA |
| Chimera 10 | CACTATAGCCCGGATGTTCCTGAGTGGTTAAGG |
In vitro transcription and tRNA purification
tRNA was synthesized by in vitro T7 RNA polymerase run-off transcription. The transcription reaction was performed at +37°C for 3 h in 40 mM Tris–HCl, pH 8.1, 22 mM MgCl2, 5 mM DTT, 2 mM spermidine, 50 μg/mL BSA, 20 mM GMP, 4 mM nucleotides (ATP, GTP, CTP, and UTP), using 60 μg/mL of the PCR product of the tRNA gene and 8 μM T7 RNA polymerase. The filtered transcription reaction was loaded onto a Resource-Q column (GE Healthcare) and the tRNA was purified using a linear gradient of NaCl (0.4–0.7 M) in 20 mM Tris–HCl, pH 8.1. The tRNA was further purified on a S200 Superdex size-exclusion column (GE Healthcare) equilibrated with 20 mM Tris–HCl, pH 8.1, and 150 mM NaCl. The purified tRNA was concentrated, flash-frozen in liquid nitrogen and stored at −80°C. The tRNAs were not subjected to denaturation at any point during purification. Elution profiles from the Resource Q and size-exclusion columns of tRNA chimeras and mutants were indistinguishable from that of WT tRNASec.
Melt curve analysis
To determine if the mutations introduced in tRNASec affected tRNA structure and fold, a melt curve analysis was performed. For the melt curve analysis, purified tRNASec samples were first treated with DNase to ensure there was no residual DNA carried over from the transcription reaction. All buffers were prepared with DEPC-treated water to ensure inhibition of RNase activity. For DNase treatment, 10 µg of each purified tRNA was resuspended in 100 µL of 1× DNase I reaction buffer (10 mM Tris–HCl, pH 7.5, 2.5 mM MgCl2, and 0.1 mM CaCl2) with a final DNase I (Thermo Fisher) concentration of 1 U/µL. The reaction was incubated at +37°C for 10 min. Subsequently, the RNA was purified using a RNA Clean & Concentrator kit (Zymo Research) and eluted in a buffer containing 20 mM Tris–HCl, pH 7.5, and 50 mM NaCl. For RNase A treatment of WT tRNASec, a 50 µL reaction containing 500 ng/µL of WT tRNASec was incubated with RNase A (100 µg/mL) in 20 mM Tris–HCl, pH 7.5, and 50 mM NaCl, at +37°C for 10 min. For tracking RNA denaturation, a 1:20,000 SYBR Green I Nucleic Acid Gel Stain (Thermo Fisher) dilution gave a superior signal-to-noise level. Practically, a 10× SYBR Green I Buffer was prepared in 100 mM Tris–HCl, pH 7.5, 20 mM MgCl2, 1.5% (v/v) Triton X-100, and 1.5 M NaCl by diluting the original dye 2000-fold. This solution could be stored at +4°C for up to a week. To prepare samples for the melt curve analysis, each tRNA sample (no RNA, WT tRNASec, mutants 1-10, and RNase A-treated WT tRNASec) was diluted in the 1× SYBR Green I Buffer to a concentration of 60 ng/µL. The melt curve analysis was performed in triplicate using a 20 µL sample volume on a Bio-Rad CFX Connect instrument with a temperature gradient of 0.5°C/s from +50 to +95°C. The Tm was best visualized as a clear peak in the negative first derivative of the melting curve (−dRFU/dT).
Denaturing TBE-urea (TBU) polyacrylamide gel electrophoresis
TBU gels were used to evaluate the purity of tRNA samples used for melt curve analysis. Two hundred nanograms of each tRNASec sample was run along with a Micro Riboready Ladder (Amresco) on a 10% TBU gel. RNA bands were visualized by staining with a 0.04% (w/v) toluidine blue solution in 300 mM sodium acetate, pH 5.0, and destained in water until the background was clear.
Electrophoretic mobility shift assay (EMSA)
EMSA assessed the ability of SerRS to bind to the mutant tRNASec constructs compared to the native tRNA substrates for SerRS (tRNASer and WT tRNASec). Samples of SerRS alone, tRNA alone, and SerRS-tRNA mixtures were prepared in 9 µL of 20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 5% (v/v) glycerol, and 0.5 mM TCEP and allowed to equilibrate for 30 min at +25°C. In all experiments, 1.5 µg of SerRS was used, whereas the amount of tRNA was adjusted so that 1:1 molar complex with SerRS is formed. In the case of tRNA samples, the same amount of tRNA that was used for preparation of complex samples (3.5 pmol) was applied on the gel. Subsequently, 1 µL of 5× Hi-Density TBE Sample Buffer (Thermo Fisher) was added to each sample and samples were electrophoresed on 6% 0.5× TBE polyacrylamide gels. Gels were stained with ethidium bromide (0.05 µg/mL) to visualize RNA, followed by Coomassie R-250 staining to visualize SerRS.
Biotinylation of SerRS
Pure SerRS-Avi was concentrated to 3 mg/mL, and then biotinylated overnight at +4°C using a commercial biotinylation kit (Avidity). The biotinylated protein was purified on a HiLoad 16/600 Superdex 200 size-exclusion chromatography column in 10 mM Tris–HCl, pH 8.0, 300 mM KCl, 0.5 mM TCEP, 5 mM MgCl2, to remove excess biotin and birA enzyme (biotin ligase). Pure biotinylated SerRS-Avi was concentrated to ∼2 mg/mL, flash-frozen in liquid nitrogen, and stored at −80°C.
Surface plasmon resonance binding assays
The binding assays were completed on a Biacore T200 instrument. The C-terminal biotinylated SerRS was immobilized on a Series S sensor chip SA (GE Healthcare) with running buffer HBS-P (10 mM Hepes, pH 7.4, 150 mM NaCl, 0.05% (v/v) surfactant P-20). Immobilization level of SerRS was ∼4800 response units (RUs); a blank surface was used as a control. The WT tRNASer, WT tRNASec, and tRNASec/Ser mutants/chimeras were applied in a series of increasing concentrations (12–1000 nM) to two channels at a flow rate of 10 μL/min at +25°C. Sensorgrams were double-referenced with blank channel and zero concentration signals, and analyzed using the Biacore T200 evaluation software v3.0. Experimental data were interpreted using a two-state binding model. Kinetics of the association and dissociation phases was analyzed separately by locally optimizing RUmax values.
In vitro aminoacylation assays
Aminoacylation assays were done as previously described (Sampson and Uhlenbeck 1988) in the presence of 20 mM Hepes pH, 7.6, 8 mM MgCl2, 150 mM ammonium chloride, 2 mM spermidine, 0.05 mM spermine, 4 mM β-ME, 2 mM ATP, 50 μM [14C]-Serine (100μCi/mL), 2.5–40 μM tRNA transcripts, and 50 nM SerRS. Kinetic parameters were extracted from nonlinear curve fitting of Michaelis–Menten curves. These values agreed with estimates of the kcat/KM values determined from linear parts of the time-course curves at low tRNA concentrations and low amounts of product formation.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the staff of the Research Resource Center (UIC-RRC) for their help with SPR data acquisition and Professor Alexander Mankin and James Marks (UIC) for help with aminoacylation assays. This work was supported by The National Institute of General Medical Sciences, National Institutes of Health grant GM097042 (to M.S.), the UIC Chancellor's Undergraduate Research Award (to J.W.L.), the UIC Honors College Research Grant (to J.W.L.), the UIC CCTS Pre-doctoral Education for Clinical and Translational Scientists Fellowship (to A.K.P.), and the UIC Provost Deiss Award (to A.K.P.).
Author contributions: K.H.M. and J.W.L. prepared SerRS and tRNA constructs. K.H.M. collected SPR and aminoacylation data and analyzed aminoacylation data. A.K.P. performed thermal shift and electrophoretic mobility assays. H.L. and M.S. analyzed and interpreted the SPR data. M.S. designed the experiments and wrote the manuscript. All authors commented on the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.061069.117.
REFERENCES
- Achsel T, Gross HJ. 1993. Identity determinants of human tRNASer: sequence elements necessary for serylation and maturation of a tRNA with a long extra arm. EMBO J 12: 3333–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara H, Himeno H, Tamura K, Nameki N, Hasegawa T, Shimizu M. 1994. Escherichia coli seryl-tRNA synthetase recognizes tRNASer by its characteristic tertiary structure. J Mol Biol 236: 738–748. [DOI] [PubMed] [Google Scholar]
- Baron C, Böck A. 1991. The length of the aminoacyl-acceptor stem of the selenocysteine-specific tRNASec of Escherichia coli is the determinant for binding to elongation factors SELB or Tu. J Biol Chem 266: 20375–20379. [PubMed] [Google Scholar]
- Biou V, Yaremchuk A, Tukalo M, Cusack S. 1994. The 2.9 Å crystal structure of T. thermophilus seryl-tRNA synthetase complexed with tRNASer. Science 263: 1404–1410. [DOI] [PubMed] [Google Scholar]
- Brigelius-Flohé R, Flohé L. 2016. Selenium and redox signaling. Arch Biochem Biophys 617: 48–59. [DOI] [PubMed] [Google Scholar]
- Bullwinkle TJ, Ibba M. 2014. Emergence and evolution. Top Curr Chem 344: 43–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba S, Itoh Y, Sekine S, Yokoyama S. 2010. Structural basis for the major role of O-phosphoseryl-tRNA kinase in the UGA-specific encoding of selenocysteine. Mol Cell 39: 410–420. [DOI] [PubMed] [Google Scholar]
- Fischer N, Neumann P, Bock LV, Maracci C, Wang Z, Paleskava A, Konevega AL, Schröder GF, Grubmüller H, Ficner R, et al. 2016. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature 540: 80–85. [DOI] [PubMed] [Google Scholar]
- Ganichkin OM, Anedchenko EA, Wahl MC. 2011. Crystal structure analysis reveals functional flexibility in the selenocysteine-specific tRNA from mouse. PLoS One 6: e20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giegé R, Sissler M, Florentz C. 1998. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res 26: 5017–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruic-Sovulj I, Jaric J, Dulic M, Cindric M, Weygand-Durasevic I. 2006. Shuffling of discrete tRNASer regions reveals differently utilized identity elements in yeast and methanogenic archaea. J Mol Biol 361: 128–139. [DOI] [PubMed] [Google Scholar]
- Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. 2014. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci 39: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckl M, Busch K, Gross HJ. 1998. Minimal tRNA(Ser) and tRNA(Sec) substrates for human seryl-tRNA synthetase: contribution of tRNA domains to serylation and tertiary structure. FEBS Lett 427: 315–319. [DOI] [PubMed] [Google Scholar]
- Hegg LA, Thurlow DL. 1990. Residual tRNA secondary structure in ‘denaturing’ 8M urea/TBE polyacrylamide gels: effects on electrophoretic mobility and dependency on prior chemical modification of the tRNA. Nucleic Acids Res 18: 2993–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibba M, Söll D. 1999. Quality control mechanisms during translation. Science 286: 1893–1897. [DOI] [PubMed] [Google Scholar]
- Ibba M, Söll D. 2000. Aminoacyl-tRNA synthesis. Annu Rev Biochem 69: 617–650. [DOI] [PubMed] [Google Scholar]
- Ibba M, Sever S, Praetorius-Ibba M, Söll D. 1999. Transfer RNA identity contributes to transition state stabilization during aminoacyl-tRNA synthesis. Nucleic Acids Res 27: 3631–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Chiba S, Sekine S, Yokoyama S. 2009. Crystal structure of human selenocysteine tRNA. Nucleic Acids Res 37: 6259–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Brocker MJ, Sekine S, Hammond G, Suetsugu S, Söll D, Yokoyama S. 2013a. Decameric SelA*tRNA(Sec) ring structure reveals mechanism of bacterial selenocysteine formation. Science 340: 75–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh Y, Sekine S, Suetsugu S, Yokoyama S. 2013b. Tertiary structure of bacterial selenocysteine tRNA. Nucleic Acids Res 41: 6729–6738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaric J, Bilokapic S, Lesjak S, Crnkovic A, Ban N, Weygand-Durasevic I. 2009. Identification of amino acids in the N-terminal domain of atypical methanogenic-type Seryl-tRNA synthetase critical for tRNA recognition. J Biol Chem 284: 30643–30651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov AV, Delgado C, Rahlfs S, Novoselov SV, Kryukov GV, Gromer S, Hatfield DL, Becker K, Gladyshev VN. 2006. The Plasmodium selenoproteome. Nucleic Acids Res 34: 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobanov AV, Hatfield DL, Gladyshev VN. 2009. Eukaryotic selenoproteins and selenoproteomes. Biochim Biophys Acta 1790: 1424–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti M, Ridge PG, Zhang Y, Lobanov AV, Pringle TH, Guigo R, Hatfield DL, Gladyshev VN. 2012. Composition and evolution of the vertebrate and mammalian selenoproteomes. PLoS One 7: e33066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T, Narihara T, Hashimoto A. 1984. Purification and properties of bovine liver seryl-tRNA synthetase. Eur J Biochem 143: 9–13. [DOI] [PubMed] [Google Scholar]
- Mizutani T, Kanaya K, Ikeda S, Fujiwara T, Yamada K, Totsuka T. 1998. The dual identities of mammalian tRNA(Sec) for SerRS and selenocysteine synthase. Mol Biol Rep 25: 211–216. [DOI] [PubMed] [Google Scholar]
- Normanly J, Ogden RC, Horvath SJ, Abelson J. 1986. Changing the identity of a transfer RNA. Nature 321: 213–219. [DOI] [PubMed] [Google Scholar]
- Normanly J, Ollick T, Abelson J. 1992. Eight base changes are sufficient to convert a leucine-inserting tRNA into a serine-inserting tRNA. Proc Natl Acad Sci 89: 5680–5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palioura S, Sherrer RL, Steitz TA, Söll D, Simonović M. 2009. The human SepSecS-tRNASec complex reveals the mechanism of selenocysteine formation. Science 325: 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayman MP. 2012. Selenium and human health. Lancet 379: 1256–1268. [DOI] [PubMed] [Google Scholar]
- Reich HJ, Hondal RJ. 2016. Why nature chose selenium. ACS Chem Biol 11: 821–841. [DOI] [PubMed] [Google Scholar]
- Rokov-Plavec J, Lesjak S, Gruic-Sovulj I, Mocibob M, Dulic M, Weygand-Durasevic I. 2013. Substrate recognition and fidelity of maize seryl-tRNA synthetases. Arch Biochem Biophys 529: 122–130. [DOI] [PubMed] [Google Scholar]
- Saks ME, Sampson JR. 1996. Variant minihelix RNAs reveal sequence-specific recognition of the helical tRNASer acceptor stem by E.coli seryl-tRNA synthetase. EMBO J 15: 2843–2849. [PMC free article] [PubMed] [Google Scholar]
- Sampson JR, Saks ME. 1993. Contributions of discrete tRNASer domains to aminoacylation by E.coli seryl-tRNA synthetase: a kinetic analysis using model RNA substrates. Nucleic Acids Res 21: 4467–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JR, Uhlenbeck OC. 1988. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc Natl Acad Sci 85: 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RL, Simonović M. 2012. Synthesis and decoding of selenocysteine and human health. Croat Med J 53: 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrer RL, Ho JM, Söll D. 2008. Divergence of selenocysteine tRNA recognition by archaeal and eukaryotic O-phosphoseryl-tRNASec kinase. Nucleic Acids Res 36: 1871–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrer RL, Araiso Y, Aldag C, Ishitani R, Ho JM, Söll D, Nureki O. 2011. C-terminal domain of archaeal O-phosphoseryl-tRNA kinase displays large-scale motion to bind the 7-bp D-stem of archaeal tRNA(Sec). Nucleic Acids Res 39: 1034–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbrenner H, Speckmann B, Klotz LO. 2016. Selenoproteins: antioxidant selenoenzymes and beyond. Arch Biochem Biophys 595: 113–119. [DOI] [PubMed] [Google Scholar]
- Wang C, Guo Y, Tian Q, Jia Q, Gao Y, Zhang Q, Zhou C, Xie W. 2015. SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis. Nucleic Acids Res 43: 10534–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XQ, Gross HJ. 1993. The long extra arms of human tRNA(Ser)Sec and tRNASer function as major identify elements for serylation in an orientation-dependent, but not sequence-specific manner. Nucleic Acids Res 21: 5589–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL. 2007. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol 5: e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Shi Y, Yang XL. 2013. Crystal structure of human Seryl-tRNA synthetase and Ser-SA complex reveals a molecular lever specific to higher eukaryotes. Structure 21: 2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Palioura S, Salazar JC, Su D, O'Donoghue P, Hohn MJ, Cardoso AM, Whitman WB, Söll D. 2006. RNA-dependent conversion of phosphoserine forms selenocysteine in eukaryotes and archaea. Proc Natl Acad Sci 103: 18923–18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, O'Donoghue P, Ambrogelly A, Gundllapalli S, Sherrer RL, Palioura S, Simonović M, Söll D. 2010. Distinct genetic code expansion strategies for selenocysteine and pyrrolysine are reflected in different aminoacyl-tRNA formation systems. FEBS Lett 584: 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.