

Abstract
Foremost among the challenges facing single molecule sequencing of proteins by nanopores is the lack of a universal method for driving proteins or peptides into nanopores. In contrast to nucleic acids, the backbones of which are uniformly negatively charged nucleotides, proteins carry positive, negative and neutral side chains that are randomly distributed. Recombinant proteins carrying a negatively charged oligonucleotide or polypeptide at the Ctermini can be translocated through a α-hemolysin (α-HL) nanopore, but the required genetic engineering limits the generality of these approaches. In this present study, we have developed a chemical approach for addition of a charged oligomer to peptides so that they can be translocated through nanopores. As an example, an oligonucleotide PolyT20 was tethered to peptides through first selectively functionalizing their N-termini with azide followed by a click reaction. The data show that the peptide-PolyT20 conjugates translocated through nanopores whereas the unmodified peptides did not. Surprisingly, the conjugates with their peptides tethered at the 5′-end of PolyT20 passed the nanopores more rapidly than the PolyT20 alone. The PolyT20 also yielded a wider distribution of blockade currents. The same broad distribution was found for a conjugate with its peptide tethered at the 3′-end of PolyT20, suggesting that the larger blockades (and longer translocation times) are associated with events in which the 5′-end of the PolyT20 enters the pore first.
Keywords: nanopore, protein sequencing, DNA thread, click addition, peptide-PolyT20 conjugate, peptide translocation
Nanopores—orifices with nanometer diameters—can function as nanofluidic channels for the flow of ions and the transport of biomolecules. When a charged molecule is electrophoretically driven through the nanopore, it partially obstructs the passage of ions and modulates the current through the pore. Parameters derived from the current blockade can be used to identify the molecule and even identify structural subunits. Nanopore techniques are emerging as a single molecule tool for sequencing DNA,1 detecting proteins,2 polysaccharides,3 and viruses,4 with possible clinical applications.5 As protein-based nanopore DNA sequencing technology makes inroads into genomic research,6-8 the question arises: can nanopores sequence proteins as well? Given the fact that even a MspA protein nanopore (which has a finer nanopore - 0.5 nm thickness and 1.2 nm diameter - than α-hemolysin) only demonstrates a four-nucleotide resolution1 with ∼ 85% accuracy,9 it seems unlikely that ion-current blockade measurements will ever resolve individual amino acids because calling each of the amino acids requires sorting 204 or 160,000 different signals. A new reading mechanism has to be developed to achieve single amino acid residue resolution for protein sequencing. Electron tunneling detection has been shown to have this capability. Recently, we have demonstrated that individual amino acids can be identified and two different peptides distinguished at a single molecule level by a technique we call recognition tunneling, which measures tunneling currents of analytes in a ∼ 2.5 nm nanogap with its two electrodes functionalized with recognition molecules.10 Kawai and coworkers have also reported the identification of amino acids and phosphorylated peptides by means of electron tunneling currents with either 0.5 or 0.7 nm nanogaps.11 Thus, one can conceive of a device that integrates a tunneling gap with a solid-state nanopore for analyzing protein sequences.
Here, we address another key roadblock to developing nanopore technology for proteomics, which is the translocation of proteins and peptides. While DNA is uniformly negatively charged along its phosphate backbone under physiological conditions, a protein can carry zero, positive or negative net charge, the sum of the charges of the positively and negatively charged side chains that are randomly distributed on its amide backbone. This makes electrophoretic translocation of the protein challenging. Furthermore, owing to the lack of a PCR-like technique for amplifying proteins, it is very difficult to even acquire direct evidences to prove protein translocation.12 Recently, Akeson and his coworkers demonstrated that a recombinant ubiquitin-like protein Smt3 bearing a polyanionic peptide at its C-terminus was unfolded and pulled through a α-hemolysin (α-HL) nanopore by the AAA+ unfoldase ClpX.13 Almost at the same time, Bayley's team reported that a thioredoxin protein tethered to a negatively charged oligonucleotide could also be unfolded and translocated through the α-HL nanopore by an applied voltage.14 These studies suggest a new approach to translocating proteins using a charged ‘pulling-string’ to draw the protein into a nanopore.
With the ultimate goal of sequencing proteins, our initial objective has focused on using a recognition tunneling nanopore to identify peptides. As a matter of fact, the most commonly used method in proteomics is the shotgun mass spectrometry, in which proteins are first digested into peptides with enzymes (such as trypsin) that generate peptides containing only one lysine or arginine residue at their C-terminus. These are separated with liquid chromatography, and injected as charged ions into a mass spectrometer for identification.15 It can also be a method to quantify proteins, for example, when applying a selected reaction monitoring (SRM) technique that uses a set of representative peptides as a surrogate for a protein.16 We are adapting the SRM strategy to our platform for analysis of proteins. While working on fabricating a fixed-gap tunnel junction in solid-state nanopores,17 we have developed a molecular threading strategy to facilitate translocation of peptides: tethering a chain molecule with a large net charge, which functions as a molecular thread, to the termini of peptides to make them all have the same sign of charge so that they can be carried over from one side to another of a nanopore by the threading molecule under a voltage bias. Thus, a basic requirement for the threading molecule is that it can readily translocate through nanopores. We chose an oligonucleotide composed of 20 thymidines (referred to as PolyT20) as a negatively charged thread because the short oligonucleotide has well-defined structure and charge distribution, its translocation has been well studied,18-20 and it is also readily available from commercial sources. In this present study, we have focused on developing a simple and effective chemistry for attaching the oligonucleotide to the N-termini of peptides, and have demonstrated the translocation of the DNA-peptide conjugates through solid-state nanopores.
Results and Discussion
Addition of PolyT20 to N-termini of peptides
We chose to functionalize the N-terminal α-amine of peptides because it is more nucleophilic than the carboxylate at C-terminus, and so susceptible to chemical modification with electrophilic reagents in physiological conditions. The challenge is to find a reagent and conditions that selectively functionalize the α-amine in the presence of other nucleophiles, such as ε-amine of lysine. Although a plethora of chemical methods for modification of proteins have been reported in literature,21 there is still lack of a universal chemistry to specifically modify the N-termini of peptides. In general, the selectivity and efficiencies of an N-terminal reaction vary with peptide sequences 22 and the amino acid residues at the end.23, 24 For example, in a very recent publication, MacDonald et al have reported that 2-pyridinecarboxaldehyde (PCA) reacted with α-amine of peptide X-ADSWAG (X = one of 20 naturally occurring amino acids) with selectivity of varying from > 50% to ∼ 100%, averaging < 90%.25 In this present study, we describe the development of a general method for functionalization of the N termini of tryptic peptides with high selectivity. Acylation is a commonly used chemical reaction in the modification of proteins with chemical reagents such as carboxylic halide, carboxylic anhydride, or an active ester.21 We chose the carboxylic anhydride—which has been reported to selectively react with the N-terminal amines of peptides under neutral or slightly basic conditions26, 27—to introduce a bioorthogonal function to Ntermini of peptides for DNA attachment. As shown in Scheme 1, an azidoacetic anhydride reagent (see Experimental Methods for its synthesis) first reacts with a peptide bearing a lysine residue at its C-terminus, generating an N-azidoacetylated peptide under slightly acidic conditions (Step 1). This is followed by reaction of the azide group with aza-dibenzocyclooctyne (ADIBO) functionalized PolyT20 through a click reaction, resulting in the desired DNA-peptide conjugate (Step 2). The ADIBO function—a strained cycloalkyne—specifically reacts with azide at a high reaction rate (k = 0.3 M−1s−1) without need of a copper catalyst28 (so called copper free click chemistry29). Due to its bioorthogonality, the ADIBO-azide click reaction has been applied to forming conjugates of DNA to protein,30 protein to protein,31 protein to drugs,32 to functionalizing gold nanoparticles,33 to labeling aptamers34 and RNA35 with fluorescent dyes, to immobilizing proteins (or antibodies),36-38 and peptides,39 and to targeting tumor cells.40 Given these advances in click chemistry, we paid more attention to the reaction of azidoacetic anhydride with peptides to optimize its selectivity. Besides the N-terminal α–amine, the acyl anhydride can react with the ε-amine of lysine, phenolate ion of tyrosine, sulfhydryl group of cysteine, aliphatic hydroxyl of serine and threonine, and the imidazolyl ring of histidine as well.41 However, the intrinsic reactivity of these groups to an electrophile and stability of their acyl derivatives are all different. In most cases, the reactive groups involved in the acylation are the α- and ε-amine, imidazolyl ring, and to a lesser extent, –SH and –OH. The thioester and ester from acylation of cysteine, tyrosine, serine and threonine residues can be reversed to the original groups.42 In an aqueous solution, these functional groups have distinguishable acid dissociation constants (pKa): For example, an average pKa value for ε-amine of lysine in proteins is 10.5, and for α-amine of the N-terminus it is 7.7.43 Because a protonated amine is not reactive, this difference in pKa creates room for us to tune the selectivity of the acylation reaction by changing the pH and, in turn, the protonation states of these amines (Step 1 in Scheme 1). We assumed that the slightly acidic conditions should achieve higher selectivity to the α-amine than the basic conditions.
Scheme 1. Chemical reactions for attaching an oligonucleotide to N-termini of peptides.
We have studied three representative short peptides for the reactions shown in Scheme 1. The sequences and calculated physicochemical properties of these peptides (designated as P-1, P-2, and P-3, respectively) are listed in Table 1. In particular, P-1 and P-3 contain different numbers of lysines (K) and no histidine (H), and P-2 has two histidines and no lysine. Each of them carries different net charge at neutral pH. These peptides have a similar width, ∼ 1 nm at their widest. In addition, they are hydrophilic and have similar diffusion coefficients. Circular Dichroism (CD) showed that they took a random conformation in aqueous solution, whereas PolyT20 adopted an organized right-handed helical conformation (Figure S1, Supporting Information) so that it may provide an entropic advantage for threading into a nanopore.
Table 1. Peptide sequences used in this study and their physicochemical properties*.
| Sequence | Mass1 | Isoelectric point (pI)1 | Net Charge at pH 71 | Hydropathy index (h)2 | Diffusion coefficient (cm2 s−1) 3 | Size4 (L/W, nm) | |
|---|---|---|---|---|---|---|---|
| P-1 | YLGEEYVK | 999.49 | 4.26 | -1 | -5.9 | 4.37 × 10−6 | 2.3/1.0 |
| P-2 | DRVYIHPFHL | 1295.68 | 7.91 | +0.2 | -2.0 | 3.89 × 10−6 | 3.0/1.0 |
| P-3 | EAIYAAPFAKKK | 1335.76 | 10.05 | +2 | -3.6 | 3.80 × 10−6 | 3.8/1.0 |
Calculated using the peptide property calculator in http://www.innovagen.com/custom-peptide-synthesis/peptide-property-calculator/peptide-property-calculator.asp;
Calculated by adding the hydropathy value for each amino acid residue together (Kyte, J.; Doolittle, R. F. J. Mol. Biol. 1982, 157, 105-132);
Calculated based on equation log D = -0.434 log MW – 4.059 (Hosoya, O.; Chono, S.; Saso, Y.; Juni, K.; Morimoto, K.; Seki, T. J. Pharm. Pharmacol. 2004, 56, 1501-1508);
Measured from the structure model calculated by molecular mechanics with Merck molecular force field (MMFF) in Spartan'14 (L = length; W = width)
We began with P-1, a peptide that mimics a trypsin digest. First, we compared the selectivity of the anhydride with another commonly used acylating reagent N-hydroxysuccinimidyl (NHS) ester. The NHS azidoactate reacted with P-1 at pH 6.7, but produced two products that were characterized as a peptide modified by one and two azidoacetyl (N3CH2CO-) groups by MALDI mass spectrometry. In general, the NHS ester may preferentially react with the lysine amine.
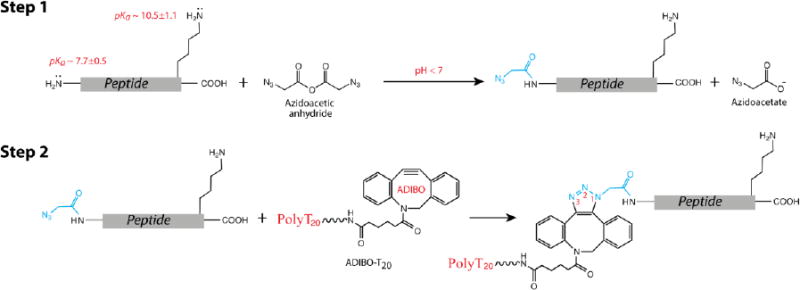
However, Mentinova et al have showed that sulfo-NHS-acetate reacts preferentially with the N-terminal α-amine only at pH ∼ 5, but did not report the reaction yield.44 These facts further motivated us to use azidoacetic anhydride as an acylating reagent. Initially, the acylation reaction was carried out with P-1 at a concentration of 0.4 mM and azidoacetic anhydride at 1.2 mM in a sodium acetate buffer, pH 6.7 at 0 °C, and monitored by reverse phase (RP) HPLC. The retention time (tR) of each starting material was determined by a separate HPLC run (Figure 1, A-i). After 15 min, three new peaks appeared in the HPLC chromatogram, labeled as 1, 2 and 3 in red (Figure 1, A-ii). Peak 1 has a shorter retention time (tR) than P-1; in contrast, both peak 2 and 3 have longer retention times compared to P-1. The ratio of these three peaks was 5:89:6, determined by their chromatographic peak areas. We separated these individual products and characterized them with MALDI and tandem mass spectrometry. Peak 1 was determined to be a product resulting from adding a 43 Da mass unit to P-1. In its CID spectrum (Figure 1, B), the mass of observed y ions (y3 to y7) matches those calculated from P-1 without modification from its C-terminus to the amino acid residue next to the N-terminus, indicating that the reaction took place at the N-terminus. The N-terminal modification was further confirmed by observed b ions (Figure 1, B), each of them matching up with the mass derived from a P-1 fragment plus an additional 42 Da. The 42 Da mass may be explained by substituting an acetyl group for one hydrogen of the α-amine (CH3CO - H). To prove this substitution, we carried out a reaction of acetic anhydride with P-1 under the same conditions, finding out that the major product (98%) had the same tR and mass as the peak 1 (Figure S2, Supporting Information). This acetyl byproduct was unexpected, albeit only ∼ 5 % in the product mixture (and not reactive in the following reaction). Further investigations are required to determine its origin. Peak 2, which was the major product, has a mass of 1083.5 Da, corresponding to mono-azidoacetylated P-1. By analyzing its CID spectrum, peak 2 was identified as a product of α-amine azidoacetylated P-1 (Figure 1, C). Peak 3 has a mass of 1166.5, corresponding to the addition of two azidoacetyl groups to P-1, and its CID spectrum indicates that it is a product of both N-terminal and ε-lysine amine azidoacetylated P-1 (Figure 1, D). The data also show that the tyrosine residue did not react with azidoacetic anhydride below neutral pH. Next, we studied effects of pH and reaction time on products by means of HPLC analysis, and the results are listed in Table 2. First of all, at a pH below 7, azidoacetic anhydride reacted with the N-terminal amine with high selectivity (> 90%). The selectivity increased by decreasing the pH, but the conversion of P-1 to products was reduced as well. Extending the reaction time increased the conversion rate of peptide to products but also reduced the selectivity, and more byproducts were produced. Overall, the reaction of azidoacetic anhydride with P-1 can achieve > 90% selectivity and a > 90% conversion rate of starting material to product. Meanwhile, we tested PCA by reacting with P-1 under the conditions reported in literature, finding that it was less reactive and selective than azidoacetic anhydride based on the MALDI mass analysis of the reaction mixtures (Figure S3).
Figure 1.
HPLC and Mass analysis of azidoacetic anhydride reacting with P-1: (A) RP HPLC chromatograms of (i) starting materials and (ii) the reaction mixture at pH 6.7; (B) Tandem mass spectrum of peak 1 in (A); (C) Tandem mass spectrum of peak 2 in (A); (D) Tandem mass spectrum of peak 3 in (A); Inserts in B, C, and D are calculated fragment ions of the corresponding peptides. See Table S1 in Supporting Information for detailed analysis of tandem mass data from collisional-induced dissociation (CID)
Table 2. Effects of reaction conditions on the selectivity of azidoacetic anhydride.
| pH | Time (min) | Conversion of P-1 | Acylating ratio* (α to ε amine) |
|---|---|---|---|
| 5.5 | 15 | 15% | 100 : 0 |
| 6.1 | 15 | 61% | 98.9 : 1.1 |
| 6.7 | 15 | 85% | 96 : 4 |
| 6.7 | 60 | 97% | 91 : 9 |
calculated based on areas of peak 2 and 3
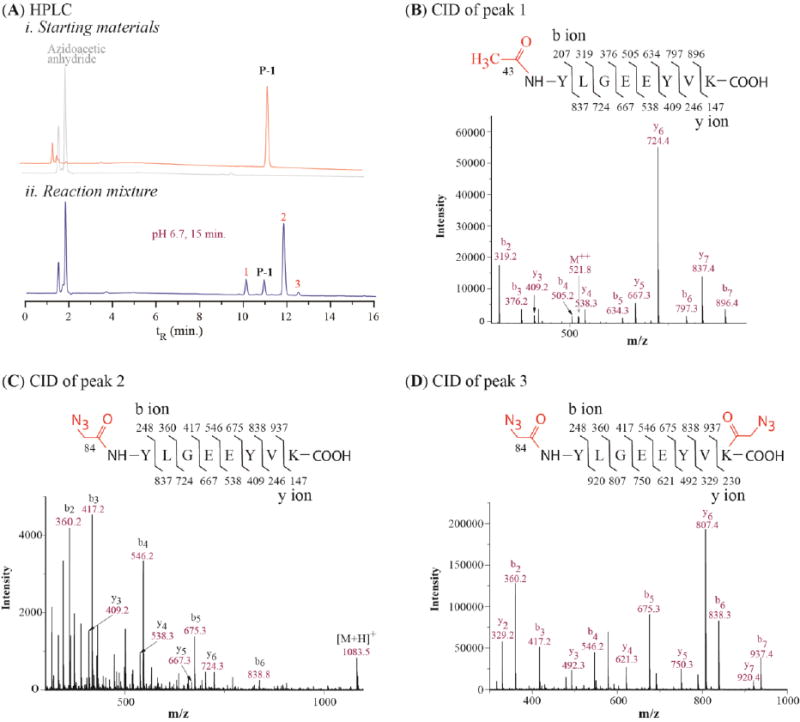
In turn, we examined the possible reaction of azidoacetic anhydride with histidine. P-2 was a peptide adopted from hormone angiotensin II, carrying two histidine residues and no lysine in the sequence. At pH 6.7, it reacted with azidoacetic anhydride, resulting in three products (Figure 2, A-i, labeled as 1, 2, 3 in red). Their ratio was determined to be 91 : 5 : 4, in which peak 1 was the major product. The CID mass analysis confirmed that the peak 1 was a product of N-terminal azidoacetylated P-2, and that both peak 2 and peak 3 were products of P-2 with one of its histidine residues azidoacetylated (see Figure S4 in Supporting Information). When lowering the reactant ratio to 3:1 between azidoacetic anhydride and P-2, the conversion rate was reduced to 17%, but the product ratio between peak 1 and 2 was 91:9 and peak 3 did not appear (Figure 2, A-ii). At pH 5.5, peak 2 became a major product (Figure 2, A-iii). This is probably because the imidazolyl ring of histidine has a lower pKa than the N-terminus amine so that it is more reactive at the low pH. Unexpectedly, the azidoacetyl group on the imidazolyl ring could not be removed by a base treatment (even with concentrated ammonia). Thus, the α-amine acylation at pH ∼ 6.7 minimizes the histidine side reaction.
Figure 2.
RP HPLC profiles of azidoacetic anhydride reacting with P-2 (A) and P-3 (B) at different pH and reaction time with temperature at 0 °C.
To further explore limitation of the acylation reaction, we studied the reaction of azidoacetic anhydride with P-3, which has three lysine residues at its C-terminus. As a result, P-3 yielded a mixture of five new products at pH 6.7, labeled as 1, 2, 3, 4, 5 in red, respectively (Figure 2, B-i). We assigned these peaks to their corresponding products in the same way as was done for P-1. The peak 1 is a product resulting from P-3 with the N-terminal α-amine acetylated, peak 2 is a product of P-3 with one lysine azidoacetylated, peak 3 is P-3 with N-terminal α-amine azidoacetylated, and peaks 4 and 5 are P-3 with two lysine azidoacetylated (Figure S5, Supporting Information). We determined the ratio of these peaks as 1:2:3:4:5 = 7.1 : 2.2 : 73.0 : 5.0 : 12.7. Again, the N-terminal azidoacetylated product (peak 3) was the major product. This result shows that the azidoacetylating reaction can selectively take place at the N-terminal α-amine of a peptide containing multiple lysine residues, but its selectivity will be reduced. We studied the pH effects on the reaction. As shown in Figure 2, B-ii, the selectivity increased to ∼ 98% at pH 5.5, but the conversion rate of the peptide-to-product was reduced to 19% from 75% at pH 6.7. Comparing the result shown in B-ii with that in B-iii of Figure 2 can deduce that extending the reaction time increases the conversion rate of peptide-to-product, and reduces the selectivity in the meanwhile. In brief, the azidoacetic anhydride reagent can rapidly react with α-amine with high selectivity under slightly acidic conditions, suitable for labeling the N-termini of trypsin digests.
Next, we studied the reaction of N-azidoacetylated peptides with PolyT20 (Step 2 in Scheme 1). ADIBO-T20 was synthesized by reacting DBCO-NHS ester with PolyT20 bearing a C12 amino modifier at its 5′-end in a phosphate buffer at pH 8, and purified by RP HPLC (see Experimental Methods and Figure S6 in Supporting Information). It spontaneously reacted with each of Nazidoacetylated peptides when they were mixed in a TEAA buffer (pH 7), resulting in the desired peptide-PolyT20 conjugates (designated as P-1-T20, P-2-T20, and P-3-T20), which were characterized by MALDI mass spectrometry. The HPLC analysis indicated that these peptides were quantitatively converted to the corresponding peptide-PolyT20 conjugates with no detectable byproducts (Figure S7, Supporting Information). Nonetheless, the times to complete these reactions were different among these peptides, 45 min for reacting with N-azidoacetylated P-1, 20 min with azidoacetylated P-2, 10 min with azidoacetylated P-3. The reaction rates seem to correlate with net charges of these peptides (see Table 1). The more positively charged P-3 reacted with the negatively charged PolyT20 fastest, the negatively charged P-1 the slowest, and P-2 (which has a smaller positive charge) was intermediate. We also noticed that the product peaks are split into two or broadened in the HPLC chromatograms. This is because the ADIBO-azide reaction produces two triazolyl regioisomers, to which a peptide is connected either at the position 1 or 3 of the triazole rings (see Scheme 1). In the same manner, we have synthesized a conjugate of P-1-T20-3′ with P-1 attached to 3′-end of Poly-T20 (Supporting Information). We also characterized these conjugates with CD spectroscopy (Figure 3). At first glance, these CD spectra are dominated by signature of DNA. Compared to polyT20 modified with ADIBO, the negative peaks (at ∼ 250 nm) of the conjugates are reduced, with their intensities in an order: ADIBO-T20 > P-2-T20 ≈ P-1-T20-3′ > P-1-T20 > P-3-T20. This may reflect the lysine residue at the C-termini interacting with the phosphate backbone when the peptide is conjugated to the 5′-end of DNA, resulting in diminished helical structure. P-3-T20, containing three lysine residues at its C-termini has the strongest interaction with PolyT20 and so the smallest intensity at 250 nm.
Figure 3.
CD spectra of polyT20-peptdie conjugates. The measurement was carried out with each analyte in a 50 μM concentration in a sodium acetate buffer, pH 6.7. Each curve was an average of 4 scans with the buffer as a reference.
Translocation of peptide-PolyT20 conjugates through solid-state nanopores
We used the setup illustrated in Figure 4-A to measure the molecular translocation, following a procedure we previously reported on measurements of DNA translocation through solid-state nanopores.45 In more detail, a silicon chip containing a nanopore drilled by TEM was mounted in a homemade PTFE/PCTFE cell and sealed with a silicone elastomer gasket to form two separate chambers. The analyte solution was loaded in the cis-side of the nanopore where the electrode was grounded, with a final concentration of ∼ 1.0 μM. All of measurements were carried out in a 0.4 M KCl electrolyte solution buffered with 1.0 mM phosphate buffer, pH 7.4. Figure 4-B shows three typical nanopores we used for the translocation measurements. Their sizes and shapes are slightly different from one another, as determined by TEM imaging. The measured conductances of pore-2 and 3 are slightly smaller than calculated values (Figure 4-B)46 probably because of their non-uniformed shapes. With each individual nanopore, we were able to finish measurements on translocation of a peptide-PolyT20 conjugate as well as its parent peptide and oligonucleotide before the pore became clogged. In a typical translocation experiment, we made measurements in the following order: PolyT20, peptide-PolyT20 conjugate, peptide, and followed by a repeated measurement of PolyT20. Between the measurements, the nanopore was rinsed with the electrolyte solution to remove any analyte residue and restore it to the original conductive state. The raw data generated by these nanopores are shown in Figure 4-C (Pore-1), D (Pore-2), and E (Pore-3), respectively. One can immediately notice that there was no translocation of peptides because neither negatively charged P-1 nor positively charged P-2 and P-3 created current blockade spikes (Figure 4, C-i, D-i, and E-i) under our measurement conditions. Although Lee and coworkers reported that charged short peptides translocated through protein nanopores and blocked the ion current,47, 48 the concentrations they used were 50 to 100 times higher than what we did here. As expected, PolyT20 was readily translocated through these nanopores (Figure 4, C-ii, D-ii, and E-ii). The peptide-PolyT20 conjugates all gave frequent blockade signals, indicating that they translocated too (Figure 4, C-iii, D-iii, and E-iii). Their translocation was further confirmed by reversing the bias across the nanopore. When doing this, we observed the blockade signals only after a significant time of translocating the conjugates in the forward direction, which strongly suggested that molecules were translocated through the pores (data not shown). Translocation was also confirmed by measuring the voltage dependence of the duration of the current blockade (see below).
Figure 4.
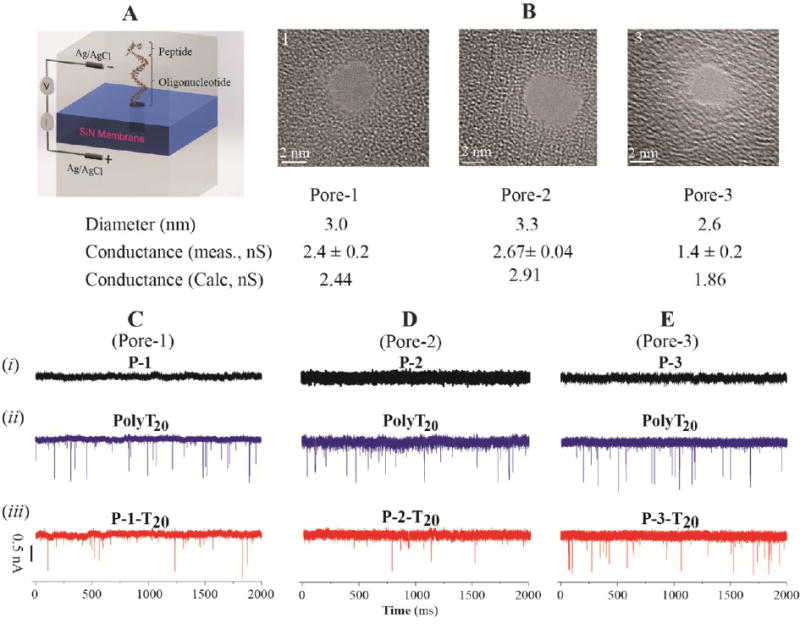
(A) Schematic illustration of a nanopore device for translocation measurements; (B) TEM images of the nanopores used for translocation and their physical parameters; (C) Ionic current traces of i: P-1, ii: PolyT20, iii: P-1-T20 in Pore-1;(D) Ionic current traces of i: P-2, ii: PolyT20, iii: P-2-T20 in Pore-2; (E) Ionic current traces of i: P-3, ii: PolyT20, iii: P-3-T20 in Pore-3 (Bias: 500 mV; Analyte concentration: ∼ 1.0 μM)
These current traces (examples are shown in Figure S8) were analyzed by means of the OpenNanopore software and each spike was assigned a dwell time and a current-blockade value. We found that the majority of translocation events were single-level blockades (> 90% for PolyT20 and ∼ 95–97% for peptide-PolyT20 conjugates) while only a small percentage of the events were multi-level blockades (∼10% for PolyT20 and ∼ 3-5% for peptide-PolyT20 conjugates). In comparison, the multi-level events occurred rarely with PolyT20 and even less so with the peptide-PolyT20 conjugates, which may be partly explained by the difference between the 5′ and 3′ end threading of the PolyT20 (further discussed below). Some typical scatter plots of dwell times vs current blockades for PolyT20 and its peptide conjugates (at 500 mV bias) are shown in the panels labeled i in Figure 5. The dwell times of the translocation events were reduced with increased voltage bias (Figure S9 in Supporting Information). This observation is also consistent with molecular translocation as the origin of these blockade events. It has been reported that for a translocation event with a favorable electric field (i.e. electrophoretically driven), the dwell time of a polymeric molecule in the nanopore decreases as the voltage increases.48, 49
Figure 5.
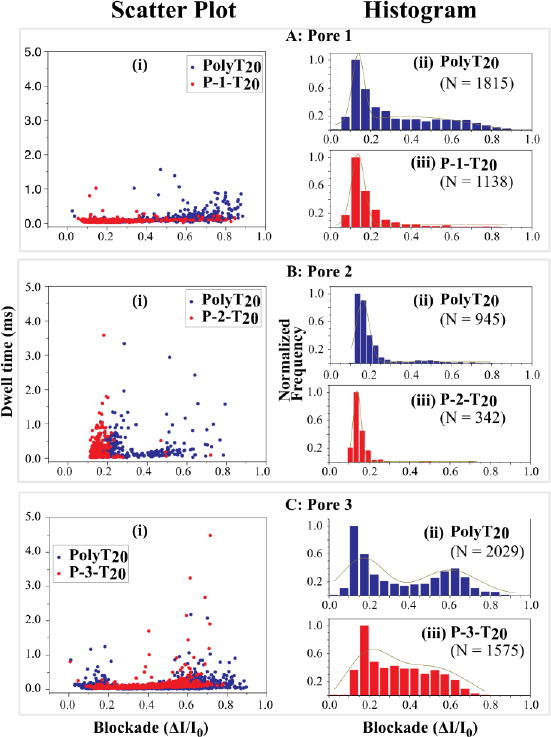
Scatter-plot of dwell time vs current blockade (i) and histograms of fractional current blockades for PolyT20 (ii) and its peptide conjugates (iii) at bias = 500 mV). (A) Translocation through Pore 1; (B) Translocation through Pore 2; (C) Translocation through Pore 3. Blue: PolyT20, Red: peptide conjugate; ΔI = I0 (open pore current) – I (blocked pore current). The dark yellow lines in histograms are the Gaussian fitting curves and N = number of events.
The distributions of dwell times for the Poly-T20 and the peptide conjugates appear to be highly overlapped. In the absence of complicating factors (such as interactions with the pore surface) the distribution of blockade times is described by a first-passage time distribution as given by Carson et al.50 We have approximated this by an exponential distribution and fitted measured data for all three pores and the various conjugates (Figure S10). There are a significant number of very long blockades, so we have also listed the mean and medians of the blockade times in Table 3. The discrepancies between the three measures of the distributions are a measure of how widely the data are distributed. In addition, the distributions change from pore to pore, as can be seen by comparing values for PolyT20 between the three pores. However, by most measures, we see the somewhat surprising result that the conjugates translocate more rapidly, or on about the same timescale, as PolyT20 alone (data for Pores 1 to 3).
Table 3. Dwell times of PolyT20 and its peptide conjugates in different nanopores.
| Pore 1 | Pore 2 | Pore 3 | Pore 4 | |||||
|---|---|---|---|---|---|---|---|---|
| Sample | P-1-T20 | PolyT20 | P-2-T20 | PolyT20 | P-3-T20 | PolyT20 | P-1-T20 | P-1-T20-3′ |
| Exp. Fit (μS) | 38±0 | 61±1 | 64±5 | 134±13 | 86± 4 | 74±1 | 75±3 | 124±3 |
| Mean (μS) | 66 | 91 | 207 | 158 | 101 | 94 | 129 | 188 |
| Median (μS) | 60 | 70 | 60 | 90 | 70 | 70 | 80 | 110 |
Data for the current blockades give a clearer picture of the differences between PolyT20 and its peptide conjugates than the widely-distributed translocation times. In order to compare data for the various samples, each blockade-current data set was normalized by its maximum value and plotted into a normalized histogram, as shown in Figure 5 (panels labeled ii for PolyT20, and panels labeled iii for its peptide conjugates). We consider first the PolyT20 data. All the data sets were fitted by a double-peaked Gaussian function with R2 > 0.90 (Panel ii's in Figure 5), with a major peak at ΔI/I0 in the range of 0.15 to 0.2 and a minor peak in the range of 0.5 to 0.6. Similar features have been reported for DNA translocation through α-hemolysin pores where they were explained by the so-called Christmas-tree effect.51,52 When a single stranded DNA is translocated, it can thread either via its 5′- or its 3′-end into a nanopore. Meller and coworkers demonstrated by all-atom molecular dynamics (MD) that DNA bases in a stretched conformation preferably tilt towards the 5′-end in a confined pore.51 This is because all of nucleosides in DNA have a β configuration, in which a nucleobase stays at the same side with the 5′-hydroxyl group of a nucleoside on the deoxyribose ring. As a result, DNA translocation with threading through the 3′-end should be more frequent and less blockaded than through the 5′-end threading (just as a Christmas tree can be moved into a door more easily from its trunk end than from its tip). Based on this hypothesis, we may assign the major blockade peaks (at smaller current values) as events for which the 3′-end threaded first. From the literature, we found a similar trend in the translocation of PolyA20 through a α-HL nanopore.53 Thus, when a short peptide is tethered to the 5′-end of PolyT20, it reduces the probability of threading through the 5′-end. This accounts for the observation that the large current blockades were diminished in the translocation of the peptide-PolyT20 conjugates, such as in Panel iii's of Figure 5-A and -B, compared to the corresponding PolyT20. P-3-T20 displays a large tail following its major peak (Panel iii in Figure 5-C) and also translocates more slowly (Table 3) than the other two conjugates just mentioned above. This might be explained by the conformational structure of the peptide-PolyT20 conjugate. The CD spectrum (Figure 3) shows that the peptide P-3 has stronger interactions with the backbone of PolyT20 in the conjugate than other two peptides, which may result in looped structures. When a conjugate with such a structure translocates through the nanopore, it would create larger blockade currents, as observed.
To further verify the Christmas-tree effect, we synthesized a conjugate P-1-T20-3′, in which P-1 is tethered to the 3′-end of PolyT20 and compared its translocation with that of P-1-T20, in which P-1 is tethered to the 5′-end of PolyT20 (illustrated in Figure 6 A), in a nanopore with a diameter of ∼ 3.2 nm (Pore 4 in Figure 6-B). From Figure 6-C, one can immediately see that P-1-T20 was translocated through the nanopore much more frequently than P-1-T20-3′. From data collected over a 2000 ms period for both conjugates, we find that that the P-1-T20 was translocated 5 fold more frequently than P-1-T20-3′ (337 vs 66 events). These two sets of data were normalized in the same manner as was done for those in Figure 5, plotted into histograms separately, and then fitted with a double peak Gaussian function (Figure 6, D). As expected, P-1-T20 shows a preference for threading into the nanopore from the 3′-end of PolyT20 with a major blockade peak at ΔI/I0 = 0.09 (Figure 6-D, i). Similarly, P-1-T20-3′ also shows a major blockade peak at ΔI/I0 = 0.11 (Figure 6-D, ii), indicating that it still entered the nanopore mainly through the 3′-end of PolyT20, despite being blocked by peptide tethered at that end. This may be explained by the Christmas-tree effect, which results in a strong tendency for DNA to translocate through the 3′-end threading. Since P-1 is relatively short and negatively charged, it would not be able to completely block the 3′-end threading and only reduced the translocation frequency when it was tethered to the 3′-end of PolyT20. Dwell times for the two conjugates are summarized under the column “Pore 4” in Table 3, distributions of which are well fitted into an exponential decay function (Figure S11). All three measures of the distribution indicate that P-1-T20 translocated faster as the conjugate entered more probably at the 3′-end. The small fraction of multi-level events may be at least partially explained by the Christmas-tree effect. The multilevel events occurred mostly in higher current blockade signals (Panel I and J in Figure S8) and were reduced from ∼10% with PolyT20 to ∼ 3-5% with the peptide-PolyT20 conjugates. Since a lower front step exists in these multi-level events, we attribute them to the resistance to entry of the pore from the 5′-end of PolyT20. Given the “Christmas-tree effect”, we conclude that the attachment of peptides to 5′-end of an oligonucleotide is an optimal choice for the nanopore translocation.
Figure 6.
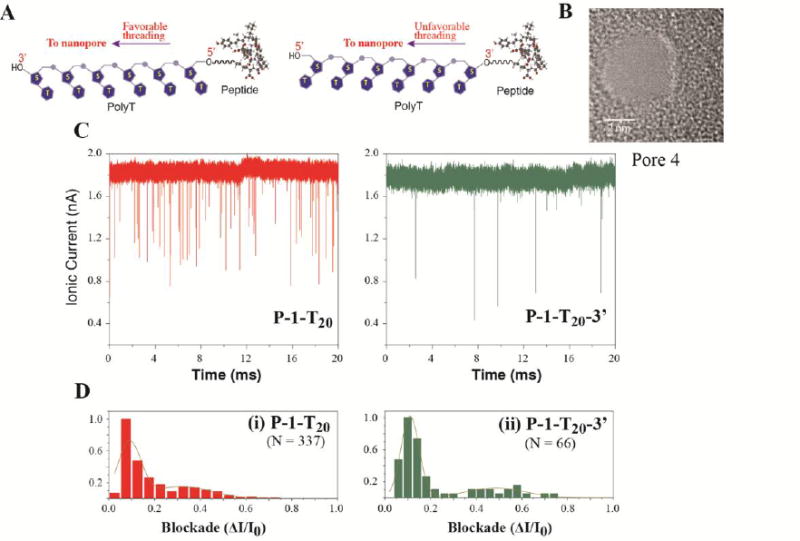
Testing the Christmas-tree effect in translocation of peptide-DNA conjugates. (A) Schematic illustration of peptide-PolyT20 conjugates; (B) TEM images of the nanopores used for translocation; (C) Ionic current traces of peptide-PolyT20 conjugates translocating through the nanopore; (D) Current blockade histograms of (i) P-1-T20 and (ii) P-1-T20-3′ with normalized data. The dark yellow lines in histograms are Gaussian fitting curves and N = number of events
Conclusions
The objective of this present study was to develop a simple and effective chemistry to functionalize peptides with a charged threading molecule to facilitate their translocation through nanopores. We have harnessed an acylation reaction for rapid introduction of an orthogonal azido function to N-termini of peptides, which allowed us to quantitatively attach a charged oligonucleotide to peptides using a click reaction without the need of separating the intermediate products. At pH ∼ 6.7 and 0 °C, azidoacetic anhydride quickly reacted with the α amine of a peptide containing one lysine residue with > 90% selectivity, and the side reaction with imidazolyl ring of histidine was reduced to a minimum. Thus, our chemistry should be practical for use in preparing peptides sample obtained from trypsin digests for nanopore analysis. Due to the reduced selectivity (∼ 80%) in reacting with a peptide containing three lysine residues, further improvements are required if the method is to be applied more widely to peptides from other sources.
Furthermore, we have shown that the peptide-PolyT20 conjugates can effectively translocate through solid-state nanopores, which lays down a foundation for us to develop a technique for analysis of proteins using nanopores. Our data indicate that an oligonucleotide, such as PolyT20 can be used as an effective molecular thread to carry peptides through solid-state nanopores, its conjugates preferentially entering the nanopore from the 3′-end. While the sensitivity of ion-current measurements is unlikely to be adequate for de novo sequencing, it is clear that structural aspects of peptides can be probed using the translocation of conjugates. We hope to integrate this technology with recognition tunneling in order to explore the possibility of sequencing with single amino acid resolution in the future.10
Methods
General information
Chemicals were purchased from Sigma-Aldrich and anhydrous organic solvents were Aldrich's Sure/Seal™. DBCO-NHS ester was purchased from Click Chemistry Tools. Peptides were custom synthesized by CPC Scientific (San Jose, C, USA) and oligonucleotides (PolyT20) with a C12 amino modifier at its 5′-end by IDT (Integrated DNA Technologies). 1H and 13C NMR spectra were recorded at 400 MHz (1H), 100 MHz (13C), respectively. Chemical shifts are given in parts per million (ppm) on the delta scale (δ) and are referred to the solvent residual peak. HPLC analysis and purification were carried out in Agilent 1100 series equipped with a UV detector and a fraction collector. A Zorbax Eclipse Plus C18 column (4.6 × 150 mm, particle size 5 μm) from Agilent was used for the reversed phase HPLC. MALDI-TOF analysis was performed on Voyager-DE STR instrument.
Azidoacetic anhydride
The synthesis was carried out following a method reported in literature with modification.54 N,N′-Dicyclohexylcarbodiimide (DCC, 204 mg, 0.98 mmol) was added to a solution of 2-azidoacetic acid (200 mg, 1.98 mmol) in anhydrous tetrahydrofuran (2 mL). The solution was stirred for 2 h and 15 min, during which the precipitate was gradually produced, and filtered. The filtrate was concentrated by rotary evaporation, giving azidoacetic anhydride (150 mg, 42%) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ: 3.85 (s, 4H); 13C NMR (100 MHz, CDCl3) δ: 168.3, 50.0.
General procedure for reaction of azidoacetic anhydride with peptides (P-1, P-2, and P-3)
A solution of azidoacetic anhydride in acetonitrile (5 mM, 3.0 μL) was added to a solution of peptide in a 50 mM sodium acetate buffer (0.5 mM, 10 μL) with a predefined pH value in an eppendorf tube. The reaction was kept at 0°C for 15 min, followed by the addition of water (10 μL), and remaining the reaction in ice for another 10 min for completely terminating the reaction. RP HPLC, with a gradient of 5 to 45% B in 25 min (solvent A: 0.1 M TEAA buffer, pH 7.0; solvent B: acetonitrile), was used to monitor the reactions and separate products.
Functionalization of PolyT20 with ADIBO
A solution of PolyT20 containing a C12 amino modifier at its 5′-end (1 mM, 10 μL) in water and DBCO-NHS ester in DMSO (15 mM, 80 μL) were mixed in a phosphate buffer (30 μL, pH 8). The solution was shaken for 20 min at room temperature. The product (ADIBO-T20) was purified by RP HPLC in a Zorbax Eclipse Plus C18 column (4.6 × 150 mm, particle size 5 μm) with a gradient of 10 to 60% B in 25 min (solvent A: 0.1 M TEAA buffer, pH 7.0; solvent B: acetonitrile). MALDI-TOF-MS calc. for [C233H304N42O143P20](M+H): m/z 6600.58; found: 6603.34.
Reaction of N-azidoacetylated P-1 with ADIBO-T20
A solution of N-azidoacetylated P-1 (30 μM, 15 μL) in a TEAA buffer (50 mM, pH 7) was mixed with ADIBO-T20 (30 μM, 15 μL) in the TEAA buffer (50 mM, pH 7), and shaken at room temperature for 45 min. RP HPLC analysis showed the starting material was consumed. The product (P-1-T20) was purified by RP HPLC in a Zorbax Eclipse Plus C18 column (4.6 × 150 mm, particle size 5 μm) with a gradient of 10 to 60% B in 25 min (solvent A: 0.1 M TEAA buffer, pH 7.0; solvent B: acetonitrile). After lyophilization, the product was given as a white powder. MALDI-TOF-MS calc. for [C282H374N54O159P20] (M+H): m/z 7683.12; found: 7684.72.
Reaction of N-azidoacetylated P-2 with ADIBO-T20
A solution of N-azidoacetylated P-2 (30 μM, 15 μL) in a TEAA buffer (50 mM, pH 7) was mixed with ADBCO-T20 (30 μM, 25 μL) in the TEAA buffer (50 mM, pH 7), and shaken at room temperature for 20 min, purified by RP HPLC under the same conditions as did for P-1-T20. After lyophilization, the product (P-2-T20) was given as a white powder. MALDI-MS calc. for [C297H394N62O158P20] (M+H): 7979.31; found: 7978.64).
Reaction of N-azidoacetylated P-3 with ADIBO-T20
A solution of N-azidoacetylated P-3 (30 μM, 15 μL) in a TEAA buffer (50 mM, pH 7) was mixed with ADIBO-T20 (30 μM, 15 μL) in the TEAA buffer (50 mM, pH 7), and shaken at room temperature for 10 min, purified by RP HPLC under the same conditions as did for P-1-T20. After lyophilization, the product (P-3-T20) was given as a white powder. MALDI-MS calc. for [C299H406N60O160P20] (M+H): m/z 8019.39; found: 8020.58.
Tandem mass spectrometry
Collisional-induced dissociation (CID) and subsequent mass analysis of modified peptides were carried out on a Bruker MaXis 4G quadrupole-time-of-flight (Q-TOF) mass spectrometer equipped with a microflow nebulizer electrospray ionization (ESI) source operated in positive ion mode. HPLC-purified peptide samples were diluted to a final concentration of approximately 20 μM in a 50/50 mixture of acetonitrile/water containing 0.1% (v/v) formic acid. Prior to analysis, the TOF mass analyzer was externally calibrated with a tuning mixture supplied by Agilent containing 10 species of varying but equally spaced masses from 118 to 2722 Da. Peptide solutions were infused into the ion source by syringe pump at a rate of 1.0 μL/min. The end plate offset and capillary were set to potentials of 500 V and 4,500 V, respectively. The nebulizer gas and the dry gas (both N2) were set to 1.4 Bar and 4.0 L/min, respectively, and the dry gas temperature was set to 220°C. The RF amplitude on ion funnel 1 and the multipole were both set to 400 Vpp. No in-source CID energy was used. Quadrupole ion energy was set to 3.0 eV. Precursor ions were selected with an m/z width of 2 and imparted enough energy (usually 10 – 40 eV) to diminish the relative abundance of the precursor ion to less than 5%. Spectral digitization was set to a rate of 4 GHz and individual TOF transients were summed and recorded at a rate of 1 Hz. After equilibration and spray stabilization, each mass spectrum was recorded for 1.5 min, and then averaged into a single spectrum.
Fabrication of nanopores
Silicon chips (5 × 5 mm) coated with silicon nitride (30 nm thick) from Norcada Inc were used for the fabrication of nanopores. First, a 250 × 250 μm SiNx window was opened from the Si backside of the chip and the thickness of free-standing SiNx membrane left was 30 nm. After standard cleaning process, nanopores were drilled using the electron beam in JEOL 2010FEG transmission electron microscope (TEM) at 200 kV. The size of the pores were controlled by the electron beam and monitored using the TEM CCD. The nanopores were imaged right after drilling.
Translocation measurements
Prior to a translocation experiment, a nanopore chip was immersed in hot piranha (H2O2 : H2SO4 = 1:3) for 10 min, and then rinsed with water. After drying with a N2 flow, the nanopore chip was assembled in a piranha-cleaned PCTFE cell to form the cis reservoir, and sealed with a quick-curing silicone elastomer gasket for reduced capacitance. The PCTFE cell with a nanopore chip was then assembled with PTFE base to form the trans reservoir. Electrolyte solution used was 0.4 M KCl in 1 mM phosphate buffer (pH 7.0), which was filtered with a Millipore 0.2 μm filter. Ag/AgCl electrodes, made from fresh Ag wires with bleach, were inserted into both cis and trans sample reservoirs for ionic current measurement. All of analytes were dissolved in the electrolyte solution for the nanopore analysis.
The electrolyte solution was used as a translocation reference. Each translocation experiment began with filling the electrolyte solution in both reservoirs and running the reference to ensure a steady baseline current and no electrical spikes. Then, an analyte solution was injected into the cis reservoir with a final concentration of 0.5 - 1 μM. A translocation bias was applied to the Ag/AgCl electrode in the trans reservoir, while the electrode in the cis reservoir was kept grounded to avoid adsorption of analyte molecules. After recording the ionic current, the cis reservoir was drained and rinsed with the buffer solution. Another base line was recorded to ensure no contaminations left in cis reservoir before a new analyte solution was injected.
Data collection and analysis
Ionic currents were collected either at a 100 kHz sampling rate with a 10 kHz low pass filter using patch clamp amplifier Axon Axopatch 200B, with digitizer DigiData 1550A from Axon Instruments Inc. PClamp 10.4 software and an in-house developed LabView program were used for recording the ionic current data. The data were analyzed using MATLAB based software OpenNanopore, developed by Laboratory of Nanoscale Biology (LBEN) of École polytechnique fédérale de Lausanne (EPFL). OpenNanopore fits abrupt stepwise signals in the presence of Gaussian noise with a level threshold of 200 pA. Statistic analysis was carried out in OriginPro 2015, in which the Levenberg–Marquardt algorithm was used for Gaussian fitting.
Supplementary Material
Acknowledgments
This work was supported in part by a DNA sequencing technology grant from the NHGRI (HG 006323). We would like to thank Professor M. Wanunu of Northeastern University for his suggestions and advice on nanopore translocation experiments.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet at http://pubs.acs.org.
Conflict of Interest: The authors declare the following competing financial interest(s): PZ, SL, and SB are named as inventors on related patent applications.
Author Contributions: The manuscript was written through contributions of all authors.
References
- 1.Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Adey A, Nova IC, Craig JM, Langford KW, Samson JM, Daza R, Doering K, Shendure J, Gundlach JH. Decoding Long Nanopore Sequencing Reads of Natural DNA. Nat Biotechnol. 2014;32:829–833. doi: 10.1038/nbt.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oukhaled A, Bacri L, Pastoriza-Gallego M, Betton JM, Pelta J. Sensing Proteins through Nanopores: Fundamental to Applications. ACS Chem Biol. 2012;7:1935–1949. doi: 10.1021/cb300449t. [DOI] [PubMed] [Google Scholar]
- 3.Fennouri A, Przybylski C, Pastoriza-Gallego M, Bacri L, Auvray L, Daniel R. Single Molecule Detection of Glycosaminoglycan Hyaluronic Acid Oligosaccharides and Depolymerization Enzyme Activity Using a Protein Nanopore. ACS Nano. 2012;6:9672–9678. doi: 10.1021/nn3031047. [DOI] [PubMed] [Google Scholar]
- 4.McMullen A, de Haan HW, Tang JX, Stein S. Stiff filamentous Virus Translocations through Solid-State Nanopores. Nat Commun. 2014;5:4171. doi: 10.1038/ncomms5171. [DOI] [PubMed] [Google Scholar]
- 5.Reiner JE, Balijepalli A, Robertson JW, Campbell J, Suehle J, Kasianowicz JJ. Disease Detection and Management via Single Nanopore-Based Sensors. Chem Rev. 2012;112:6431–6451. doi: 10.1021/cr300381m. [DOI] [PubMed] [Google Scholar]
- 6.Pennisi E. DNA Sequencers Still Waiting for the Nanopore Revolution. Science. 2014;343:829–830. doi: 10.1126/science.343.6173.829. [DOI] [PubMed] [Google Scholar]
- 7.Mikheyev AA, Tin MM. A First Look at the Oxford Nanopore MinION Sequencer. Mol Ecol Resour. 2014;14:1097–1102. doi: 10.1111/1755-0998.12324. [DOI] [PubMed] [Google Scholar]
- 8.Ashton PM, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, Wain J, O'Grady J. MinION Nanopore Sequencing Identifies the Position and Structure of a Bacterial Antibiotic Resistance Island. Nat Biotechnol. 2015;33:296–300. doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 9.Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved Data Analysis for the MinION Nanopore Sequencer. Nat methods. 2015;12:351–356. doi: 10.1038/nmeth.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao Y, Ashcroft B, Zhang P, Liu H, Sen S, Song W, Im J, Gyarfas B, Manna S, Biswas S, Borges C, Lindsay S. Single-Molecule Spectroscopy of Amino Acids and Peptides by Recognition Tunnelling. Nat Nanotechnol. 2014;9:466–473. doi: 10.1038/nnano.2014.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ohshiro T, Tsutsui M, Yokota K, Furuhashi M, Taniguchi M, Kawai T. Detection of Post-Translational Modifications in Single Peptides Using Electron Tunnelling Currents. Nat Nanotechnol. 2014;9:835–840. doi: 10.1038/nnano.2014.193. [DOI] [PubMed] [Google Scholar]
- 12.Krasniqi B, Lee JS. RNase A Does Not Translocate the Alpha-Hemolysin Pore. PLoS One. 2014;9:e88004. doi: 10.1371/journal.pone.0088004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nivala J, Marks DB, Akeson M. Unfoldase-Mediated Protein Translocation Through an Alpha-Hemolysin Nanopore. Nat Biotechnol. 2013;31:247–250. doi: 10.1038/nbt.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez-Larrea D, Bayley H. Multistep Protein Unfolding during Nanopore Translocation. Nat Nanotechnol. 2013;8:288–295. doi: 10.1038/nnano.2013.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steen H, Mann M. The ABC's (and XYZ's) of Peptide Sequencing. Nat Rev Mol Cell Biol. 2004;5:699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- 16.Picotti P, Aebersold R. Selected Reaction Monitoring–Based Proteomics: Workflows, Potential, Pitfalls and Future Directions. Nat methods. 2012;6:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- 17.Pang P, Ashcroft BA, Song W, Zhang P, Biswas S, Qing Q, Yang J, Nemanich RJ, Bai J, Smith JT, et al. Fixed-Gap Tunnel Junction for Reading DNA Nucleotides. ACS Nano. 2014;8:11994–12003. doi: 10.1021/nn505356g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Venta K, Shemer G, Puster M, Rodriguez-Manzo JA, Balan A, Rosenstein JK, Shepard K, Drndic M. Differentiation of Short, Single-Stranded DNA Homopolymers in Solid-State Nanopores. ACS Nano. 2013;7:4629–4636. doi: 10.1021/nn4014388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Si W, Sha J, Liu L, Qiu Y, Chen Y. Effect of Nanopore Size on Poly(dT)30 Translocation through Silicon Nitride Membrane. Sci China Technol Sc. 2013;56:2398–2402. [Google Scholar]
- 20.Lee MH, Kumar A, Park KB, Cho SY, Kim HM, Lim MC, Kim YR, Kim KB. A Low-Noise Solid-State Nanopore Platform Based on a Highly Insulating Substrate. Sci Rep. 2014;4:7448. doi: 10.1038/srep07448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundblad RL. Chemical Reagents for Protein Modification. Fourth. CRC Press; 2014. [Google Scholar]
- 22.Chan AO, Ho CM, Chong HC, Leung YC, Huang JS, Wong MK, Che CM. Modification of N-Terminal Alpha-Amino Groups of Peptides and Proteins Using Ketenes. J Am Chem Soc. 2012;134:2589–2598. doi: 10.1021/ja208009r. [DOI] [PubMed] [Google Scholar]
- 23.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. N-Terminal Protein Modification through a Biomimetic Transamination Reaction. Angew Chem Int Ed. 2006;45:5307–5311. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 24.Witus LS, Netirojjanakul C, Palla KS, Muehl EM, Weng CH, Iavarone AT, Francis MB. Site-Specific Protein Transamination Using N-Methylpyridinium-4-Carboxaldehyde. J Am Chem Soc. 2013;135:17223–17229. doi: 10.1021/ja408868a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacDonald JI, Munch HK, Moore T, Francis MB. One-Step Site-Specific Modification of Native Proteins with 2-Pyridinecarboxyaldehydes. Nat Chem Biol. 2015;11:326–331. doi: 10.1038/nchembio.1792. [DOI] [PubMed] [Google Scholar]
- 26.Song J, Kim HJ. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Peptide Sequencing Utilizing Selective N-Terminal Bromoacetylation. Anal Biochem. 2012;423:269–276. doi: 10.1016/j.ab.2011.11.022. [DOI] [PubMed] [Google Scholar]
- 27.Koehler CJ, Arntzen MO, Strozynski M, Treumann A, Thiede B. Isobaric Peptide Termini Labeling Utilizing Site-Specific N-Terminal Succinylation. Anal Chem. 2011;83:4775–4781. doi: 10.1021/ac200229w. [DOI] [PubMed] [Google Scholar]
- 28.Debets MF, van Berkel SS, Schoffelen S, Rutjes FP, van Hest JC, van Delft FL. Aza-Dibenzocyclooctynes for Fast and Efficient Enzyme PEGylation via Copper-Free (3+2) Cycloaddition. Chem Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]
- 29.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-Free Click Chemistry for Dynamic in vivo Imaging. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nojima T, Konno H, Kodera N, Seio K, Taguchi H, Yoshida M. Nano-Scale Alignment of Proteins on a Flexible DNA Backbone. PLoS One. 2012;7:e52534. doi: 10.1371/journal.pone.0052534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Witte MD, Cragnolini JJ, Dougan SK, Yoder NC, Popp MW, Ploegh HL. Preparation of Unnatural N-to-N and C-to-C Protein Fusions. Proc Natl Acad Sci U S A. 2012;109:11993–11998. doi: 10.1073/pnas.1205427109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zimmerman ES, Heibeck TH, Gill A, Li X, Murray CJ, Madlansacay MR, Tran C, Uter NT, Yin G, Rivers PJ, et al. Production of Site-Specific Antibody-Drug Conjugates Using Optimized Non-Natural Amino Acids in a Cell-Free Expression System. Bioconjugate Chem. 2014;25:351–361. doi: 10.1021/bc400490z. [DOI] [PubMed] [Google Scholar]
- 33.Heuer-Jungemann A, Kirkwood R, El-Sagheer AH, Brown T, Kanaras AG. Copper-Free Click Chemistry as an Emerging Tool for the Programmed Ligation of DNA-Functionalised Gold Nanoparticles. Nanoscale. 2013;5:7209–7212. doi: 10.1039/c3nr02362a. [DOI] [PubMed] [Google Scholar]
- 34.Subramanian N, Sreemanthula JB, Balaji B, Kanwar JR, Biswas J, Krishnakumar S. A Strain-Promoted Alkyne-Azide Cycloaddition (SPAAC) Reaction of a Novel EpCAM Aptamer-Fluorescent Conjugate for Imaging of Cancer Cells. Chem Commun. 2014;50:11810–11813. doi: 10.1039/c4cc02996h. [DOI] [PubMed] [Google Scholar]
- 35.Winz ML, Samanta A, Benzinger D, Jaschke A. Site-Specific Terminal and Internal Labeling of RNA by Poly(A) Polymerase Tailing and Copper-Catalyzed or Copper-Free Strain-Promoted Click Chemistry. Nucleic Acids Res. 2012;40:e78. doi: 10.1093/nar/gks062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuzmin A, Poloukhtine A, Wolfert MA, Popik VV. Surface Functionalization Using Catalyst-Free Azide-Alkyne Cycloaddition. Bioconjugate Chem. 2010;21:2076–2085. doi: 10.1021/bc100306u. [DOI] [PubMed] [Google Scholar]
- 37.Hui JZ, Al Zaki A, Cheng Z, Popik V, Zhang H, Luning Prak ET, Tsourkas A. Facile Method for the Site-Specific, Covalent Attachment of Full-Length IgG onto Nanoparticles. Small. 2014;10:3354–3363. doi: 10.1002/smll.201303629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotagiri N, Li Z, Xu X, Mondal S, Nehorai A, Achilefu S. Antibody Quantum Dot Conjugates Developed via Copper-Free Click Chemistry for Rapid Analysis of Biological Samples Using a Microfluidic Microsphere Array System. Bioconjugate Chem. 2014;25:1272–1281. doi: 10.1021/bc500139u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prim D, Rebeaud F, Cosandey V, Marti R, Passeraub P, Pfeifer ME. ADIBO-Based “Click” Chemistry for Diagnostic Peptide Micro-Array Fabrication: Physicochemical and Assay Characteristics. Molecules. 2013;18:9833–9849. doi: 10.3390/molecules18089833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koo H, Lee S, Na JH, Kim SH, Hahn SK, Choi K, Kwon IC, Jeong SY, Kim K. Bioorthogonal Copper-Free Click Chemistry in Vivo for Tumor-Targeted Delivery of Nanoparticles. Angew Chem Int Ed. 2012;51:11836–11840. doi: 10.1002/anie.201206703. [DOI] [PubMed] [Google Scholar]
- 41.Gounaris AD, Perlmann GE. Succinylation of Pepsinogen. J Biol Chem. 1967;242:2739–2745. [PubMed] [Google Scholar]
- 42.Hermanson GT. Bopconjugate Techniques. Second. Elsevier; 2008. [Google Scholar]
- 43.Grimsley GR, Scholtz JM, Pace CN. A Summary of the Measured pK Values of the Ionizable Groups in Folded Proteins. Protein Sci. 2009;18:247–251. doi: 10.1002/pro.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mentinova M, Barefoot NZ, McLuckey SA. Solution versus Gas-Phase Modification of Peptide Cations with NHS-Ester Reagents. J Am Soc Mass Spectrom. 2012;23:282–289. doi: 10.1007/s13361-011-0291-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krishnakumar P, Gyarfas B, Song W, Sen S, Zhang P, Krstic P, Lindsay S. Slowing DNA Translocation through a Nanopore Using a Functionalized Electrode. ACS Nano. 2013;7:10319–10326. doi: 10.1021/nn404743f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wanunu M. Nanopores: A Journey towards DNA Sequencing. Phys Life Rev. 2012;9:125–158. doi: 10.1016/j.plrev.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stefureac R, Long Yt, Kraatz HB, Howard P, Lee JS. Transport of α-Helical Peptides through α-Hemolysin and Aerolysin Pores. Biochemistry. 2006;45:9172–9179. doi: 10.1021/bi0604835. [DOI] [PubMed] [Google Scholar]
- 48.Meng H, Detillieux D, Baran C, Krasniqi B, Christensen C, Madampage C, Stefureac RI, Lee JS. Nanopore Analysis of Tethered Peptides. J Pept Sci. 2010;16:701–708. doi: 10.1002/psc.1289. [DOI] [PubMed] [Google Scholar]
- 49.Christensen C, Baran C, Krasniqi B, Stefureac RI, Nokhrin S, Lee JS. Effect of Charge, Topology and Orientation of the Electric Field on the Interaction of Peptides with the α -Hemolysin pore. J Pept Sci. 2011;17:726–734. doi: 10.1002/psc.1393. [DOI] [PubMed] [Google Scholar]
- 50.Carson S, Wilson J, Aksimentiev A, Wanunu M. Smooth DNA Transport through a Narrowed Pore Geometry. Biophys J. 2014;107:2381–2393. doi: 10.1016/j.bpj.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mathe J, Aksimentiev A, Nelson DR, Schulten K, Meller A. Orientation Discrimination of Single-Stranded DNA Inside the Alpha-Hemolysin Membrane Channel. Proc Natl Acad Sci U S A. 2005;102:12377–12382. doi: 10.1073/pnas.0502947102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Muzard J, Martinho M, Mathe J, Bockelmann U, Viasnoff V. DNA Translocation and Unzipping Through a Nanopore: Some Geometrical Effects. Biophys J. 2010;98:2170–2178. doi: 10.1016/j.bpj.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ying YL, Li DW, Liu Y, Dey SK, Kraatz HB, Long YT. Recognizing the Translocation Signals of Individual Peptide-Oligonucleotide Conjugates Using an α-Hemolysin Nanopore. Chem Commun. 2012;48:8784–8786. doi: 10.1039/c2cc32636a. [DOI] [PubMed] [Google Scholar]
- 54.Grajkowski A, 'lak JC, Gapeev A, Schindler C, Beaucage SL. Convenient Synthesis of a Propargylated Cyclic (3′-5′) Diguanylic Acid and Its “Click” Conjugation to a Biotinylated Azide. Bioconjugate Chem. 2010;21:2147–2152. doi: 10.1021/bc1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.