

Abstract
Recent breakthroughs in cryo-electron microscopy imaging technology provide an unprecedented view of biology at the nanoscale. To complement these technical advances, here we demonstrate the use of tunable substrates to streamline the isolation of biological entities from human cells. We have tested the capacity of tunable microchip devices using a variety of samples including virus assemblies and the breast cancer susceptibility protein (BRCA1) produced in cancer cells. Overall, microchip applications may shed light on ill-defined clinical issues related to molecular disease mechanisms.
Introduction
Understanding the properties of molecular assemblies can provide important insight to decode human health and disease processes. Transmission electron microscopes (TEMs) are important tools to view the intricate details of disease-related tissues, cells, and proteins. By preparing EM specimens under frozen-hydrated conditions, we can preserve biological features in a near-native environment [1]. This form of preservation embeds the starting material in thin films of vitreous ice. By convention, amorphous carbon is the most common support film used to prepare specimens for cryogenic (cryo)-EM imaging. Micron-sized holes engineered into carbon support films provide a transparent background to see individual proteins contained in the surrounding ice [2]. Factors that limit spatial resolution in ice are specimen charging, beam-induced movements, and other noise-producing artifacts [3]. Recent advances in the EM field are aimed at minimizing resolution-liming effects. Devices that spurred these efforts include automated freezing units, phase-plates, direct electron detectors, and in-column energy filters. Each of these tools offers improved technology for recording images of biological and clinical samples. Correspondingly, the next generation of specimen support materials must also be developed to best utilize these new tools.
With the progression of the U.S. Materials Genome Initiative and other worldwide ventures to produce new substances, the EM field is presented with a prime opportunity to test alternative substrates as EM support films. Some examples of alternative substrates include conductive titanium-silicon metal glass (Ti88Si12) [4], silicon carbide (cryomesh) [5], graphene [6], and silicon nitride (SiN) [7]. A major benefit of using alternative substrates is their versatile surface properties in comparison to carbon-based films. One example of this versatility for SiN microchips is the ability to modify their surface and tether biological complexes to the pristine, flat microchip prior to specimen preservation. Taking this process a step further, by decorating microchips with specific adaptor molecules, we can create “tunable” devices for the intended purpose of harvesting proteins from human cells for clinical studies. Here, tunable devices are engineered before use to target a particular protein or process under investigation, rather than being tuned during use.
Traditionally, protein isolation schemes involve lengthy steps that employ harsh chemicals to isolate fragile proteins. During the several days required to complete chromatographic separations, multi-subunit protein assemblies can easily dissociate, which is detrimental to studying their structural attributes. In an effort to minimize these negative effects and to streamline the isolation of native assemblies from human cells, we tested the capacity of SiN microchips decorated with specific antibody adaptors. In each test case, the microchips effectively isolated the target proteins from human cells, nuclear material, or pre-fractionated samples. For cryo-EM applications, we refer to SiN microchips as “Cryo-SiN” [7]. Recently developed protocols for using Cryo-SiN permitted us to recruit protein assemblies from patient-derived cancer cells under mild conditions [8, 9]. This approach represents a viable alternative to extensive protein purification techniques and can be applied to a multitude of proteins. Therefore, a major advantage to using SiN microchips is the cost and time savings in comparison to conventional pursuits. To demonstrate these advances, in this article we describe cryo-EM information recently published for both viruses and protein assemblies isolated from eukaryotic cells using tunable microchip devices [7-9].
Materials and Methods
Rotavirus preparation
Rotavirus (strain SA11-4F) double-layered particles (DLPs) were prepared as described previously [7] by the laboratory of Dr. Sarah M. McDonald located at the Virginia Tech Carilion Research Institute. Aliquots (3 μL each) of purified DLPs (0.1 mg/mL) contained in 50 mM HEPES buffer (pH, 7.5) supplemented with 150 mM NaCl, 10 mM CaCl2 and 10 mM MgCl2 were applied to antibody-decorated EM grids (Figure 1a, b) or SiN microchips (Figure 1c) and incubated for 2 minutes prior to plunge-freezing.
Figure 1. A comparison of rotavirus assemblies prepared using different substrates and methods.
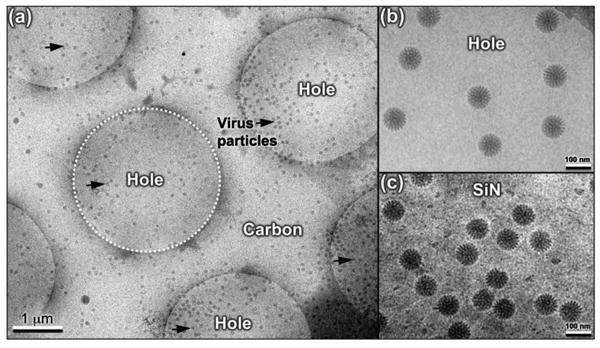
(a) Rotavirus particles (black arrows) are primarily found in holes (white dashed circle) systematically engineered into carbon support films that were plunge-frozen into liquid ethane for cryo-preservation. (b) Close-up view of rotavirus particles located in the holes of carbon support film. (c) Image of rotavirus specimens prepared in the same manner and optimized on silicon nitride (SiN) [7].
BRCA1 protein assemblies isolated from breast cancer cells
We separated the nuclear contents of primary ductal carcinoma cells that express wild type BRCA1 (HCC70 line; ATCC) using the commercially available NE-PER extraction kit (Thermo Scientific). Transcriptional assemblies from the cells' nucleus can be naturally separated by interacting with immobilized-metal affinity matrices [8-10]. The following components were contained in the separated nuclear material and were collectively enriched by incubating with nickel-nitrilotriacetic acid (Ni-NTA) agarose beads: 1) active RNA Polymerase II (RNAP II); 2) the breast cancer susceptibility protein (BRCA1); 3) the BRCA1 binding partner, BARD1. These enriched components makeup an active BRCA1-transcriptional complex that we were able to isolate using SiN microchips.
Preparation of tunable microchips
To produce tunable SiN microchips, we used commercially available microchips (TEMwindows) that are hydrophobic in nature. The microchips were decorated with a layered system that is used to captures specific complexes of interest. Microchips were coated with lipid monolayers containing Ni-NTA-functionalized phosphatidyl head groups (Avanti Polar Lipids) as previously described [7-9]. Briefly, lipid stocks of both Ni-NTA lipids and DLPC lipid were constituted to 1 mg/mL (w/v) in chloroform. The Ni-NTA lipid allows for capture of the assemblies, while the DLPC lipid acts as a spacer. Lipid mixtures were cast over 15-μL aliquots of Milli-Q water placed on parafilm, then sealed in a petri dish, and incubated on ice for 1 hour. For negatively stained specimens, 5% Ni-NTA lipid layers were used. For cryo-EM specimens, 25% Ni-NTA lipid layers were used, each of which also contained 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC) filler lipids (Avanti Polar Lipids). The cleaned SiN microchips were placed on top each monolayer droplet, and the microchips were gently lifted off of each droplet. The coated microchips were incubated for 1 minute with aliquots (3-μL each) of adaptor proteins including His-tagged protein A (AbCam) and IgG antibodies raised against the N-terminal (RING) or C-terminal (BRCT) domains of BRCA1. The antibody-decorated microchips were then incubated for 2 minutes with aliquots of enriched nuclear material derived from breast cancer cells prior to plunge freezing [8, 9].
Results
Using tunable substrates for cryo-EM applications
The use of tunable substrates to prepare biological samples for cryo-EM analysis was recently demonstrated using Rotavirus double-layered particles (DLPs) as a model system. Applying functionalized coatings to holey carbon film allowed us to observe a sufficient amount of virus particles in the desired holes, rather than the surrounding carbon (Figure 1a, b). The same processes and coatings were then used to capture DLPs upon SiN microchips [7]. Surprisingly, the microchip specimens provided enhanced visual contrast in cryo-EM images (Figure 1c) in comparison to the holey carbon specimens. These insights provided the inspiration to develop tunable devices in which interchangeable adaptor molecules can be used to harvest healthy or dysfunctional proteins from human cancer cells for cryo-EM studies [8-9].
Harvesting BRCA1 from breast cancer cells
Major limitations in our understanding of the BRCA1 tumor suppressor are due to insufficient structural studies on human samples. We recently utilized SiN microchips to harvest native BRCA1 proteins from patient-derived breast cancer cells [8, 9] (Figure 2). Specifically, cancer cells (HCC70 line) were cultured and approximately 2,000,000 cells were collected for biochemical and structural analysis of the internal protein assemblies. The nuclear material was separated from the other cellular components, and the nuclear fraction was further enriched for the RNA Polymerase II (RNAP II) core complex (Figure 2a). The RNAP II complex is responsible for transcribing DNA to mRNA messages in all human cells, and it is known to interact with BRCA1 in the cell's nucleus [10]. The RNAP II-enriched fractions were applied to microchips decorated with antibodies against BRCA1 (Figure 2b). This step selects for active RNAP II assemblies associated with BRCA1. The microchips samples were then plunge-frozen for cryo-EM imaging (Figure 2c).
Figure 2. BRCA1 assemblies formed in human cancer cells were isolated using SiN microchips.
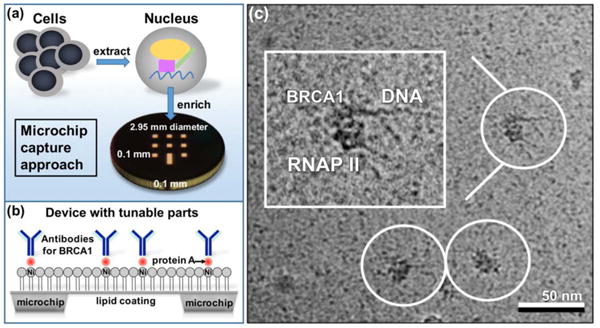
(a) Cancer cells were extracted and the nuclear material was used to enrich for protein assemblies containing BRCA1. Enriched nuclear material was added to the microchip devices. (b) Microchip devices were coated with a lipid layer doped with Ni-NTA functionalized lipids. Tunable parts added to the devices included His-tagged protein A and antibodies for recruiting BRCA1 complexes. (c) Cryo-EM image of BRCA1-RNAP II assemblies poised on DNA [8].
Analysis of BRCA1 complexes tethered to SiN microchips
Specimens were examined using a FEI Spirit BioTwin Transmission Electron Microscope (TEM) (FEI Company) equipped with a LaB6 emitter operating at 120kV under low-dose conditions (∼5 electrons / Å2). We collected EM images and selected >20,000 individual particles from the images using the program PARTICLE (http://www.image-analysis.net/EM/). The selected particles were exported into the RELION software package [11] that was used to compute and refine 3D reconstructions of the frozen-hydrated assemblies.
Based on computed comparisons between the experimental particle images and that of the RNAP II initial model (pdb code, 4A93 [12]), multiple structures were output by RELION independent of the starting parameters. As each reconstruction was highly similar in its 3D features, we calculated a composite structure by combining all of the particles (Figure 3a). The resolution of the final structure was ∼2.2 nm using the 0.5-Fourier Shell Correlation (FSC) criteria [11]. In comparing the composite structure to the RNAP II model, we confirmed two extra densities in the EM map that were not present in the RNAP II structure lacking BRCA1 (Figure 3, yellow [12]). In conjunction with BRCA1 antibody-labeling results, each of the additional densities in the 3D map were attributed to either the BRCA1 N-terminal (RING) or C-terminal (BRCT) domains. The unoccupied density proximal to the C-terminus of RNAP II was assigned to the BRCT domain (Figure 3b, gray; pdb code, 1JNX) [13]. Representative cross-sections (1 – 6) through the BRCT region assigned in the EM map show that the atomic model fits well within the density. As one traverses through the cross-sections, the atomic model and EM density disappear at the same rate. This effect indicates that the atomic model fully occupies its assigned location and that there is no unassigned density leftover, an indicator of a suitable model fit in the density map.
Figure 3. Structural biology interpretation of the isolated BRCA1 assemblies.
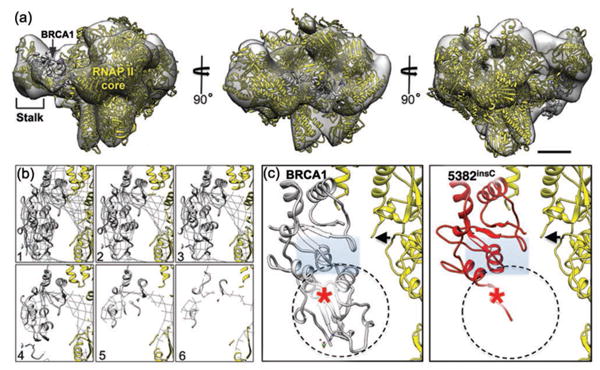
(a) The 3D density map calculated from cryo-EM images of BRCA1 assemblies shown in different orientations. The structure contains the RNAP II core (yellow; pdbcode, 4A93) [12] and the BRCA1 C-terminal domain (gray; pdbcode, 1JNX) [13] adjacent to the RNAP II stalk. Scale bar is 10 nm. (b) Cross-sections (1 – 6) are shown in the region of the density map that contains the BRCT domain. (c) A close-up view of the BRCA1 C-terminal domain (gray) in the left panel shows the peptide-binding pocket (gray rectangle). The homology model of the mutated BRCA15382insC protein (red) reveals the binding pocket is significantly disrupted in the mutated domain. A red star indicated the location of the mutated amino acid adjacent to the RNAP II core (black arrow) [8].
Improving our knowledge of how the BRCT domain interacts with other proteins is important considering many cancer-related mutations in BRCA1 reside in this region of the protein. Therefore, we calculated a homology model for the notable BRCA1 frame-shift mutation, BRCA15382insC. Comparing the structure of the wild type BRCT (Figure 3c, left panel) with a homology-based model of the mutated BRCA15382insC (Figure 3c, right panel), we found S1755 was mutated to L1755 (Figure 3, red star) due to a frame-shift in the DNA sequence [8]. Likewise, we found no secondary structure beyond G1763, and the BRCA1 binding pocket was significantly disrupted in the mutated model compared to the wild type binding site (Figure 3c). These modeling results suggested that the BRCA15382insC mutation alters interactions with proteins that bind in this region, including RNAP II [9,10]. With this new information in hand we expect the continued investigation of these new structures, along with complementary biochemical analysis, to shed light on the molecular underpinnings of BRCA1 disease mechanisms in the growing field of Structural Oncology.
Discussion
We have demonstrated the use of a new support material to enhance specimen preparation techniques for EM investigations in the clinical sciences. Here, we described recent applications of the tunable microchip approach to dissect molecular disease states for 3D structural analysis. While this new platform is currently being implemented to directly see and reconstruct native BRCA1 assemblies formed in human breast cancer cells, there are many other areas of scientific inquiry that can benefit from the use of this technology. For instance, alternative substrates can also be used to dissect active protein components in a number of other disease conditions related to infectious pathogens, cardiac deficiencies, or neurological disorders. Broader applications for specialized microchips may include their integration into wearable technology (i.e., an Apple Watch) for the detection of diagnostic or clinical biomarkers. Overall, having versatile molecular tools to determine new hallmarks of disease states can also contribute important information to improve the design of drug therapies.
Acknowledgments
This work was supported in part by funds from the Commonwealth Health Research Board [2080914], the Concern Foundation [303872], and NIH/NCI [R01CA193578] to D.F.K. C.W. is funded through the ICTAS Doctoral Scholar's program at Virginia Tech and the Medical Research Scholar's program at the Virginia Tech Carilion Research Institute.
References
- 1.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schutlz P. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 2.Taylor KA, Glaeser RM. J Struct Biol. 2008;163:214–23. doi: 10.1016/j.jsb.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brilot AF, Chen JZ, Cheng AC, Pan JH, Harrison SC, Potter CS, Carragher B, Henderson R, Grigorieff N. J Struct Biol. 2012;177:630–37. doi: 10.1016/j.jsb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhinow D, Kuhlbrandt W. Ultramicroscopy. 2008;108:698–705. doi: 10.1016/j.ultramic.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Yoshioka C, Carragher B, Potter CS. Microscopy and Microanalysis. 2010;16:43–53. doi: 10.1017/S1431927609991310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russo CJ, Passmore LA. Nat Methods. 2014;11:649–52. doi: 10.1038/nmeth.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tanner JR, Demmert AC, Dukes MJ, Melanson LA, McDonald SM, Kelly DF. J Anal Mol Tech. 2014;1(1):1–6. doi: 10.13188/2474-1914.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilmore BL, Winton CE, Demmert AC, Tanner JR, Bowman S, Karageorge V, Patel K, Sheng Z, Kelly DF. Sci Rep. 2015;5:14440. doi: 10.1038/srep14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winton CE, Gilmore BL, Demmert AC, Karageorge V, Sheng Z, Kelly DF. npj Breast Cancer. 2016;2:16016. doi: 10.1038/npjbcancer.2016.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scully R, Anderson SF, Chao DM, Wei W, Ye L, Young RA, Livingston DM, Parvin JD. Proc Natl Acad Sci U S A. 1997;94:5605–5610. doi: 10.1073/pnas.94.11.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheres SH. J Struct Biol. 2012;180:519–30. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walmacq C, Cheung AC, Kireeva ML, Lubkowska L, Ye C, Gotte D, Strathern JN, Carell T, Cramer P, Kashlev M. Mol Cell. 2012;46:18–29. doi: 10.1016/j.molcel.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams RS, Green R, Glover JN. Nat Struct Biol. 2001;8:838–842. doi: 10.1038/nsb1001-838. [DOI] [PubMed] [Google Scholar]