

Abstract
Introduction
TGF-β is an important target for many cancer therapies under development. In addition to suppressing anti-tumor immunity, it has pleiotropic direct pro- and anti-tumor effects. The actions of increased endogenous TGF-β production remain unclear, and may affect the outcomes of anti-TGF-β cancer therapy. We hypothesize that tumor-derived TGF-β (td-TGF-β) plays an important role in maintaining tumor remission by controlling tumor proliferation in vivo, and that decreasing td-TGF-β in the tumor microenvironment will result in tumor progression. The aim of this study was to examine the effect of TGF-β in the tumor microenvironment on the balance between its anti-proliferative and immunosuppressive effects.
Methods
A murine BALB/c spontaneous colon adenocarcinoma cell line (CT26) was genetically engineered to produce increased active TGF-β (CT26-TGF-β), a dominant-negative soluble TGF-β receptor (CT26-TGF-β-R), or the empty neomycin cassette as control (CT26-neo). In vitro proliferation rates were measured. For in vivo studies, the three cell lines were injected into syngeneic BALB/c mice, and tumor growth was measured over time. Immunodeficient BALB/c nude mice were used to investigate the role of T and B cells.
Results
In vitro, CT26-TGF-β-R and CT26-TGF-β cells showed increased and suppressed proliferation, respectively, compared to control (CT26-neo), confirming TGF-β has direct anti-tumor effects. In vivo, we found that CT26-TGF-β-R cells displayed slower growth compared to control, likely secondary to reduced suppression of anti-tumor immunity, as this effect was ablated in immunodeficient BALB/c nude mice. However, CT26-TGF-β cells (excess TGF-β) exhibited rapid early growth compared to control, but later failed to progress. The same pattern was shown in immunodeficient BALB/c nude mice, suggesting the effect on tumor growth is direct, with minimal immune system involvement. There was minimal effect on systemic antitumor immunity as determined by peripheral antigen-specific splenocyte type 1 cytokine production and tumor growth rate of CT26-neo on the contralateral flank of the same mice.
Conclusion
Although TGF-β has opposing effects on tumor growth, this study showed that excessive td-TGF-β in the tumor microenvironment renders the tumor non-proliferative. Depleting excess td-TGF-β may release this endogenous tumor suppressive mechanism, thus triggering the progression of the tumor. Therefore, our findings support cautions against using anti-TGF-β strategies in treating cancer, as this may tip the balance of anti-immunity vs. anti-tumor effects of TGF-β, leading to tumor progression instead of remission.
Keywords: Tumor microenvironment, TGF-β, proliferation, immune suppression, colon cancer
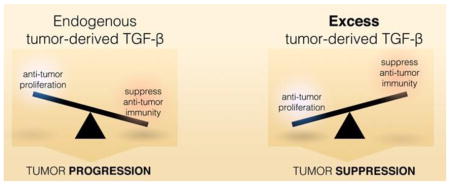
Graphical Abstract
The impact of tumor-derived TGF-β on the balance of suppressing pro-tumor proliferation versus anti-tumor immunity
1. Introduction
The mortality of advanced stage colon cancer remains high despite advances in chemotherapy. Clinical outcomes of patients with colon cancer depend on the balance of tumor proliferation and antitumor immunity; dominance of proliferation will lead to tumor progression, while dominance of anti-tumor immunity will result in tumor regression. Transforming growth factor-β (TGF-β) is produced by the majority of solid tumors, including colon adenocarcinoma, and has potent autocrine anti-proliferative and paracrine immunosuppressive effects. Thus, it plays a complex dual role in tumorigenesis, with both pro- and anti-tumor effects.
Studies on many epithelial cancers, including colorectal, breast, prostate, pancreatic, lung, skin, and ovarian carcinomas, have clearly shown that TGF-β has both tumor suppressor and tumor promoter effects [1]. The current paradigm is that td-TGF-β suppresses host innate and adaptive anti-tumor immunity [2, 3], favoring tumor progression. TGF-β interferes with generation of tumor-specific cytotoxic T lymphocytes [2]. More specifically, TGF-β has been linked with CD4+CD25+ Treg-mediated suppression of antigen-activated T cells [4]. Highly immunogenic tumor cells transfected with murine TGF-β cDNA did not stimulate CTL responses in vitro or in vivo, and thus evaded eradication in mice [5]. Through its modulation of the immune response, TGF-β facilitates tumor escape from immune surveillance. Thus, anti-TGF-β strategies are being developed to counter this effect [6]. However, various methods of TGF-β inhibition have shown mixed results (as reviewed by Neuzillet et al. [7]), suggesting that under certain pathological conditions, inhibition of TGF-β may promote tumor progression.
In addition, TGF-β has been shown to have a role in tumor promotion, with induction of epithelial-to-mesenchymal transformation (EMT) [8, 9] as well as increased cell migration in vitro [10], which may cause increased tumor invasion and metastasis [11]. Overexpression of TGF-β in human cancers correlates with increased tumor angiogenesis, progression, metastases, and poor outcomes [12].
However, TGF-β is also known to act as a tumor suppressor via inhibition of cell growth and induction of apoptosis [13]. In one study, anti-TGF-β antibodies stimulated growth in human breast cancer cells [14]. In another study, over-expression of endogenous TGF-β in human malignant oral keratinocytes led to growth inhibition in vitro and in vivo; expression of a dominant-negative TGF-β type II receptor in these cells enhanced growth in vitro and diminished the tumor suppressor effect in vivo [15].
To further complicate matters, mutation of surface TGF-β receptors and signaling components within the TGF-β pathway, such as the SMAD family, aid further tumor progression by mitigating the autocrine anti-proliferative effect of td-TGF-β [11]. Interestingly, in colon carcinomas, loss of the TGFBR2 receptor and therefore TGF-β signaling is associated with both suppressed growth of early stage tumors and dissemination of late stage tumors [16]. This mutation is associated with progression of late adenomas to colon carcinomas with MSI [17], but longer survival in patients with MSI colon carcinoma [18].
Many TGF-β studies to date have used exogenous TGF-β [19]; these observations may not be applicable to tumor-derived TGF-β, which may act locally but not systemically. We hypothesize that td-TGF-β in the local microenvironment has dual effects on both suppression and promotion of tumor proliferation in addition to inhibition of anti-tumor immunity. The balance of these effects may depend on the amount of TGF-β secreted by tumor cells, thus affecting the outcome of anti-TGF-β therapies. Td-TGF-β may play an important role in maintaining tumor remission by controlling tumor progression in vivo, and decreasing TGF-β in the tumor microenvironment could potentially result in tumor progression. Therefore, we hope to clarify the consequences of over- and under-expression of TGF-β by tumor cells.
This study aims to further define and differentiate between direct/local and immune effects of TGF-β by comparing its effects in vitro and in vivo. To test our hypothesis, CT26, a BALB/c colon adenocarcinoma cell line, was transformed into three different cell lines: CT26-neo, CT26-TGF-β, and CT26-TGF-β-R, which expressed baseline, 10× baseline, and significantly reduced biologically active amounts of TGF-β, respectively. This allowed us to create tumors that expressed different amounts of TGF-β. The direct effect of TGF-β on tumor cells was evaluated by measuring cell proliferation in each of the cell lines in vitro. To examine the role of TGF-β on immune response and tumor growth, we compared the action of TGF-β in tumors injected into syngeneic BALB/c mice, BALB/c nude mice, and allogeneic C57Bl/6 mice. We also examined whether effects of td-TGF-β are predominantly local or systemic.
Our results demonstrated that endogenous td-TGF-β aids in suppressing local anti-tumor immunity, resulting in tumor growth, whereas excess td-TGF-β initially results in more rapid tumor proliferation and later growth suppression, independent of anti-tumor immunity. This supports cautions against using anti-TGF-β therapies, especially in tumors producing excess td-TGF-β, as depleting excess TGF-β may tip the balance towards suppression of anti-tumor immunity, leading to tumor progression.
2. Materials and Methods
2.1 Cell culture and media
All cell lines were maintained in complete medium (CM), which consisted of RPMI 1640 with 10% heat-inactivated FCS, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/mL streptomycin. CT26, a murine colon carcinoma cell line [20] from a BALB/c mouse, was propagated in CM and incubated at 37 degrees C. Cells were trypsinized when they reached confluency and were resuspended at half the original concentration in fresh CM approximately every other day. Cells were seeded at 105 cells/mL CM for all in vitro studies. Cells were stored at −80 degrees C in FBS with 5% DMSO.
2.2 Generation of a TGF-β-expressing CT26 cell line
A CT26 cell line stably transfected to express a constitutively active form of TGF-β1 (CT26-TGF-β) was generated using a retrovirus that expressed TGF-β and a neomycin resistance gene, kindly provided by Dr. G. Nabel (Vaccine Research Center, National Institutes of Health, Bethesda, MD) (28). A control CT26 cell line (CT26-neo) was generated similarly using the retrovirus backbone containing the neomycin resistance gene alone. An adenovirus encoding human TGF-β receptor type II fused to the Fc region of human IgM was used to express a soluble form of the neutralizing TGF-β receptor (TGF-β-R), which has been shown to cross-react with mouse TGF-β [21]. We have previously demonstrated that the soluble TGF-β-R neutralizes TGF-β produced by tumor cells [22]. A CT26 cell line that hence produces below physiologic levels of TGF-β (CT26-TGF-β-R) was generated using a retrovirus that expressed TGF-β-R and the neomycin resistance gene. The three CT26 cell lines were grown in a 12-well plates in 1mL of CM per well. Cells were counted daily using a hemocytometer for 4 days.
2.3 TGF-β and TGF-β-R production by CT26 tumor cell lines
Tumor cell supernatant was collected and stored immediately at 20°C until further use. Bioactive TGF-β secretion was determined by ELISA (R&D Systems). Supernatant was acidified according to the manufacturer’s instructions: to activate TGF-β, 20uL 1N HCl was added per 100uL of sample, the samples were incubated for 10 minutes at room temperature, and then 20uL 1.2N NaOH/0.5M HEPES per 100uL of sample were added to neutralize the pH.
To confirm the ability of TGF-β-R to capture TGF-β, we designed an in vitro assay that borrowed reagents from a TGF-β ELISA kit (R&D Systems). This modified ELISA estimates the TGF-β-R concentration by measuring the amount of TGF-β bound to TGF-β-R, and has previously been described in further detail [22].
2.4 Animal studies
Mice were housed at the Animal Maintenance Facility at the University of Michigan Health System (Ann Arbor, MI). All animal experiments were reviewed and approved by the University Committee on Use and Care of Animals at the University of Michigan. All 3 CT26 cell lines were implanted into syngeneic BALB/c mice to examine the in vitro effects of endogenous td-TGF-β on tumor growth. A similar study was performed in BALB/c nude mice to isolate T and B cell-independent effects in vivo. To understand whether the effect of endogenous td-TGF-β is predominantly local or systemic, a dual tumor study was performed where control CT26 tumor cells were implanted into the mice in which each of the three CT26 cell lines had been injected into the contralateral flank. Finally, CT26-neo and CT26-TGF-β cells were injected into allogeneic C57Bl/6 mice to investigate the action of increased td-TGF-β secretion under high immune pressure.
2.5 Implantation of CT26 tumor cell lines into BALB/c mice
Female BALB/c mice aged 6–8 wks were purchased from The Jackson Laboratory (Bar Harbor, ME). Experiments were conducted at age 10–14 wks. 1×106 CT26-neo, CT26-TGF-β, or CT26-TGF-β-R cells suspended in 100 ul PBS were injected intraperitoneally into the left flank. Tumors were measured with a pair of Vernier calipers three times weekly and size was calculated according to the following equation: volume = b2 * a/2, where a is the longer diameter of the tumor and b the shorter diameter. Mice were sacrificed after 21 days.
Splenocytes from tumor-bearing mice were co-cultured with CT26-neo lysate (5ng/mL) for 72 hours in CM and production of Th1 type cytokines IFN-g and TNF-a were measured by ELISA.
2.6 Immunodeficient BALB/c nude mouse studies
To determine the role of T and B cells in mediating the effects of td-TGF-β, female Nude BALB/c mice aged 6–8 wks were purchased from The Jackson Laboratory. Experiments were conducted at age 10–14 wks. Tumor cells were injected and measured as above.
2.7 BALB/c mouse dual tumor studies
To determine whether TGF-β has a systemic effect on tumor growth, female BALB/c mice aged 6–8 wks were purchased from The Jackson Laboratory (Bar Harbor, ME). Experiments were conducted at age 10–14 wks. 1×106 CT26-neo, CT26-TGF-β, or CT26-TGF-β-R cells suspended in 100 ul PBS were injected into the left flank. In addition, 1×106 CT26-neo cells suspended in 100 ul PBS were injected into the right flank. Volume of both tumors was measured as above and mice were sacrificed on day 19.
2.8 C57Bl/6 mouse studies
To determine whether increased production of td-TGF-β is sufficient to enable immune escape and tumor growth under increased immune pressure a similar experiment, in which tumor cells were implanted and measured as above, was performed on allogeneic female C57B/6 mice aged 6–8 wks purchased from The Jackson Laboratory.
2.9 Statistical analysis
Unpaired Student’s t test analyses and 1-way ANOVA were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, San Diego CA). Results with P < 0.05 were considered statistically significant.
3. Results
3.1 Generation of CT26 cell lines producing excess, endogenous, or decreased TGF-β
We genetically engineered parental CT26 BALB/c colon adenocarcinoma cells to overexpress an active form of TGF-β1 (CT26-TGF-β) and to deplete TGF-β in the tumor microenvironment (CT26-TGF-β-R) (Figure 1A). To confirm that CT26-TGF-β cells produce excess amounts of td-TGF-β, we measured by ELISA the concentration of the active (acidified) form of TGF-β in serum-free supernatant after four days of culture. The highest concentration of TGF-β was measured in CT26-TGF-β, followed by CT26-neo and CT26-TGF-β-R (Figure 1B). To confirm that CT26-TGF-β-R cells produce TGF-β-R, we measured TGF-β-R in the supernatant after 4 days of growth using ELISA. Only CT26-TGF-β-R cells produced TGF-β-R, while the other two cell lines produced no detectable TGF-β-R (Figure 1C).
Figure 1.

Generation of CT26 cell lines producing excess, endogenous, or decreased TGF-β.
A CT26 colon cancer cell line was genetically engineered (see Methods) to express TGF-β (CT26-TGF-β) or TGF-β dominant-negative receptor (CT26-TGF-β-R). Neomycin empty cassette was used to generate the control cell line, CT26-neo. A) Schematic representation of the in vitro experiments performed. B) The concentration of the active form (acidified) of TGF-β present in the tumor cell culture media measured by ELISA. C) The concentration of TGF-β-R in the tumor culture media measured by ELISA.
3.2 Tumor-derived TGF-β directly inhibits CT26 cell proliferation in vitro
To determine the effect of tumor-derived TGF-β in vitro, CT26-neo (endogenous TGF-β production), CT26-TGF-β (excessive TGF-β production), and CT26-TGF-β-R (expression of a water-soluble dominant-negative TGF-β receptor, which results in TGF-β depletion) cells were grown in CM for 4 days and cell number measured by cytometer. CT26-TGF-β cells exhibited significantly slower growth than the other two cell lines, while CT26-TGF-β-R cells showed the most rapid growth (Figure 2B, 2C). This suggests that TGF-β exerts a direct suppressive effect on tumor growth.
Figure 2.

Tumor-derived TGF-β directly inhibits CT26 cell proliferation in vitro.
A) The micrographs of the cell confluence of tumor cultures at day 4. B) The concentrations of tumor cells measured by hemocytometer. The data shown represent results obtained from three independent experiments. * P<0.05, **P<0.01.
3.3 Endogenous tumor-derived TGF-β promotes CT26 tumor growth in vivo
We next examined the endogenous effect of tumor-derived TGF-β in vivo by comparing the growth rate of implanted CT26-neo vs CT26-TGF-β-R. Both CT26-neo and CT26-TGF-β-R implanted Balb/c mice developed measureable tumors on day 10 and the tumor growth rate of CT26-TGF-β-R was significantly lower than the growth rate of CT26-neo tumors (Figure 3A, 3B).
Figure 3.

Endogenous td-TGF-β promotes tumor growth locally but not systemically.
In vivo depletion of TGF-β in the CT26 tumor microenvironment resulted in the suppression of CT26 tumor growth without a significant increase in systemic antitumor immunity. CT26-neo or CT26-TGF-β-R were implanted (106/mouse) in the right flank of syngeneic BALB/c mice (n=5). A) Tumor growth curve and B) Day 17 tumor volumes are shown (See Methods). C) Splenocytes from tumor-bearing mice were co-cultured with CT26-neo lysate (5ng/mL) for 72 hours and production of Th1 type cytokines were measured by ELISA. D) To investigate the role of tumor-derived TGF-β in the tumor microenvironment on systemic antitumor immunity, CT26-neo (Group 1) or CT26-TGF-β-R (Group 2) were implanted in the right flank, labeled as Tumor A, and the control CT26-neo tumor was simultaneously implanted in the left flank of BALB/c mice labeled as Tumor B. E) Tumor volume comparison between Tumor A and Tumor B of each group (left, Group 1; right, Group 2). F) Comparison of tumor volume of Tumor B between the two groups (* P<0.05).
3.4 Endogenous td-TGF-β promotes tumor growth locally but not systemically
To assess whether the difference in the observed growth rate is due to local or systemic influence of td-TGF-β, splenocytes from CT26-neo or CT26-TGF-β-R tumor-bearing mice were co-cultured with CT26-neo lysate for 72 hours and the production of type 1 cytokines IFN-g and TNF-a were measured by ELISA and indicated no significant difference in peripheral anti-tumor immune reactivity (Figure 3C). To further confirm that the observed difference is limited to the tumor microenvironment and does not impact systemic anti-tumor immunity, mice were simultaneously implanted with control CT26-neo on the contralateral flank to assess the impact of td TGF-β-R on the tumor growth at a distant site (Figure 3D). Despite different tumor growth rates observed in CT26-neo vs CT26-TGF-β-R in the right flank of mice (Figure 3E), there was no significant difference in tumor growth at the distant site (contralateral left flank) (Figure 3F). These results indicate that the effect of TGF-β-R is limited to the tumor microenvironment rather than on systemic anti-tumor immunity.
3.5 Endogenous td-TGF-β modulates tumor growth via modification of anti-tumor adaptive immunity
To examine dependence of tumor growth rate differences between CT26-neo and CT26-TGF-β-R on changes in adaptive anti-tumor immunity, we injected the 2 cell lines into BALB/c nude mice lacking T and B cell mediated immunity. Unlike in adaptive immune sufficient mice, CT26-TGF-β-R grew faster than CT26-neo implanted tumors (Figure 4), indicating that the effect of endogenous tumor-derived TGF-β acts predominantly via suppression of adaptive antitumor immunity.
Figure 4.

Endogenous td-TGF-β modulates tumor growth via modification of anti-tumor adaptive immunity.
An accelerated growth curve was observed in immunodeficient BALB/c nude mice implanted with CT26-TGF-β-R compared to control CT26-neo, revealing the impact of tumor-derived TGF-β on anti-tumor immunity. CT26-neo or CT26-TGF-β-R were implanted (106/mouse) in the right flank of nude mice (n=5 per group). The tumor growth curve measured by tumor volume at each indicated time point post tumor implantation is shown. * P<0.05.
3.6 Excess tumor-derived TGF-β predominantly inhibits CT26 growth in vivo
To study the effect of excess tumor-derived TGF-β in vivo, we compared the growth rate of implanted CT26-neo vs CT26-TGF-β. CT26-TGF-β implanted BALB/c mice developed measurable tumors on day 3, compared to day 10 for CT26-neo implanted BALB/c mice. The growth curve of the CT26-TGF-β tumors showed a later plateau, whereas the CT26-neo tumors continued to grow exponentially (Figure 5A). The CT26-TGF-β implanted mice grew significantly smaller tumors (Figure 5B), thus demonstrating a predominantly anti-proliferative effect.
Figure 5.

Excess tumor-derived TGF-β predominantly inhibits CT26 growth in vivo locally, but not systemically.
In vivo forced-expression of tumor-derived TGF-β on CT26 cell proliferation resulted in an early establishment of tumor without apparent progression in syngeneic mice. A) CT26-neo or CT26-TGF-β were implanted (106/mouse) in the right flank of syngeneic BALB/c mice and early tumor establishment without tumor progression was observed only in CT26-TGF-β and not in CT26-neo mice (n=5 per group. B) Splenocytes from tumor-bearing mice were co-cultured with CT26-neo lysate (5ng/mL) for 72 hours and production of Th1 type cytokines were measured by ELISA. C) To investigate the role of tumor-derived TGF-β in the tumor microenvironment on systemic antitumor immunity, CT26-neo (Group 1) or CT26-neo TGF-β (Group 2) were implanted in the right flank, labeled as Tumor A, and the control CT26-neo tumor was simultaneously implanted in the left flank of BALB/c mice labeled as Tumor B. D) Tumor volume comparison between Tumor A and Tumor B of Group 2. E) Comparison of tumor volume of Tumor B between the two groups. * P<0.05.
3.7 Excess td-TGF-β modifies tumor growth locally but not systemically
To assess whether the difference in the observed growth rate is due to local or systemic influences of excess td-TGF-β, splenocytes from CT26-neo or CT26-TGF-β tumor-bearing mice were co-cultured in CM with CT26-neo lysate for 72 hours and the production of type 1 cytokines IFN-g and TNF-a were measured by ELISA. Cytokine levels were not significantly different, and indicated no significant difference in peripheral anti-tumor immunity (Figure 5C).
To confirm the observed difference is limited to the tumor microenvironment without effects on systemic anti-tumor immunity, mice were simultaneously implanted with control CT26-neo in the contralateral flank to assess the impact of td-TGF-β on the tumor growth at a distant site (Figure 5D). Despite different tumor growth rates observed in CT26-neo vs CT26-TGF-β in the right flank of mice (Figure 5E), there was no significant difference in tumor growth at the distant site (contralateral left flank) (Figure 5F). This indicates that effects of excess td-TGF-β are limited to the tumor microenvironment rather than on systemic anti-tumor immunity.
3.8 Excess td-TGF-β modulates tumor growth independently of adaptive immune function
To examine the role of the adaptive immune system in growth rate differences between CT26-neo and CT26-TGF-β, we injected the 2 cell lines into BALB/c nude mice lacking T and B cell mediated immunity. The early growth and later plateau for CT26-TGF-β remained the same as before, and the CT26-TGF-β tumors were significantly smaller than the control tumors (P<0.05) (Figure 6). These observations imply that td-TGF-β acts on tumor cells directly, independent of host adaptive immune response.
Figure 6.

Excess td-TGF-β modulates tumor growth independently of adaptive immune function.
A similar early tumor establishment without progression was observed in immunodeficient nude mice implanted with CT26-TGF-β compared to control CT26-neo, confirming that the failure of CT26-TGF-β progression is not due to anti-tumor immunity, but rather likely suppression of tumor growth by TGF-β. CT26-neo or CT26-TGF-β were implanted (106/mouse) in the right flank of nude mice (n=5 per group). The tumor growth curve measured by tumor volume at each indicated time point post tumor implantation is shown. * P<0.05.
3.9 Increased td-TGF-β allows tumor growth under high immune pressure, with later growth suppression and eradication
To further characterize the early growth seen in CT26-TGF-β tumors, CT26-neo and CT26-TGF-β cell lines were injected into allogeneic C57Bl/6 mice. Tumors were measured for 21 days. Mice injected with CT26-neo cells showed no visible tumor growth, whereas CT26-TGF-β-R tumors showed measurable tumor growth before diminishing in volume. Eradication occurred by day 21 (Figure 7). This implies that increased td-TGF-β is sufficient to allow tumor growth, even under high immune pressure in which tumors producing normal amounts of TGF-β cannot initiate growth. However, later predominantly anti-proliferative effects of TGF-β combined with increased immune pressure led to eradication of tumors.
Figure 7.

Increased td-TGF-β allows tumor growth under high immune pressure, with later growth suppression and eradication.
CT26-neo or CT26-TGF-β cells were implanted (106/mouse) in the right flank of allogeneic C57B/6 mice and early tumor establishment was observed only in CT26-TGF-β and not in CT26-neo mice (n=5 per group). However, tumors were eradicated by day 21.
4. Discussion
This study is the first to combine in vivo and in vitro observations on the actions of td-TGF-β on tumor growth. The novel aspect of the study involved the use of genetically engineered CT26 cell lines that over- or under-produce TGF-β. The in vitro experiments allowed observation of direct actions of TGF-β on tumor cell proliferation independent of anti-tumor immunity. On the other hand, the in vivo experiments utilizing these CT26 cell lines permitted evaluation of the effects of td-TGF-β in the local tumor microenvironment.
Our results showed that endogenous td-TGF-β suppresses local anti-tumor immunity, allowing for more rapid tumor growth. Excess td-TGF-β initially results in more rapid tumor proliferation and later predominant suppression of growth, independent of anti-tumor immunity. Importantly, this evidence of the anti-proliferative role of excess td-TGF-β supports cautions against using anti-TGF-β therapies in tumors producing excess td-TGF-β, as depleting excess TGF-β may tip the balance towards suppression of anti-tumor immunity, leading to tumor progression.
In vitro, excess td-TGF-β suppressed tumor cell proliferation. On the other hand, decreased td-TGF-β promoted tumor cell proliferation. These observations suggest that under in vitro conditions, td-TGF-β has a direct suppressive effect on tumor growth. This supports the current understanding that TGF-β is a potent tumor suppressor early in carcinogenesis [23, 24], which primarily occurs via inhibition of cell cycle progression, induction of apoptosis, and suppression of growth factor production [25].
As expected, reduced TGF-β production resulted in more rapid tumor cell proliferation, consistent with knowledge that loss of the TGF-β signaling pathway in tumor cells is associated with aggressive cancers and poor prognosis [26, 27], suggesting a tumor suppressor role. This most often occurs through loss of function mutations in either TGF-β receptor or SMAD family genes or inactivation of the TGF-β receptor-SMAD pathway [28]. In colon carcinogenesis, the most common mechanism resulting in alteration of TGF-β pathway signaling is a mutation in TGF-βRII, with an incidence of about 30% in CRC [29]. In addition, TGF-βRII frameshift mutations occur in about 80% of MSI + colon cancers [30].
In contrast, under in vivo conditions, excess tumor-derived TGF-β resulted in early tumor establishment compared to control in syngeneic BALB/c mice. However, the growth quickly reached a plateau by day 5, while the control tumors continued to grow exponentially. This early growth in tumors producing excess td-TGF-β may be due to induction of epithelial-to-mesenchymal transition [31, 32]. As tumor size increases and more TGF-β is produced, this effect is either lost or is overcome by pro-apoptotic or cytostatic effects [13, 33, 34], resulting in a growth plateau after day 5. Apoptotic effects of TGF-β have also been demonstrated in human cancer cells in vitro [35, 36].
Comparatively, tumors producing reduced amounts of TGF-β (CT26-TGF-β-R) showed slower growth in WT mice compared to control, but maintained a similar growth curve. However, in BALB/c nude mice, these tumors proliferated faster than control, suggesting involvement of T and B cells at these physiologic and lower concentrations of TGF-β. It is well known that TGF-β inhibits T cell reactivity and promotes immune tolerance [37, 38]. TGF-β reduces tumor-specific CTL function and generation [2], and blocking TGF-β signaling in these cells results in more rapid tumor surveillance [39]. In addition, highly immunogenic tumor cells transfected with murine TGF-β cDNA were able to evade eradication in mice due to failure to stimulate CTL responses [5].
The effect of td-TGF-β appears to be local rather than systemic, as tumor cells displayed identical growth curves regardless of the level of TGF-β production by tumor cells injected into the contralateral flank. Furthermore, splenocytes taken from each group of mice stimulated with CT26-neo lysate did not produce significantly different levels of IFN-g and TNF-a. Thus, serum levels of TGF-β may not be an accurate predictor of td-TGF-β production, which in turn appears to be a critical influencer of tumor growth.
When allogeneic C26Bl/6 mice were injected with control versus tumor cells producing increased td-TGF-β, the control group demonstrated no visible tumor growth, whereas the group injected with CT26-TGF-β cells showed tumor growth. However, compared to growth in syngeneic BALB/c mice, the tumors regressed and were eliminated by day 21. This shows that while increased td-TGF-β is sufficient to allow tumor proliferation under high immune pressure, the balance is tipped towards tumor eradication. It is then conceivable that tumors producing excessive TGF-β may actually be eliminated with sufficient immunotherapy.
Numerous studies have investigated effects of exogenous TGF-β on tumor growth [19, 40–42]; however, there is relatively sparse literature on effects of excess tumor-derived TGF-β on tumor growth. In this study, because TGF-β was produced by tumor cells, its concentration in the local microenvironment may be higher than in studies in which it is administered exogenously. The difference in local TGF-β concentration may account for this study’s different results; the balance of its pro-tumor and anti-tumor effects may be shifted towards direct anti-tumor growth inhibiting effects and away from its pro-tumor immunosuppressive effects. Thus, the sequelae of exogenous administration of TGF-β may not reflect the true effects and actions of tumor-derived TGF-β.
5. Conclusions
In conclusion, by creating a model system to examine the in vivo microenvironment and altering the levels of TGF-β, we demonstrated that endogenous TGF-β may also promote tumor progression through suppressing the host’s innate immune response. Reducing TGF-β to below basal level may enhance T cell dependent antitumor immunity. However, excessive TGF-β may act directly on the tumor to inhibit tumor growth. This suggests that the effects of TGF-β on tumor growth hinge on the fine balance of its effect on the immune system and its direct tumor growth suppression. It is therefore conceivable that if anti-TGF-β is administered as cancer therapy on a tumor overexpressing TGF-β, the balance might be tipped, leading to tumor growth instead of eradication.
Acknowledgments
Grant support: Research reported in this publication was supported by NIDDK of the National Institutes of Health under award number R01 DK087708 (JYK) and American Gastroenterological Association Student Summer Fellowship Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We appreciate helpful input and suggestions regarding our manuscript from Wei-ping Zou and Venkat Keshamouni.
Abbreviations
- td-TGF-β
tumor-derived TGF-β
- EMT
epithelial-to-mesenchymal transformation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Padua D, Massagué J. Roles of TGFbeta in metastasis. Cell Res. 2009;19(1):89–102. doi: 10.1038/cr.2008.316. [DOI] [PubMed] [Google Scholar]
- 2.Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol. 2006;177(2):896–904. doi: 10.4049/jimmunol.177.2.896. [DOI] [PubMed] [Google Scholar]
- 3.Gigante M, Gesualdo L, Ranieri E. TGF-beta: a master switch in tumor immunity. Curr Pharm Des. 2012;18(27):4126–34. doi: 10.2174/138161212802430378. [DOI] [PubMed] [Google Scholar]
- 4.Chen ZF, Xu Q, Ding JB, Zhang Y, Du R, Ding Y. CD4+CD25+Foxp3+ Treg and TGF-beta play important roles in pathogenesis of Uygur cervical carcinoma. Eur J Gynaecol Oncol. 2012;33(5):502–7. [PubMed] [Google Scholar]
- 5.Torre-Amione G, Beauchamp RD, Koeppen H, Park BH, Schreiber H, Moses HL, Rowley DA. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc Natl Acad Sci U S A. 1990;87(4):1486–90. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arjaans M, Oude Munnink TH, Timmer-Bosscha H, Reiss M, Walenkamp AM, Lub-de Hooge MN, de Vries EG, Schröder CP. Transforming growth factor (TGF)-β expression and activation mechanisms as potential targets for anti-tumor therapy and tumor imaging. Pharmacol Ther. 2012;135(2):123–32. doi: 10.1016/j.pharmthera.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A. Targeting the TGFβ pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. doi: 10.1016/j.pharmthera.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-beta. Future Oncol. 2009;5(8):1145–68. doi: 10.2217/fon.09.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao J, Zhu Y, Nilsson M, Sundfeldt K. TGF-β isoforms induce EMT independent migration of ovarian cancer cells. Cancer Cell Int. 2014;14(1):72. doi: 10.1186/s12935-014-0072-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17(1–2):29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 12.Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17(1–2):41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 13.Sánchez-Capelo A. Dual role for TGF-beta1 in apoptosis. Cytokine Growth Factor Rev. 2005;16(1):15–34. doi: 10.1016/j.cytogfr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Arteaga CL, Coffey RJ, Dugger TC, McCutchen CM, Moses HL, Lyons RM. Growth stimulation of human breast cancer cells with anti-transforming growth factor beta antibodies: evidence for negative autocrine regulation by transforming growth factor beta. Cell Growth Differ. 1990;1(8):367–74. [PubMed] [Google Scholar]
- 15.Paterson IC, Davies M, Stone A, Huntley S, Smith E, Pring M, Eveson JW, Robinson CM, Parkinson EK, Prime SS. TGF-beta1 acts as a tumor suppressor of human malignant keratinocytes independently of Smad 4 expression and ligand-induced G(1) arrest. Oncogene. 2002;21(10):1616–24. doi: 10.1038/sj.onc.1205217. [DOI] [PubMed] [Google Scholar]
- 16.Jung B, Staudacher JJ, Beauchamp D. Transforming Growth Factor β Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology. 2017;152(1):36–52. doi: 10.1053/j.gastro.2016.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grady WM, Rajput A, Myeroff L, Liu DF, Kwon K, Willis J, Markowitz S. Mutation of the type II transforming growth factor-beta receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998;58(14):3101–4. [PubMed] [Google Scholar]
- 18.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344(16):1196–206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dumont N, Arteaga CL. Transforming growth factor-beta and breast cancer: Tumor promoting effects of transforming growth factor-beta. Breast Cancer Res. 2000;2(2):125–32. doi: 10.1186/bcr44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 21.Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L) Science. 1998;282(5394):1714–7. doi: 10.1126/science.282.5394.1714. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Berndt BE, Chen JJ, Kao JY. Expression of a soluble TGF-beta receptor by tumor cells enhances dendritic cell/tumor fusion vaccine efficacy. J Immunol. 2008;181(5):3690–7. doi: 10.4049/jimmunol.181.5.3690. [DOI] [PubMed] [Google Scholar]
- 23.Akhurst RJ, Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11(11):S44–51. doi: 10.1016/s0962-8924(01)02130-4. [DOI] [PubMed] [Google Scholar]
- 24.Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR, Muller WJ, Moses HL. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65(6):2296–302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- 25.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220–7. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moses HL, Yang EY, Pietenpol JA. TGF-beta stimulation and inhibition of cell proliferation: new mechanistic insights. Cell. 1990;63(2):245–7. doi: 10.1016/0092-8674(90)90155-8. [DOI] [PubMed] [Google Scholar]
- 27.Massagué J. TGFbeta in Cancer. Cell. 2008;134(2):215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775(1):21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Lampropoulos P, Zizi-Sermpetzoglou A, Rizos S, Kostakis A, Nikiteas N, Papavassiliou AG. TGF-beta signalling in colon carcinogenesis. Cancer Lett. 2012;314(1):1–7. doi: 10.1016/j.canlet.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 30.Takayama T, Miyanishi K, Hayashi T, Sato Y, Niitsu Y. Colorectal cancer: genetics of development and metastasis. J Gastroenterol. 2006;41(3):185–92. doi: 10.1007/s00535-006-1801-6. [DOI] [PubMed] [Google Scholar]
- 31.Miettinen PJ, Ebner R, Lopez AR, Derynck R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: involvement of type I receptors. J Cell Biol. 1994;127(6 Pt 2):2021–36. doi: 10.1083/jcb.127.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 33.Chen CR, Kang Y, Siegel PM, Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110(1):19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 34.Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massagué J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3(4):400–8. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- 35.Tao MZ, Gao X, Zhou TJ, Guo QX, Zhang Q, Yang CW. Effects of TGF-β1 on the Proliferation and Apoptosis of Human Cervical Cancer Hela Cells In Vitro. Cell Biochem Biophys. 2015;73(3):737–41. doi: 10.1007/s12013-015-0673-x. [DOI] [PubMed] [Google Scholar]
- 36.Ha Thi HT, Lim HS, Kim J, Kim YM, Kim HY, Hong S. Transcriptional and post-translational regulation of Bim is essential for TGF-β and TNF-α-induced apoptosis of gastric cancer cell. Biochim Biophys Acta. 2013;1830(6):3584–92. doi: 10.1016/j.bbagen.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 37.Li MO, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134(3):392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delisle JS, Giroux M, Boucher G, Landry JR, Hardy MP, Lemieux S, Jones RG, Wilhelm BT, Perreault C. The TGF-β-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 2013;14(2):115–26. doi: 10.1038/gene.2012.63. [DOI] [PubMed] [Google Scholar]
- 39.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limón P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massagué J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103(2):197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welch DR, Fabra A, Nakajima M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci U S A. 1990;87(19):7678–82. doi: 10.1073/pnas.87.19.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portella G, Cumming SA, Liddell J, Cui W, Ireland H, Akhurst RJ, Balmain A. Transforming growth factor beta is essential for spindle cell conversion of mouse skin carcinoma in vivo: implications for tumor invasion. Cell Growth Differ. 1998;9(5):393–404. [PubMed] [Google Scholar]