

Visual Abstract

Key Words: heart, phosphatase, phospholamban
Abbrevations and Acronyms: CaMKII, calmodulin kinase II; ER, endoplasmic reticulum; GFP, green fluorescent protein; I/R, ischemia/reperfusion; ISO, isoproterenol; NRVM, neonatal rat ventricular myocyte; PLN, phospholamban; PP1, protein phosphatase 1; PP2A, protein phosphatase 2A; PP2Ce, protein phosphatase 2C on ER membrane; RyR2, ryanodine receptor; SERCA2a, sarcolemma-endoplasmic reticulum calcium ATPase 2a; SR, sarcoplasmic reticulum; TUNEL, transferase dUTP nick end labeling
Highlights
-
•
PP2Ce is Ser-Thr phosphatase specifically localized on SR and expressed in cardiomyocytes.
-
•
PP2Ce has specific phosphatase activity to dephosphorylate Thr-17 site of phospholamban.
-
•
PP2Ce expression is induced upon pathological stress, including beta-AR stimulation and ROS.
-
•
PP2Ce induction suppresses cardiomyocyte calcium cycling, reduces beta-AR-induced contractility, and promotes oxidative ischemia/reperfusion injury.
-
•
PP2Ce is a new molecular component of stress-mediated cardiomyocyte calcium regulation.
Summary
Phospholamban (PLN) is a key regulator of sarcolemma calcium uptake in cardiomyocyte; its inhibitory activity to sarcolemma-endoplasmic reticulum calcium ATPase is regulated by phosphorylation. PLN hypophosphorylation is a common molecular feature in the failing heart. The current study provided evidence at the molecular, cellular, and whole-heart levels to implicate a sarcolemma membrane-targeted protein phosphatase, PP2Ce, as a specific and potent PLN phosphatase. PP2Ce expression was elevated in failing human heart and induced acutely at protein level by β-adrenergic stimulation or oxidative stress in cardiomyocytes. PP2Ce expression in mouse heart blunted β-adrenergic response and exacerbated ischemia/reperfusion injury. Therefore, PP2Ce is a new regulator for cardiac function and pathogenesis.
Heart disease is a leading cause of death in the United States. Defects in intracellular Ca2+ homeostasis have been implicated in the onset and progression of heart failure 1, 2, 3, 4, 5, 6, which lead to a blunted response to adrenergic stimulation and loss of contractility 7, 8, 9. Cardiac sarcoplasmic reticulum (SR) Ca2+ cycling, involving Ca2+-induced Ca2+ release from SR through the ryanodine receptor (RyR2) during systole, and Ca2+ uptake via SR calcium-ATPase 2a (SERCA2a), is central to cardiac contraction and relaxation (10). This process is tightly regulated by reversible phosphorylation of an SR-targeted protein phospholamban (PLN), a key negative regulator for SERCA2a activity (11). PLN phosphorylation by PKA at the Ser-16 site and by calmodulin kinase II (CaMKII) at the Thr-17 site releases its inhibition to SERCA2a activity, resulting in elevated SR Ca2+ uptake and accelerated intracellular Ca2+ decline during relaxation. This is a major mechanism contributing to the enhanced contractility in response to adrenergic and other stimulation (12). In addition to its fundamental role in mediating myocyte contraction, PLN-mediated Ca2+ regulation also plays a crucial role in cardiomyocyte cell death regulation, especially following myocardial infarction (MI) (13). Depressed SR Ca2+ cycling in the post-MI heart contributes to intracellular Ca2+ overload, which in turn leads to myocyte necrosis and apoptosis 14, 15. Compared with our extensive studies of the protein kinases involved in PLN regulation, our insights into the PLN-specific phosphatases responsible for its dephosphorylation are relatively limited. Previous studies have revealed that protein phosphatase-1 (PP1), a relatively nonselective ser/thr protein phosphatase, contributes to a significant level of PLN phosphorylation regulation at both Ser-16 and Thr-17 sites 11, 16, 17, 18. In addition, protein phosphatase 2A (PP2A) subunit B56 also mediates targeted dephosphorylation of PLN Ser-16 in addition to other cardiac proteins. PLN hypophosphorylation is a common molecular feature in the failing hearts and mutations of PLN that affect its phosphorylation and interaction with SERCA2a lead to cardiomyopathy in humans 16, 17, 19, 20, 21, 22, 23, 24, 25. Therefore, there is a need to better understand the underlying mechanism for PLN dephosphorylation and the relevant proteins.
In this report, we characterized an ER-targeted member of the protein phosphatase 2C family, PP2Ce, in the heart. PP2Ce messenger ribonucleic acid (mRNA) was detected in the heart. In cardiomyocytes and a cell-free system, PP2Ce showed high specificity toward the Thr-17 site of PLN, and its expression significantly suppressed β-adrenergic stimulation-induced calcium transients and exacerbated cell death upon oxidative stress. Expression of PP2Ce mRNA was found to be elevated in human cardiomyopathy hearts, and its protein was shown to be acutely induced in myocytes by pathological stressors via modulation of protein homeostasis. Cardiac-specific expression of PP2Ce in a transgenic mouse model led to hypophosphorylation of PLN associated with exacerbated cardiac dysfunction and injury following ischemia/reperfusion (I/R). Therefore, PP2Ce is a novel SR membrane-targeted and specific phosphatase for PLN in cardiomyocytes, and its induction under pathological conditions may have a significant contribution to PLN hypophosphorylation, cardiomyocyte dysfunction and death.
Methods
Generation of cardiac-specific and inducible PP2Ce mice (PP2Ce TG)
Double transgenic PP2Ce mice were generated by crossing PP2Ce single transgenic line to a strain carrying the tetracycline-controlled transactivator gene (tTA), which is under the regulation of the alpha-myosin heavy chain promoter. Enhanced PP2Ce expression was achieved upon withdrawal of doxycycline. Control animals used in this study were littermate mice carrying single transgenic tTA. Male mice between age 10 to 15 weeks were used in the present study. All experiments were conducted in accordance with University of California, Los Angeles, animal welfare guidelines.
Echocardiography
Mice were anesthetized with 1% isoflurane. Echocardiography was quantified on 2-dimensional M-mode along the short-axis of the left ventricle using the Vevo 770 system (VisualSonics, Toronto, Ontario, Canada) and analyzed using the software suite provided by the vendor.
Isolation of adult cardiomyocytes and measurement of Ca2+ transients
Rat adult ventricular cardiomyocytes were enzymatically isolated in the following procedure. Hearts were quickly removed and retrogradely perfused with Ca2+-free Tyrode’s buffer containing collagenase II until hearts became softer (∼10 min). Calcium transients recording and measurement were performed as previously described (26).
I/R in isolated heart
The ex vivo I/R model was performed as previously described (27). Briefly, the heart was perfused in the Langendorff system at a constant pressure of 80 mm Hg by gravity. A fluid-filled balloon was inserted into the left ventricle and connected to a pressure transducer for continuous recording of left ventricular pressure and heart rate. After 30 min of stabilization, hearts were subjected to 30 min of global normothermic (37ºC) ischemia followed by up to 120 min of reperfusion. Heart tissues were collected for further studies. Infarct size measurement was performed by triphenyltetrazolium chloride (TTC) staining, and apoptosis-positive cells were characterized by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining.
Complementary deoxyribonucleic acid synthesis and quantitative real-time polymerase chain reaction analysis
Total RNA was extracted from the left ventricle with TRIzol reagent (Invitrogen, Carlsbad, California) according to the manufacturer’s instructions. Five micrograms of total RNA were used to reverse-transcribe the first-strand complementary deoxyribonucleic acid with a Superscript first-strand synthesis kit (Invitrogen). Complementary deoxyribonucleic acid transcripts were quantified by the iCycler iQ real-time polymerase chain reaction detection system using iQ SYBR Green Supermix (Bio-Rad, Hercules, California). Glyceraldehyde 3-phosphate dehydrogenase mRNA was used for normalization. Each reaction was performed in duplicate.
Western blot and immunoprecipitation assay
Western blots were performed as described previously (26). A total of 10 to 50 μg of total protein from ventricles and myocytes was loaded on 4% to 12% Bis-Tris gel, followed by electric transfer to nitrocellulose membranes. All protein signals were digitally recorded and quantified based on intensities, presented as average ± SD.
In vitro dephosphorylation assay
Left ventricle tissue extract was prepared in lysis buffer (50 mmol/l Tris-HCl [pH 7.4], 150 mmol/l NaCl, 1% Nonidet P-40, and 1 mmol/l phenylmethylsulfonyl fluoride) with complete protein inhibitors (Roche Diagnostics, Indianapolis, Indiana) and incubated with 1 ug of monoclonal antiphospholamban antibody (Thermo Scientific, Waltham, Massachusetts) for 2 h at 4ºC. The tissue lysate then was incubated with 60 μg of protein A/G-agarose for 2 h at 4ºC. After washing 4× with washing buffer (20 mmol/l Tris-HCl (pH 7.4), 150 mmol/l NaCl, and 1% Nonidet P-40), the beads were incubated with 2 μg of glutathione S-transferase (GST) or GST fusion protein in 20 μl of reaction buffer containing 20 mmol/l 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid (pH 7.4), 20 mmol/l MgCl2, 2 mmol/l dithiothreitol, and 200 μmol/l ATP at 37ºC for 1 h. The reaction was terminated by adding NuPAGE LDS sample buffer (Invitrogen) containing 10% β-mercaptoethanol and boiling for 10 min. Samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis.
Assessment of infarct area
After 120 min of reperfusion, hearts were perfused with a TTC 1% solution for 5 min and frozen at −20ºC. The hearts were sliced into 6 (1-mm thickness/slice) cross sections. Viable myocardium showed red, and the infarct area showed pale white. Each slice image was recorded digitally using a camera mounted on a dissecting scope. The total and infarct area of ventricle of each slice were visually demarcated and measured using SPOT image analysis software (Sterling Heights, Michigan). The relative infarction was calculated as the percentage of infracted area over the total ventricular area.
Tunel staining
To detect apoptotic cells, a TUNEL assay was performed using the Apoptag kit (Invitrogen). Cross sections (5 μm) from frozen hearts (6 sections/heart) were prepared. Nuclei were counterstained with 4,6-diamidino-2-phenylindole, cardiomyocytes were labeled with α-actin, and the percentage of TUNEL-positive cardiomyocytes cells was calculated from total TUNEL-positive myocyte nuclei versus total 4,6-diamidino-2-phenylindole–labeled myocyte nuclei.
MTT assay
Neonatal rat ventricular myocytes (NRVMs) were plated out in 100 ul of medium at a concentration of 5 × 104 cells in 96-well culture plates and cultured in an incubator at 37°C in an atmosphere of 5% CO2 in air. After 36 h of adenovirus infection, NRVM were treated with hydrogen peroxide (H2O2: 20, 50, or 100 μmol/l for 16 h) or 100 μmol/l of H2O2 for 2, 8, and 16 h. A total of 10 ul of 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT 5 mg/ml, dissolved in phosphate-buffered saline) was added to each well. After 3 h incubation in the dark, NRVMs were washed and lysed to release formazan, which was subsequently dissolved in 100 μl of dimethyl sulfoxide. The absorbance was recorded in a microplate reader (Synergy H1, BioTek Instruments, Winooski, Vermont) at a wavelength of 570 nm. Results were normalized for the control group.
Statistical analysis
Data are presented as mean ± SD. Means between 2 experimental groups were compared with an unpaired Student t test or Mann-Whitney U test. Multigroup comparisons were performed by Kruskal-Wallis test, 2-way analysis of variance, and repeated measure analysis of variance. Multiple comparisons were performed by Stell method and Bonferroni correction. A p value <0.05 was considered statistically significant.
Results
Identification of PP2Ce as an ER/SR membrane-targeted protein phosphatase in cardiomyocytes
In previous studies, we reported that PP2Ce had targeted localization on the endoplasmic reticulum (ER) membrane 28, 29 (Figure 1A). In fat cells and mammary glands, PP2Ce possesses highly specific activity toward IRE1, a key signal molecule for ER stress response and serves as a negative feedback modulator to ER stress signaling, with a significant role in adiposity and lactating activity 28, 29. By Northern blot analysis from tissues of adult mice, PP2Ce mRNA was also detected at substantial levels in the brain, heart, and diaphragm (Figure 1B). Using immunofluorescent microscopy, we confirmed that the full-length PP2Ce protein was specifically targeted on the SR membrane in adult rat cardiomyocytes, colocalizing with the SERCA2a (Figure 1C). Furthermore, PP2Ce the mRNA level was elevated in human ischemia cardiomyopathy or dilated cardiomyopathy hearts comparing with the nonfailing control subjects (Figure 1D). However, overexpressing PP2Ce in cultured NRVMs did not significantly affect ER stress response pathway, including phosphor-PERK and phosphor-elF2α, as well as BiP and Xbp1 expression under basal or oxidative stress (Figure 2B), although there was a modest reduction in CHOP and activated caspase 3.
Figure 1.

PP2Ce is an ER Membrane-Targeted Protein Phosphatase and Detected in Adult Mouse Hearts
(A) Schematic description of protein phosphatase 2C on endoplasmic reticulum (ER) membrane (PP2Ce) protein with signal peptide sequence (aa20-40), transmembrane domain, and conserved PP2C phosphatase activity domain highlighted. (B) Northern-blot analysis of PP2Ce messenger ribonucleic acid (mRNA) expression in different tissues from adult mice with significant expression detected in in the brain, heart, and diaphragm. (C) Immunohistochemistry results displayed PP2Ce (green) was colocalized with sarcolemma-endoplasmic reticulum calcium ATPase (SERCA) 2a (red) in adult rabbit ventricular cardiomyocytes as supported in merged image and line-scanning intensity analysis. (D) Relative expression of PP2Ce mRNA detected by quantitative real-time polymerase chain reaction in heart samples from ischemic cardiomyopathy (ICM) (n = 3) and dilated cardiomyopathy (DCM) (n = 3) compared with non-heart failure hearts (normal, n = 3). *p < 0.05 versus normal.
Figure 2.


PP2Ce Expression on ER Stress Signaling Pathway in NRVM
(A) Representative immunoblot of ER stress regulators as indicated in NRVM expressing GFP and PP2Ce in response to 50 μmol/l H2O2 treatment at specific time points as indicated. The 5 μg/ml tunicamycin-treated NRVM was used as a positive control. (B) Quantification of immunoblot signals with independent experimental replicate number labeled in each column. **p < 0.01. ER = endoplasmic reticulum; GFP = green fluorescent protein; NRVM = neonatal rat ventricular myocyte; PP2Ce = protein phosphatase 2C on ER membrane.
Function of PP2Ce in cardiomyocytes as a PLN-specific phosphatase
Considering the fact that PP2Ce is an SR-targeted protein phosphatase with substantial cardiac expression, we investigated the effect of PP2Ce expression on the phosphorylation of several SR membrane proteins in cardiomyocytes under basal or post-isoproterenol (ISO) treatment. As shown in Figures 3A and 3B, PP2Ce expression did not markedly affect RyR2 phosphorylation at either PKA (Ser-2808) or CaMKII (Ser-2814)–targeted sites or the phosphorylation levels of other cytosolic proteins, such as troponin I. In contrast, PP2Ce expression potently blocked ISO-induced PLN phosphorylation at the Thr-17 residue, with a relatively more modest effect on its Ser-16 phosphorylation under the same condition. To evaluate whether PP2Ce-induced PLN Thr-17 dephosphorylation was achieved by inhibiting CaMK activity or by directly targeting PLN, we tested the effect of PP2Ce expression on CaMK activity. As shown in Figures 3A and 3B, PP2Ce expression did not markedly affect the basal or the ISO-induced autophosphorylation levels of CaMKII. Furthermore, when we coexpressed PP2Ce and a constitutively active form of CaMKII (CA) in NRVM, the PLN Thr-17 phosphorylation induced by the CaMKII-CA expression was markedly abolished (Figure 3C), further supporting PLN as a molecular target of PP2Ce. To demonstrate the direct phosphatase activity of PP2Ce toward PLN, we expressed a phosphatase dead mutant (D302A) (Figure 4A) and deletion mutant PP2Ce without ER targeting ability (Δ1-57) in NRVM (Figure 4B), and these PP2Ce mutants showed no marked effect on PLN phosphorylation under ISO stimulation. These data suggest the PP2Ce-mediated PLN dephosphorylation requires both phosphatase activity and ER membrane localization. To further support its direct function and specificity, we generated recombinant GST-fusion proteins for the wild-type and a phosphatase dead mutant of PP2Ce, and incubated the recombinant PP2Ce or PP2Ce-mutant proteins with PLN proteins prepared by immunoprecipitation from ISO-treated mouse left ventricle tissues where both Ser-16 and Thr-17 sites were highly phosphorylated. In this cell-free system, PP2Ce did not affect PLN phospho-Ser-16 levels, but effectively diminished the phospho-Thr-17 level (Figure 4C), supporting that dephosphorylating the PLN-Thr-17 is a direct and specific intrinsic activity of PP2Ce. Finally, by coimmunoprecipitation assay, we demonstrated protein–protein interaction between PLN and PP2Ce in intact mouse hearts (Figure 4D). All of these data suggest that PP2Ce is a novel PLN phosphatase with a highly specific intrinsic phosphatase activity toward phosphor-Thr-17.
Figure 3.


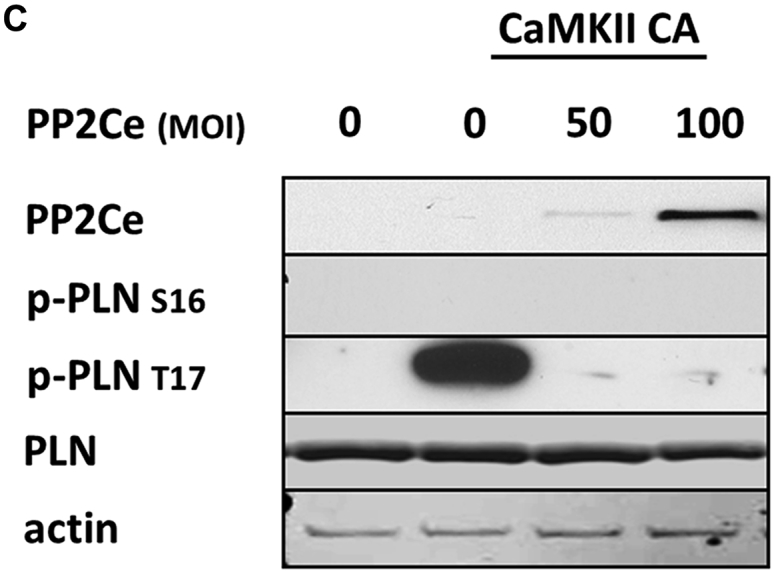
PP2Ce is a Specific Phosphatase for PLN Thr-17 Phosphorylation Site
(A) Representative immunoblot from NRVM-expressing GFP or PP2Ce were treated with 1 μmol/l isoproterenol for 0.5 and 2 h as indicated. (B) Quantification of immunoblot signals with independent experimental replicate number labeled in each corresponding column. (C) Western blot for NRVM coexpressing active CaMKII (CaMKII-CA) and increasing amount of PP2Ce as indicated. *p < 0.05; **p < 0.01 versus untreated; and #p < 0.05; between PP2Ce and GFP at each time point. PLN = phospholamban; other abbreviations as in Figure 2.
Figure 4.

Specificity of PP2Ce Activity to PLN Thr-17 Phosphorylation
(A) Immunoblot of PP2Ce, GFP, and PLN phosphorylation (Ser-16 and Thr-17) in NRVM expressing GFP or PP2Ce wild-type or PP2Ce phosphatase dead mutant (D320A) following 1 μmol/l isoproterenol treatment at different time points as indicated. (B) ISO-induced PP2Ce expression and PLN phosphorylation in NRVM-expressing PP2Ce truncation mutant (⊿1–57), which has lost ER targeting capacity 28, 29. (C) PP2Ce–GST fusion protein was incubated with PLN isolated from NRVM with or without ISO treatment, followed by immunoblot for total and phosphorylation levels of PLN at Ser-16 and Thr-17. (D) Coimmunoprecipitation of PP2Ce and PLN in mouse heart using PP2Ce TG and PP2Ce KO as positive and negative control, respectively. ISO = isoproterenol; TG = transgenic; other abbreviations as in Figures 2 and 3.
Post-transcriptional regulation of PP2Ce expression in cardiomyocytes
Because there is no commercial antibody sufficiently sensitive to detect the endogenous PP2Ce protein in cardiomyocytes, we followed the expression of PP2Ce-Flag fusion protein introduced into NRVM (Figures 3, 4, and 5). Stimulation of NRVM with ISO significantly and consistently induced PP2Ce protein level as early as 2 h after treatment (Figure 3B). This induction is specific to PP2Ce, as ectopic expression of green fluorescent protein (GFP) or another PP2C family member, PP2Cα, using the same expression vectors were not affected by ISO treatment within the same time period (Figures 3B and 5A). The ISO-mediated induced PP2Ce protein was significantly blunted by protein synthesis inhibition (Figures 5B and 5C) but further increased by short-term proteasome inhibition (Figures 5B and 5C). Therefore, ISO-mediated PP2Ce induction involved protein synthesis and degradation. Direct induction of cAMP by forskolin treatment was sufficient to induce the same pattern of PP2Ce protein increase (Figure 5D). However, PKA inhibition by treating cells with H89 markedly blocked ISO-mediated PP2Ce induction (Figure 5E). In addition to β-adrenergic stimulation, oxidative stress induced by superoxide treatment also increased PP2Ce protein level in cardiomyocytes as early as 8 h (Figures 2A and 2B). This evidence suggests that PP2Ce expression is subjected to dynamic regulation at protein level in response to pathological stressors in cardiomyocytes. Such induction can serve as an intrinsic and local negative modulator for β-adrenergic signaling by specifically targeting PLN Thr-17 phosphorylation.
Figure 5.

ISO-Mediated Regulation of PP2Ce Protein in Cardiomyocytes
(A) PP2Ce and PP2Cα expression in NRVM following ISO treatment. (B) PP2Ce expression in NRVM treated with ISO (1 μmol/l) with or without cotreatment of proteasome inhibitor MG132 (10 μmol/l) or lysosomal inhibitor BAF (50 nmol/l) as indicated. (C) Quantification of PP2Ce expression in NRVM treated with ISO with or without additional treatment of cycloheximide (CHX), MG132, or BAF as indicated. (D) PP2Ce expression in NRVM-treated ISO or forskolin (FSK) at 0.1, 1, and 10 μmol/l concentrations as indicated. (E) PP2Ce expression in NRVM treated with FSK (1 μmol/l) with or without PKA inhibitor H89 (1 μmol/l) as indicated. n indicates the number of total experimental replicates. *p < 0.05; **p < 0.01. Abbreviations as in Figures 2, 3, and 4.
Functional effect of PP2Ce in cardiomyocytes
As we established PLN as a specific substrate of PP2Ce and a key modulator of SR calcium homeostasis, we investigated the physiological effect of PP2Ce on SR calcium cycling in cardiomyocytes. We measured the effect of PP2Ce expression on SR calcium cycling in isolated adult rat ventricular cardiomyocytes. As shown in Figure 6, expression of PP2Ce did not affect basal calcium transient but significantly reduced the intracellular peak calcium amplitude and slowed the calcium transient decline rate following ISO stimulation, supporting the functional effect of PP2Ce expression on SR calcium cycling consistent with its targeted dephosphorylation of PLN.
Figure 6.

Functional Effect of PP2Ce Expression on SR Calcium Homeostasis in Adult Rat Cardiomyocytes
(A) Representative images of calcium transients from control rat myocytes at basal and following ISO (0.1 μmol/l) treatment. (B) Representative recording of calcium signal (F/F0) at basal and post-ISO treatment in LacZ-expressing rat adult cardiomyocytes. (C) Representative recording of calcium signal at basal and post-ISO treatment in PP2Ce-expressing rat adult cardiomyocytes. (D) Summary data of changes in calcium amplitude and decay time in adult cardiomyocytes expressing LacZ (n = 5) versus PP2Ce (n = 3) between basal and post-ISO treatment. *p < 0.05; **p < 0.01 LacZ versus PP2Ce, respectively. ⊿ = the difference between post- and pre-ISO treatment; other abbreviations as in Figures 2, 3, and 4.
To further determine the functional role of PP2Ce in the intact heart, we developed a transgenic mouse model with cardiac-specific expression of PP2Ce (PP2Ce-TG) (Figure 7A). Western blots revealed that PP2Ce expression was induced in the transgenic heart specifically in double transgenic animals (Figure 7B). Consistent with previous in vitro observations at the basal level, the PP2Ce-TG mice exhibited normal cardiac phenotype at morphological and functional levels (Table 1). Also, consistent with the in vitro results, the basal PLN phosphorylation at Thr-17 was significantly inhibited, although in this case, Ser-16 phosphorylation was also reduced (Figures 7C and 7D). In contrast, the phosphorylation levels of RyR2 at both PKA- and CaMKII-dependent sites and the phosphorylation levels of troponin I were not changed, nor were the total protein levels for RyR2, troponin I, SERCA-2a, and sodium-calcium exchanger 1 (Figures 7C and 7D). Notably, PP1 expression was not affected by PP2Ce expression. In isolated intact left ventricular cardiomyocytes loaded with Fluo-4 AM (Ca2+ fluorescent dye) and paced at 1 Hz by an electric stimulator (Figure 7E), the 50% decay time of Ca2+ transients was significantly prolonged in the PP2Ce-TG myocytes (average 159.4 ms) relative to the non-TG control subjects (average 135.9 ms; p < 0.05). However, there were no significant differences in the peak Ca2+ transient amplitude or the time to peak of Ca2+ transients between the PP2Ce-TG myocytes and control subjects (Figure 7E).
Figure 7.



Effect of PP2Ce on Calcium Homeostasis in Intact Mouse Heart
(A) Schematic drawing of PP2Ce-transgenic mouse with tissue-specific expression of PP2Ce in heart. (B) PP2Ce protein expression in mouse hearts from 4 genotypes carrying either none, single, or double transgenes of αMHC-tTA expressing tet-off suppressor (tTA) and αHMC-TetO-PP2Ce (iPP2Ce). PP2Ce expression was detected only from double transgenic hearts in the absence of tetracycline. PC is the positive control from NRVM-expressing adv-PP2Ce. (C) Representative immunoblot analysis. (D) Quantification of key proteins involved in calcium homeostasis regulation in control and PP2Ce transgenic (TG) hearts. Number of replicates is labeled in each column. (E) Calcium transient peak amplitudes (F/F0), time to peak and decay measured from isolated cardiomyocytes of control (n = 8) and PP2Ce TG (n = 8) hearts. *p < 0.05; **p < 0.01. Abbreviations as in Figures 2, 3, and 4.
Table 1.
Echocardiogram Parameters Measured From Nontransgenic and PP2Ce Transgenic Hearts
| HR (beats/min) | LVPW(s) (mm) | LVPW(d) (mm) | LVID(s) (mm) | LVID(d) (mm) | FS (%) | |
|---|---|---|---|---|---|---|
| Control (n = 41) | 484 ± 86 | 1.00 ± 0.12 | 0.66 ± 0.12 | 2.86 ± 0.34 | 4.13 ± 0.33 | 31.1 ± 4.8 |
| TG (n = 33) | 479 ± 36 | 1.06 ± 0.14∗ | 0.68 ± 0.15 | 2.70 ± 0.23∗ | 4.11 ± 0.23 | 34.0 ± 4.7† |
Values are mean ± SEM.
(d) = diastolic; FS = fractional shortening; HR = heart rate; LVID = left ventricle internal dimension; LVPW = left ventricle posterior wall thickness; (s) = systolic; TG = transgenic.
p < 0.05; †p < 0.01 between control and TG groups.
PP2Ce expression impairs inotropic response to isoproterenol
To directly examine the functional effect of PP2Ce expression on cardiac contractile function and isoproterenol response, we measured left ventricular pressure in isolated perfused hearts from nontransgenic and PP2Ce transgenic mice. As shown in Figure 8, the PP2Ce transgenic hearts had similar contractile function at the basal condition (Figure 8, Table 2) but showed a blunted response to isoproterenol stimulation compared with wild-type control hearts based on systolic pressure, dP/dTmax, and dP/dT min, as well as left ventricle developed pressure (Figure 8B). This is consistent with in vitro calcium measurement observed in isolated myocytes (Figure 7).
Figure 8.

PP2Ce Expression Impairs ISO Stimulated Enhancement of Contractility
(A) Representative left ventricle pressure (LVP) measured from isolated hearts of wildtype or PP2Ce TG mice at basal or perfused by 1 μmol/l of ISO. (B) Hemodynamic parameters from control (n = 9) and PP2Ce TG (n = 6) hearts at basal or treated by 10 nmol/l ISO. *p < 0.05; **p < 0.01 between basal and I/R within control or PP2Ce TG groups. #p < 0.05 between corresponding control and PP2Ce TG. dP/dt max = maximal systolic pressure derivative; dP/dt min = maximal diastolic pressure derivative; HR = heart rate; I/R = ischemia/reperfusion; LVDP = left ventricle developed pressure (Pmax − Pmin); Pmax = systolic pressure; Pmin = diastolic pressure; Tau = relaxation index; other abbreviations as in Figures 2, 3, and 4.
Table 2.
Hemodynamic Parameters
| Control (n = 9) |
TG (n = 6) |
|||
|---|---|---|---|---|
| Basal | I/R | Basal | I/R | |
| HR, beats/min | 403.5 ± 96.4 | 334.7 ± 87.2 | 390.0 ± 74.6 | 281.8 ± 49.9 |
| Pmax, mm Hg | 91.22 ± 20.67 | 95.50 ± 10.60 | 98.55 ± 17.72 | 94.35 ± 10.42 |
| Pmin, mm Hg | 3.68 ± 2.30 | 39.71 ± 12.52∗ | 4.28 ± 2.44 | 55.03 ± 11.26∗† |
| dP/dt max, mm Hg/s | 4,794.9 ± 446.4 | 2,906.0 ± 490.8∗ | 4,942.2 ± 871.5 | 1,839.8 ± 411.0∗‡ |
| dP/dt min, mm Hg/s | 3,031.6 ± 359.8 | 1,867.3 ± 235.3∗ | 3,156.9 ± 435.6 | 1,225.6 ± 344.6∗‡ |
| LVDP, mm Hg | 87.54 ± 18.76 | 55.79 ± 8.31∗ | 94.27 ± 18.45 | 39.33 ± 11.64∗ |
Values are mean ± SEM. Cardiac performance was measured from isolated nontransgenic (control, n = 9) and the PP2Ce TG (n = 6) hearts under basal and post-30-min global ischemia and 45-min reperfusion (I/R).
dP/dt max = maximal systolic pressure derivative; dP/dt min = maximal diastolic pressure derivative; LVDP = left ventricle developed pressure (Pmax − Pmin); Pmax = systolic pressure; Pmin = diastolic pressure; other abbreviations as in Table 1.
p < 0.01 between basal and I/R within control or TG groups.
p < 0.05; ‡p < 0.01 between corresponding control and TG groups.
PP2Ce expression reduced function recovery post-I/R injury
CaMKII-mediated Thr-17 phosphorylation of PLN has been implicated in I/R injury and cardiomyocyte death regulation 13, 14, 15. Therefore, we investigated the effect of PP2Ce expression in the intact PP2Ce-TG hearts during I/R injury. Using a Langendorff preparation, the isolated mouse hearts from the PP2Ce-TG and the control mice were subjected to 30 min of global no-flow ischemia followed by 60 to 120 min of reperfusion (I/R) while contractility was continuously monitored via a conductance catheter for left ventricle pressure (Figure 9A). As shown in Figure 9B, the functional recovery following reperfusion was significantly impaired in the PP2Ce-TG transgenic hearts compared with the controls. Particularly, after 45 min of reperfusion, systolic function as measured from left ventricle developed pressure (PP2Ce-TG vs. control: 39.33 ± 11.64 mm Hg vs. 55.79 ± 8.3 mm Hg; p < 0.01) (Figure 9B) and dP/dTmax (PP2Ce-TG vs. control: 1,839.78 ± 410.98 mm Hg/s vs. 2,905.99 ± 490.76 mm Hg/s; p < 0.01) was significantly reduced in the PP2Ce-TG hearts compared with the controls (Figures 9A to 9D, Table 2). In addition, cardiac relaxation as measured from dP/dT min (PP2Ce-TG vs. control: 1,274.42 ± 239.59 mm Hg/s vs. 1,707.4 ± 190.53 mm Hg/s; p < 0.05) (Figure 9D) and Tau (PP2Ce-TG vs. control: 46.7 ± 21.4 ms vs. 23.3 ± 5.5 ms; p < 0.05) (Figure 9E) were also significantly impaired in the PP2Ce-TG hearts compared with the control hearts at 60 min post-reperfusion. These data demonstrated exacerbated systolic and diastolic dysfunction following I/R injury on PP2Ce expression in the heart. Along with impaired functional recovery, myocardial infarct size as measured by TTC staining (infarct area vs. area at risk) at 120 min post-reperfusion was significantly larger in the PP2Ce-TG hearts compared with the control hearts (infarct area/area at risk: 59.82 ± 6.10% vs. 40.06 ± 6.71%; p < 0.01) (Figures 10A and 10B). Additionally, using TUNEL staining, more apoptotic cells were detected in the PP2Ce9-TG heart than the control hearts (Figure 10C). To further support the status of impaired cardiac protection by PP2Ce expression, mitochondrial-mediated apoptotic cell death signaling as represented by cleaved caspase 9 was detected to be significantly induced in the PP2Ce-TG hearts compared with control hearts following I/R (Figures 10D and 10E).
Figure 9.

PP2Ce Expression Impairs Function Recovery Following Ischemia/Reperfusion Injury
(A) Left ventricle dP/dt max during 15-min global ischemia followed by 60-min reperfusion in isolated hearts from nontransgenic control subjects (control, n = 9) and PP2Ce TG (TG, n = 6) mice. Left ventricle function parameters measured as (B) left ventricular developed pressure (LVDP), (C) systolic pressure increase (dP/dt max), (D) diastolic pressure decrease (dP/dt min), and (E) relaxation time index (Tau) in nontransgenic control subjects (control, n = 9) and PP2Ce TG hearts (n = 6) at basal and 45 min after reperfusion. **p < 0.01 between control and PP2Ce TG groups. #p < 0.05; ##p < 0.01 between corresponding control and PP2Ce TG groups at each time point.
Figure 10.

Induced PP2Ce Expression Promotes Cardiomyocyte Death in Response to I/R Injury
(A) Representative TTC staining cross section images from 1 PP2Ce TG and 1 control heart following 15 min of ischemia and 120 min of reperfusion (I/R). (B) Infarct sizes of control (n = 5) and PP2Ce TG (n = 6) hearts as measured from (A). (C) Apoptotic index measured by TUNEL staining in control (n = 4) and PP2Ce TG (n = 4) hearts. (D) Representative immunoblot for level of activated caspase 9 in control (n = 10) and PP2Ce TG (n = 8) hearts following the same I/R protocol. (E) Quantification of level of cleaved caspase 9 signal from immunoblots. *p < 0.05 between control and PP2Ce TG groups. **p < 0.01 between control and PP2Ce TG groups. Abbreviations as in Figures 2, 3, and 4.
PP2Ce expression promotes oxidative stress-induced cell death in cardiomyocytes
We next investigated whether PP2Ce expression directly affects myocyte survival under oxidative stress in vitro. NRVMs showed dosage-dependent cell death following H2O2 treatment at 20, 50, and 100 mol/l for up to 16 h based on MTT assay. Following PP2Ce expression, the NRVM showed significantly reduced cell viability upon H2O2 or ionomycin treatment compared with the GFP-expressing cells (Figures 11A and 11B). To explore the signaling mechanism involved in this process, we examined AKT and mitogen-activated protein kinase pathways. As shown in Figures 11C and 11D, PP2Ce expression significantly reduced pro-survival AKT and extracellular signal-regulated kinase activity following oxidative stress without significantly affecting pro-death stress MAPK activities, including p38 and c-jun N-terminal kinase. In line with the role of PP2Ce in SR calcium regulation, calcium overload-induced cell death triggered by ionomycin treatment for 24 h was also significantly enhanced by PP2Ce expression (Figure 11B). Taken together, PP2Ce expression promotes oxidative injury via enhanced myocyte death associated with disturbance in intracellular signaling.
Figure 11.


PP2Ce Expression Promotes Myocytes Death and Perturbs Prosurvival Signaling Following Oxidative Injury
(A) NRVM viability measured by MTT assay in NRVM expressing GFP or PP2Ce following H2O2 treatment for 16 h at different doses as indicated. Replicate number is labeled in each corresponding column. (B) NRVM viability expressing GFP (n = 8) or PP2Ce (n = 8) under untreated or following 24 h of treatment of 2 μmol/l ionomycin. (C) Immunoblot analysis of intracellular signaling molecules as indicated in NRVM-expressing GFP or PP2Ce with 50 μmol/l H2O2 treatment at different time points as indicated. Tunicamycin (5 μg/ml for 4 h) was used as a control. (D) Quantification of signaling molecules as indicated with independent experimental replicate number labeled in each column. **p < 0.01 versus untreated. ##p < 0.01 between GFP and PP2Ce groups at same doses. Abbreviations as in Figures 2, 3, and 4.
Discussion
In this report, we characterized a newly identified protein phosphatase PP2Ce in cardiomyocytes in terms of its molecular target and cellular/physiological function in the heart. Our findings establish that PP2Ce is localized on the SR membrane overlapping with SR calcium ATPase 2a. PP2Ce expression is significantly induced at the mRNA level in human failing hearts, whereas PP2Ce protein, when expressed in cardiomyocytes, is acutely induced by β-adrenergic stimulation and oxidative stress. From extensive molecular and cellular studies, we find that PP2Ce is a highly specific PLN phosphatase with intrinsic activity toward PLN Thr-17. Expression of PP2Ce in cardiomyocytes showed a significant effect on SR calcium homeostasis, particularly in calcium uptake, in line with its targeted dephosphorylation of PLN. In addition, PP2Ce expression both in vitro and in vivo promoted oxidative injury and myocyte death. Therefore, PP2Ce functions as an uncharacterized modulator for cardiac calcium cycling and has a potentially significant contribution to stress adaption in the heart in response to β-adrenergic stimulation and oxidative injury.
PP2Ce protein is increased within 2 h in response to acute β-adrenergic stimulation at the protein level. PP2Ce protein is also affected by short-term proteasome inhibition alone, and ISO-induced PP2Ce protein induction requires de novo protein synthesis. Therefore, PP2Ce protein homeostasis is modulated by both synthesis and proteasome-dependent degradation at the basal state. The underlying mechanism of its turnover is unclear, and the molecular signaling participated in ISO-mediated PP2Ce protein induction remains to be established, although our data suggest that it is a PKA-dependent process. Interestingly, similar induction was observed in oxidative stressed myocytes, suggesting such induction can be triggered by different stressors through different pathways. Induction of a PLN-specific phosphatase in cardiomyocytes following pathological stress offers an intriguing mechanism of negative feedback regulation in cardiomyocytes to maintain SR calcium homeostasis following stress stimulations (Figure 12). Significant induction of PP2Ce expression detected in human failing hearts may represent a chronic maladaptation similar to β-adrenergic desensitization.
Figure 12.

Illustration of PP2Ce Function in Cardiac Myocytes
We showed that, in a cell-free system, PP2Ce specifically dephosphorylated the Thr-17 site of PLN but not the Ser-16 site, and PP2Ce expression did not affect the phosphorylation status of other ER membrane targeted proteins, such as RyR. This implies that PP2Ce possesses a very high degree of intrinsic substrate specificity. This contrasts with PP1, a major PLN phosphatase with specificity toward both Ser-16 and Thr-17 sites, as well as other substrates. In addition, PLN dephosphorylation can also be carried out by PP2A with its regulatory subunit B56. However, the specificity of PP2A/B56 is also not limited to PLN. In intact heart, however, PP2Ce expression affected both Ser-16 and Thr-17 sites, suggesting that either PP2Ce acquires new specificity in adult cardiomyocytes due to a complex interaction that is yet to be characterized or indirectly regulates Ser-16 phosphorylation. Nevertheless, targeted PLN dephosphorylation in vivo is consistent with the phenotype observed in the PP2Ce-TG hearts, which displayed relatively normal basal contractile function but showed blunted inotropic response to ISO stimulation and more susceptibility to I/R-induced injury. In fact, our observation is in good agreement with previous findings that CaM kinase-mediated PLN regulation (through Thr-17 phosphorylation) affects cardiac injury following I/R. PP2Ce is also reported to regulate IRE1-mediated unfold-protein response by specifically interacting with and dephosphorylating IRE1 without cross-reactivity toward PERK. In the heart, basal IRE1 expression is low (undetectable with current antibodies, data not shown), whereas the PP2Ce expression is relatively abundant. Therefore, PP2Ce function in heart may involve both substrates or other substrates that are yet to be characterized. Further molecular studies will be needed to determine all possible downstream targets of PP2Ce in the heart and to uncover the underlying molecular basis for such a high substrate specificity.
Conclusions
Our extensive molecular, cellular, and molecular analyses have revealed a previously uncharacterized signaling regulator in the heart: an ER membrane-targeted protein phosphatase with high specificity toward PLN Thr-17 phosphorylation. Its induction in human failing hearts and our in vivo evidence from intact mouse hearts implicate its potential role in the pathogenesis of cardiac injury through modulating SR calcium uptake and homeostasis.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Defects in cardiomyocyte calcium homeostasis and cycling significantly contribute to cardiac dysfunction. PLN is a key regulator of SERCA, and its function is modulated by PKA and CaMKII-mediated protein phosphorylation; loss of PLN phosphorylation is a molecular hallmark associated with heart failure. PP2Ce is a newly identified phosphatase with potent and specific activity to dephosphorylate PLN in cardiomyocytes.
TRANSLATIONAL OUTLOOK: PP2Ce expression is induced rapidly after pathological stress, and PP2Ce induction has negative effects on cardiac contractile function and cardiomyocyte viability. Therefore, PP2Ce induction may represent a novel mechanism for heart failure, and its inactivation can serve as a potential therapeutic strategy.
Footnotes
This work is supported in part by National Institutes of Health grants R01HL108186 (to Drs. Wang and Ruan) and R01 HL070079 (to Dr. Wang); as well as grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan and the MEXT Supported Program for the Strategic Research Foundation at Private Universities (to Drs. Akaike and Minamisawa); and the Vehicle Racing Commemorative Foundation and Jikei University Graduate Research Fund (to Dr. Minamisawa). All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
Contributor Information
Yibin Wang, Email: yibinwang@mednet.ucla.edu.
Hongmei Ruan, Email: hmruan@ucla.edu.
References
- 1.Thomas N.L., George C.H., Lai F.A. Role of ryanodine receptor mutations in cardiac pathology: more questions than answers? Biochem Soc Trans. 2006;34:913–918. doi: 10.1042/BST0340913. [DOI] [PubMed] [Google Scholar]
- 2.Jiang D., Xiao B., Yang D. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci U S A. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Periasamy M., Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 4.Tiso N., Stephan D.A., Nava A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Genet. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 5.Periasamy M., Reed T.D., Liu L.H. Impaired cardiac performance in heterozygous mice with a null mutation in the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 (SERCA2) gene. J Biol Chem. 1999;274:2556–2562. doi: 10.1074/jbc.274.4.2556. [DOI] [PubMed] [Google Scholar]
- 6.Hovnanian A. SERCA pumps and human diseases. Subcell Biochem. 2007;45:337–363. doi: 10.1007/978-1-4020-6191-2_12. [DOI] [PubMed] [Google Scholar]
- 7.MacLellan W.R. Advances in the molecular mechanisms of heart failure. Curr Opin Cardiol. 2000;15:128–135. doi: 10.1097/00001573-200005000-00002. [DOI] [PubMed] [Google Scholar]
- 8.Elliott P., McKenna W.J. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. doi: 10.1016/S0140-6736(04)16358-7. [DOI] [PubMed] [Google Scholar]
- 9.Bers D.M., Despa S., Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- 10.Eisner D. Calcium in the heart: from physiology to disease. Exp Physiol. 2014;99:1273–1282. doi: 10.1113/expphysiol.2013.077305. [DOI] [PubMed] [Google Scholar]
- 11.MacLennan D.H., Kranias E.G. Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 12.Haghighi K., Bidwell P., Kranias E.G. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J Mol Cell Cardiol. 2014;77:160–167. doi: 10.1016/j.yjmcc.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwanaga Y., Hoshijima M., Gu Y. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest. 2004;113:727–736. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama H., Chen X., Baines C.P. Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. 2007;117:2431–2444. doi: 10.1172/JCI31060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vittone L., Mundina-Weilenmann C., Mattiazzi A. Phospholamban phosphorylation by CaMKII under pathophysiological conditions. Front Biosci. 2008;13:5988–6005. doi: 10.2741/3131. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez P., Kranias E.G. Phospholamban: a key determinant of cardiac function and dysfunction. Arch Mal Coeur Vaiss. 2005;98:1239–1243. [PubMed] [Google Scholar]
- 17.Chu G., Kranias E.G. Phospholamban as a therapeutic modality in heart failure. Novartis Found Symp. 2006;274:156–171. discussion 172–5, 272–6. [PubMed] [Google Scholar]
- 18.Waggoner J.R., Kranias E.G. Role of phospholamban in the pathogenesis of heart failure. Heart Fail Clin. 2005;1:207–218. doi: 10.1016/j.hfc.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Minamisawa S., Sato Y., Tatsuguchi Y. Mutation of the phospholamban promoter associated with hypertrophic cardiomyopathy. Biochem Biophys Res Commun. 2003;304:1–4. doi: 10.1016/s0006-291x(03)00526-6. [DOI] [PubMed] [Google Scholar]
- 20.Schmitt J.P., Kamisago M., Asahi M. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003;299:1410–1413. doi: 10.1126/science.1081578. [DOI] [PubMed] [Google Scholar]
- 21.Haghighi K., Kolokathis F., Pater L. Human phospholamban null results in lethal dilated cardiomyopathy revealing a critical difference between mouse and human. J Clin Invest. 2003;111:869–876. doi: 10.1172/JCI17892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haghighi K., Kolokathis F., Gramolini A.O. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeWitt M.M., MacLeod H.M., Soliven B., McNally E.M. Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J Am Coll Cardiol. 2006;48:1396–1398. doi: 10.1016/j.jacc.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 24.Haghighi K., Chen G., Sato Y. A human phospholamban promoter polymorphism in dilated cardiomyopathy alters transcriptional regulation by glucocorticoids. Hum Mutat. 2008;29:640–647. doi: 10.1002/humu.20692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Medin M., Hermida-Prieto M., Monserrat L. Mutational screening of phospholamban gene in hypertrophic and idiopathic dilated cardiomyopathy and functional study of the PLN -42 C>G mutation. Eur J Heart Fail. 2007;9:37–43. doi: 10.1016/j.ejheart.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Ruan H., Mitchell S., Vainoriene M. Gi alpha 1-mediated cardiac electrophysiological remodeling and arrhythmia in hypertrophic cardiomyopathy. Circulation. 2007;116:596–605. doi: 10.1161/CIRCULATIONAHA.106.682773. [DOI] [PubMed] [Google Scholar]
- 27.Ruan H., Li J., Ren S. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J Mol Cell Cardiol. 2009;46:193–200. doi: 10.1016/j.yjmcc.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 28.Ren S., Lu G., Ota A. IRE1 phosphatase PP2Ce regulates adaptive ER stress response in the postpartum mammary gland. PLoS One. 2014;9:e111606. doi: 10.1371/journal.pone.0111606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu G., Ota A., Ren S. PPM1l encodes an inositol requiring-protein 1 (IRE1) specific phosphatase that regulates the functional outcome of the ER stress response. Molecular metabolism. 2013;2:405–416. doi: 10.1016/j.molmet.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]