

Abstract
Background
Omphalocele is a congenital midline ventral body wall defect that can exist as isolated malformation or as part of a syndrome. It can be considered one of the major and most frequent clinical manifestation of Beckwith-Wiedemann Syndrome (BWS) in case of loss of methylation at KCNQ1OT1: Transcription Star Site-Differentially Methylated Region (TSS-DMR) or in presence of CDKN1C mutations. The isolated form of the omphalocele accounts approximately for about the 14% of the total cases and its molecular etiology has never been fully elucidated.
Methods
Given the tight relationship with BWS, we hypothesized that the isolated form of the omphalocele could belong to the heterogeneous spectrum of the BWS associated features, representing an endophenotype with a clear genetic connection. We therefore investigated genetic and epigenetic changes affecting BWS imprinted locus at 11p15.5 imprinted region, focusing in particular on the KCNQ1OT1:TSS DMR.
Results
We studied 21 cases of isolated omphalocele detected during pregnancy or at birth and identified the following rare maternally inherited variants: i) the non-coding variant G > A at nucleotide 687 (NR_002728.3) at KCNQ1OT1:TSS-DMR, which alters the methylation pattern of the imprinted allele, in one patient; ii) the deletion c.624-629delGGCCCC at exon 1 of CDKN1C, with unknown clinical significance, in two unrelated cases.
Conclusions
Taken together, these findings suggest that KCNQ1OT1:TSS-DMR could be a susceptibility locus for the isolated omphalocele.
Electronic supplementary material
The online version of this article (10.1186/s12881-017-0470-z) contains supplementary material, which is available to authorized users.
Keywords: Abdominal wall defects, Beckwith-Wiedemann syndrome, CDKN1C, Genomic imprinting, KCNQ1OT1:TSS-DMR, Omphalocele
Background
Omphalocele is a congenital midline ventral body wall defect characterized by the extrusion of abdominal viscera through the base of the umbilical cord. This malformation is related to the failure of the midgut loop to return to the peritoneal cavity after its herniation into the umbilical cord [1]. The incidence of omphalocele diagnosed during pregnancy is approximately 1/5000; however, this falls to 0.8/10,000 live births because of pregnancy termination [2].
The definition of abdominal wall defects can be difficult, since a ruptured omphalocele may be misdiagnosed as gastroschisis and an umbilical cord hernia can be confused with a small omphalocele. Gastroschisis is caused by vascular defects, does not involve the insertion site of the umbilical cord, and is generally localized to the right of the umbilicus [3]. Conversely, omphalocele results from an enlargement of the umbilical ring due to the migration failure of cephalic and/or lateral folds and abdominal bands during early embryogenesis [4, 5].
Omphalocele may be syndromic or isolated. Of syndromic cases, 54–57% are caused by aneuploidy, with a prevalence of trisomy 18 [6]. The remaining cases are associated with a number of genetic conditions including Beckwith-Wiedemann syndrome (BWS; MIM 130650), pentalogy of Cantrell (OMIM 313850), and OEIS (Omphalocele, Exstrophy, Imperforate anus, Spinal defects; OMIM 258040) [7–9]. Of these, BWS is the most common syndrome associated with omphalocele [10]. Indeed, isolated omphalocele can be the first manifestation of a mild clinical form of BWS, in a similar way to isolated hemihyperplasia, which can be predictive of BWS [11, 12]. BWS is genetically heterogeneous and may be caused by genomic, epigenetic, or genetic alterations at the 11p15.5 chromosomal locus. This region harbors two imprinted domains, IGF2/H19 and KCNQ1/CDKN1C, regulated by H19/IGF2:IG-DMR (InterGenic - Differentially Methylated Region) and KCNQ1OT1:TSS-DMR (Transcription Start Site – Differentially Methylated Region), respectively. In healthy individuals, H19/IGF2:IG-DMR is methylated on the paternally derived allele and unmethylated on the maternally inherited allele, resulting in the preferential expression of the paternal IGF2 and the maternal H19. At KCNQ1OT1:TSS-DMR, the opposite methylation pattern is observed [11, 12]. The KCNQ1OT1:TSS-DMR regulates the expression of CDKN1C and KCNQ1, as well as other genes, including PHLDA and SLC22A18. KCNQ1OT1:TSS-DMR is located within intron 10 of KCNQ1 and encompasses the promoter of the non-coding RNA KCNQ1OT1, which is orientated in the antisense direction relative to KCNQ1. Fig. 1a shows a schematic representation of the imprinted locus. Physiological expression of KCNQ1OT1 from the unmethylated paternal allele results in the bi-directional silencing of its flanking imprinted genes [13]. Among them, CDKN1C encodes P57 (alias KIP2), a tight-binding inhibitor of several G1 cyclin/Cdk complexes, acting as a negative regulator of cell proliferation [14]. The CDKN1C gene contains three exons and 2 GC-rich introns; however, only exons 1 and 2 encode the functional P57 protein. P57 is composed of 316 amino acids and consists of three structurally distinct domains: (i) the N-terminal domain (aa 1–110), which has significant similarity to the CdK-inhibitors, p21Cip1 and p27Kip1, and is necessary for CdK inhibition; (ii) the central highly polymorphic hexanucleotide repeat domain, encoding a series of proline-alanine repeats (PAPA-repeats, aa 156–213) involved in Mitogen-Activated Protein Kinase (MAPK) phosphorylation [15]; and (iii) the highly conserved C-terminal region (QT domain) that exhibits homology with p27Kip1 [16].
Fig. 1.
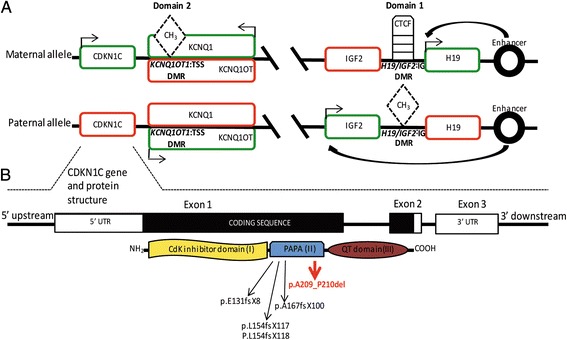
Epigenetic and genetic regulation of the 11p15.5 imprinted locus. As shown in (a), the 11p15.5 imprinted locus presents with 2 domains (Domain 1 and Domain 2) differentially regulated by H19/IGF2-IG and KCNQ1OT1:TSS DMRs, respectively. Transcribed genes are depicted in green, silenced genes in red. On the maternal allele, the effect of the enhancer promotes the expression of H19, the most proximal gene, but not of IGF2. This is the consequence of the CTCF binding to H19/IGF2-IG DMR located between the two genes. On the paternal allele, methylation of this DMR prevents the binding of CTCF, allowing the expression of IGF2 and the silencing of H19. At KCNQ1OT1:TSS DMR the opposite methylation pattern is observed. This DMR is methylated on the maternal allele and not on the paternal one. Thus, maternal repression of KCNQ10T allows the expression of CDKN1C and KCNQ1, while the expression of the KCNQ10T on the paternal allele inhibits the transcription of the flanking genes CDKN1C and KCNQ1. CDKN1C gene and protein structure are depicted in (b). White striped boxes identify the 5′- and 3′-UTRs and the black portion represents the coding sequence of the gene. CDKN1C encodes for P57 protein (alias KIP2), composed of the three functional domains depicted with different colors. Black arrows indicate previously identified truncating mutations inside the PAPA domain; red arrow points out the susceptibility variant p.A209_P210del identified in this paper
Decreased CDKN1C activity due to genetic variants, loss of KCNQ1OT1:TSS-DMR maternal methylation, or changes in gene copy number is associated with BWS. Forty to 50 % of BWS cases present with loss of KCNQ1OT1:TSS-DMR maternal methylation [17]. Abdominal wall defects are very common in cases with hypomethylation: 42.1% of patients have omphalocele, 20% present with umbilical hernia, and approximately 12% with diastasis recti [18].
Concerning genetic variants, causative CDKN1C point mutations are maternally inherited and cause protein loss-of-function. Point mutations of CDKN1C are present in up to 5% of sporadic and 50% of familial cases of BWS [19]. Most frequently, CDKN1C mutations are truncating or frameshift and are distributed along the entire coding region; conversely, missense mutations are mainly localized in the CDK-like domain [14]. Almost 90% of BWS patients with CDKN1C mutations have abdominal wall defects [18]; in particular, omphalocele occurs in 71.6% of these cases [18]. Finally, structural chromosome alterations such as deletions/duplications (1–2% of BWS cases) and uniparental disomy (UPD) at 11p15.5 involving CDKN1C (10–20% of BWS cases) [15] cause umbilical hernias in about half of patients, whereas in others no abdominal wall defects are described [18]. Thus, omphalocele can be considered one of the major and most frequent clinical manifestations of BWS in cases of KCNQ1OT1:TSS-DMR loss of methylation or CDKN1C mutations [20, 21].
A well-defined (epi)genotype–phenotype correlation exists for omphalocele in BWS: it is caused by KCNQ1OT1:TSS-DMR loss of methylation in 86% of the cases, by CDKN1C mutation in 7%, and by UPD or H19/IGF2:IG-DMR gain of methylation in <10% of patients [18].
Approximatively 14% of all cases of omphalocele can be classified as isolated [2]. Despite several studies investigating the genetic causes [22–25] or environmental risk factors [24] involved in this malformation, the etiology of isolated omphalocele has not been fully elucidated. Kanagawa et al. [23] described an extensive family with vertical transmission of isolated omphalocele, with nine subjects over three generations affected, suggesting an autosomal dominant pattern of inheritance. This finding was in agreement with five other previously reported pedigrees demonstrating vertical transmission of isolated omphalocele [25]. The same authors also described five families with an inheritance consistent with an autosomal recessive pattern [25]. Despite these observations, inherited alterations have never been identified in these families. More recently, a genomic duplication was found in a large pedigree with autosomal dominant transmission of isolated omphalocele [22]. Genetic analysis revealed that all affected individuals were positive for a duplication at chromosome 1p31.3, suggesting the involvement of one or more genes in the duplicated region, including FOXD3, ALG6, ITGB3BP, KIAA1799, DLEU2L, PGM1, and the proximal portion of ROR1 [22].
Methods
The early genetic characterization of newborns with omphalocele initially defined as isolated, together with a careful follow-up, can help pre- and/or post-natal clinical management and possibly enable definition of recurrence risk.
We studied 21 cases with isolated omphalocele diagnosed during pregnancy or at birth and we investigated molecular changes associated with BWS to determine whether isolated omphalocele could belong to the heterogeneous spectrum of BWS associated features. We did not consider other conditions (Pentalogy of Cantrell and OEIS) since the affected individuals usually present with complex and more severe phenotypes. Finally, we investigated whether genomic alterations at 1p31 or at other chromosome regions could be associated with this clinical manifestation.
Participants
The study included 21 patients (10 females and 11 males) with isolated omphalocele confirmed at birth, examined from 2013 to 2015 at the Department of Pediatric Surgery of Fondazione IRCCS Ca′ Granda Ospedale Maggiore Policlinico, Milan (Italy). Patients came to the attention of the pediatric clinicians of our hospital for diagnostic purposes in the period specified above. They (or their parents, in case of patients were underage) gave their consent to the possible future use of their genetic material for research purposes.
The clinical features of the population are described in Table 1. In details, seven cases were diagnosed at birth and 14 during prenatal ultrasound screening (US). Gestational age at diagnosis, in these latter cases, ranged from 12 to 26 Weeks of gestation (Wog).
Table 1.
Clinical features of the population involved in the study
| Case Nr | Conception | Omphalocele sonographic diagnosis (Wog) | Other sonographic features (Wog) | Delivery Type (Wog) | Omphalocele features | APGAR SCORE 1′/5’ | Other main clinical findings | Birth Weight (Centile) | Birth Lenght (Centile) | Birth OFC (Centile) | Follow-up | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age (Months) | Height/lenght (Centile) | Weight (Centile) | |||||||||||
| 1 | S | 20 | no | CS (37+5) | minor | 9/10 | no | 75° | 75° | 75° | 12 | 75° | 75°-90° |
| 2 | ICSI, egg donation | 20 | no | V (38) | medium | 9/10 | slight lingual protrusion; folded helix | 50°-75° | 75° | nd | 60 | 50°-75° | 25°-50° |
| 3 | S | ND | no | CS (40) | minor | 9/10 | ileal atresia | 75°-90° | 95° | 75° | 18 | 75° | 75° |
| 4 | S | 20 | no | CS (39) | medium | 9/10 | hepatic cyst | 3°-5° | <<3° | 10° | 60 | 25°-50° | 3° |
| 5 | S | ND | no | V (37) | minor | 8/9 | no | 10° | 10°-25° | 10° | 66 | 10°-25° | 25° |
| 6 | ICSI | 26 | no | CS (34) | minor | 6/7 | neonatal hypoglycemia (20 mg/dl) | 25°-50° | 25° | 10° | 60 | 10°-25° | 25° |
| 7 | S | 20 | no | CS (38) | minor | 9/10 | no | 10° | 10°-25° | na | 54 | 50° | 25°-50° |
| 8 | S | 12 | no | V (40) | minor | 9/10 | no | 50° | 50°-75° | na | 29 | 25°-50° | 25°-50° |
| 9 | S | ND | no | V (39) | minor | 9/10 | no | 10°-25° | 10°-25° | 10°-25° | 84 | 50° | 25°-50° |
| 10 | S | ND | no | CS (39) | medium | 9/10 | right bowel atresia | 50° | 10° | 50°-75° | 5 | 50° | 10° |
| 11 | IVF (twin pregnancy) | ND | funicular cyst (16th) | V (37) | minor | 9/10 | uracal anomalies | 5°-10° | na | na | 6 | 50° | 25°-50° |
| 12 | S | 22 | no | CS (38) | minor | 9/10 | Meckel’s diverticulum | 75° | na | na | 22 | 10° | 25° |
| 13 | S | 13 | no | CS (38) | giant | 5/8 | interatrial defect | 50° | na | na | 22 | 25° | <3° |
| 14 | S | 12 | no | CS (38) | giant | 8/9 | no | 50° | na | na | 3 | 25° | 25° |
| 15 | S | 20 | no | V (36) | minor | 9/10 | slight left foot nails hypoplasia | 5°-10° | na | na | 12 | 50° | 50° |
| 16 | S | 19 | no | V (39) | minor | 9/10 | no | 50° | na | na | 8 | 50° | 75° |
| 17 | S | ND | no | V (39) | minor | 10/10 | no | 90° | na | na | 8 | 10°-25° | 10°-25° |
| 18 | S | 12 | no | CS (36) | medium | 4/8 | no | <5° | na | na | 6 | 10°-25° | 25° |
| 19 | S | 16 | no | CS (37) | minor | 8/10 | no | 3–10° | 10° | 10° | 3 | 10°-50° | 10°-50° |
| 20 | S | 20 | multiple cysts of umbilical cord | CS (33) | minor | 8/9 | multiple cysts of umbilical cord | 10–25° | na | na | 15 | 50° | <3° |
| 21 | S | ND | no | CS (39) | minor | 9/10 | no | 10° | 10–25° | 25° | 6 | 50°-75° | 50°-75° |
For each of the 21 cases involved in the study, table summarizes the type of conception, the time of the US omphalocele diagnosis, the presence of other US features, time and type of the delivery, classification of the omphalocele, the APGAR score at birth, the presence of other clinical findings, the weight, the length and the occipital frontal circumference at birth expressed as percentile, and the follow up data
CS Caesarian Section, ICSI Intra-Cytoplasmic Sperm Injection, IVF In Vitro Fertilization, S Spontaneous, V Vaginal, Wog weeks of gestation, OFC Occipital Frontal Circumference
ND Not Detected, NP Not Performed
The omphaloceles were classified as minor (diameter < 5 cm), medium (diameter = 5 cm) or “giant” (diameter > 5 \cm), according to literature [26]. Of the 15 cases with minor omphalocele, seven were diagnosed at birth. Only two fetuses (C11 and C20) were detected with both omphalocele and umbilical cord abnormalities (funicular cyst and multiple cysts) (Table 1).
Conception was spontaneous in 19 cases. Two cases, C2 and C6, were conceived by ICSI (intracytoplasmic sperm injection), with egg donation in C2.
Twenty of 21 pregnancies were singleton. One, C20, was a monochorionic diamniotic twin gestation in which only one twin had omphalocele and the other twin died at 32 weeks of gestation (Intrauterine Fetal Death) without exhibiting omphalocele or other evident malformations; the cause of death was unknown.
Consanguinity was reported in case 16 (the parents were first cousins).
Since omphalocele can be isolated at birth but additional signs may develop during the postnatal period, we followed up all babies for 3–84 months (Table 1). An accurate follow-up is very important and helps the correct diagnosis. Three newborns presented at birth with additional signs, but these spontaneously resolved or were considered aspecific features at the follow-up. In details, C13 had a small interatrial defect that closed spontaneously during the first year of life. Given the high prevalence of non-syndromic atrial defects (4.1:1000 live births), we considered this as a chance association, rather than an indication of a syndromic presentation [27]. C2 presented with slight lingual protrusion without true macroglossia, and helix indentation. Follow-up at 48 months did not reveal any additional signs, and the lingual protrusion spontaneously resolved. C6 had neonatal hypoglycemia, mild neonatal hypotonia, and a mild delay in the acquisition of motor skills (first steps at 19 months); at 60 months, neurological development was normal. Importantly, none of patients showed additional clinical signs associated with BWS. Maternal and paternal age at the conception ranged from 26 to 41 and from 28 to 48 years old, respectively. Other known environmental risk factors [10] were also considered and reported in the Additional file 1.
In study design/implementation STROBE guidelines were followed.
DNA extraction
Genomic DNA was extracted from amniotic fluid (case 15) or peripheral blood lymphocytes (PBL; all other cases and controls), using the QIAamp DNA Mini Kit (Qiagen, Redwood City, CA, USA) according to the manufacturer’s protocol. We also analyzed DNA from PBL of 50 healthy controls, to investigate Variants of Unknown Significance (VUS) in CDKN1C, and from relatives of cases 16, 19, and 20, to investigate the inheritance pattern of genetic variants found in the probands. DNA from the deceased twin of case 20 was extracted from formalin-fixed paraffin-embedded (FFPE) material obtained at autopsy, using a FFPE Nucleic Acid Extraction and Purification Kit (Covaris, Woburn, MA, USA), following the manufacturer’s instructions. DNA was quantified using a NanoDrop ND1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and stored at −20 °C until use.
DNA methylation analysis
We investigated the methylation level of H19/IGF2:IG-DMR and KCNQ1OT1:TSS-DMR (four CpG sites each) at the imprinted locus on chromosome 11p15.5, according to previous reports [28, 29]. Sodium bisulfite conversion of DNA samples (500 ng) was performed using the EZ DNA Methylation-Gold Kit (Zymo Research Corporation, Orange, CA, USA). PCRs were carried out using 40 ng of bisulfite-converted DNA and 200 nM forward and reverse primers, one of which was biotinylated [29]. Quantitative DNA methylation analyses were performed using a Pyro Mark ID instrument (Biotage AB, Uppsala, Sweden) in the PSQ HS 96 System (Biotage), with a PyroGold SQA reagent kit (Biotage). Hypermethylated, hypomethylated, and normal DNA samples were included in each experiment as controls. Raw data were analyzed using Q-CpG software v1.0.9 (Biotage), which calculates the ratio of converted to unconverted cytosines at each CpG, to determine the percentage of methylation. Results are presented as mean values from the analyzed CpG sites ± standard deviation (SD). Reported methylation levels represent the mean of at least two independent experiments. The statistical significance of differences in methylation among samples was determined using the Student’s t-test. We considered as “normal” H19/IGF2:IG-DMR and KCNQ1OT1: TSS-DMR methylation ranges previously reported by our group [28, 29].
CDKN1C and KCNQ1OT1:TSS-DMR sequencing
For CDKN1C mutation analysis, primers were designed to amplify the two coding exons, the intron between them, and the 5′ and 3′ UTRs. PCR conditions and primers sequences have been previously described [30].
Sequencing of KCNQ1OT1:TSS-DMR in case 19 was performed with primers (Forward, 5′-CACGCTGTCCATAAGGTGCA-3′; Reverse, 5′-TTCTCCCAGACT Control Region 2, between the genomic positions chr11:2,699,231–2,699,440. Fragments were amplified at an annealing temperature of 55 °C, using 100 ng of template genomic DNA in 1× One Taq GC reaction buffer, 0.2 μM each primer, 0.2 mM each dNTP, and 1.25 U of One Taq DNA Polymerase (New England BioLabs, Ipswich, MA, USA). Taq High GC Enhancer One was added to the PCR to a final concentration of 20%. PCR products were analyzed by 2% agarose gel electrophoresis and purified using an UltraClean PCR Clean-Up Kit (MO BIO laboratories, Carlsbad, CA, USA), according to the manufacturer’s instructions.
Cycle sequencing reactions of both CDKN1C and KCNQ1OT1:TSS-DMR were carried out using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s protocol and analyzed on a 3130xl Genetic Analyzer automated 16 capillary sequencer (Applied Biosciences, Foster City, CA, USA), after purification with a ZR DNA Sequencing Clean-up Kit (Zymo Research, Irvine, CA, USA). Primers used for sequencing reactions were the same as those used for PCR. Sequences were compared to reference genomic sequence using DNA Dynamo software (Blue Tractor Software), and identified sequence variants were named according to Human Genome Variation Society recommendations (http://www.hgvs.org/mutnomen/). Nucleotide changes were numbered according to Ensembl reference sequence ENST00000414822 for CDKN1C and NCBI reference sequence NR_002728.3 for KCNQ1OT1:TSS-DMR. Functional in silico prediction of CDKN1C VUS was carried out using Mutation taster (www.mutationtaster.org) and PROVEAN (Protein Variation Effect Analyzer) databases.
Array CGH
Array CGH was performed using the Agilent Technologies Platform (Santa Clara, CA, USA) with a SurePrint G3 Human CGH Microarray harboring 180,000 oligonucleotide probes.
Labeling, purification, and hybridization of DNA samples were carried out according to the manufacturer’s protocol (Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis, version 7.3). Slides were scanned using a DNA Microarray Scanner (Agilent Technologies), and TIFF images were obtained from Agilent Scan Control software. Raw data were generated using Agilent Feature extraction and analyzed by Agilent Cytogenomics 3.0. Copy number variation analysis was performed using the ADAM2 algorithm. The aberration filter was set to detect a minimum number of three consecutive probes/region, and the minimum absolute average Log Ratio (MAALR) was ±0.25.
Results
Analysis of methylation of chromosome 11p15.5
In all samples, the methylation percentage at H19/IGF2:IG-DMR was within the normal range (41–51%; Table 2). At KCNQ1OT1:TSS-DMR, we found that 19 of 21 cases exhibited normal methylation (range: 39–50%), whereas case 2 had slight hypomethylation (38%) and case 19 (Fig. 2a) displayed a non-homogeneous methylation pattern at the fourth CpG site of the analyzed region (Fig. 2b). In the latter case we hypothesized the presence of a nucleotide variant generating an arrest in the pyrosequencing reaction of the altered strand. To explore this possibility, we performed KCNQ1OT1:TSS-DMR sequencing and found a heterozygous non-coding variant G > A at nucleotide 687 (NR_002728.3) (Fig. 2c). This nucleotide change affects the guanine of the fourth CpG site investigated by pyrosequencing, abolishing it and altering the global KCNQ1OT1:TSS-DMR methylation pattern. On the basis of the pyrosequencing methylation pattern, we hypothesized that the modified allele was of maternal origin. The pattern of inheritance was indeed confirmed in parental DNA samples by Sanger sequencing (Fig. 2a, c). The variant was also found in the healthy maternal aunt and not in her healthy daughter (Fig. 2a); thus the clinical significance of this alteration remains unknown. In addition, the same nucleotide change was identified by the 1000 Genomes project, and is described in dbSNP (ID rs547284149) with a minor allele frequency of 0.05%. Using a cut-off value of 1%, we can, therefore, define this nucleotide change as a rare variant.
Table 2.
Methylation and sequencing analysis of 11p15.5 locus. Methylation percentage at the two 11p15.5 Differentially Methylated Regions, CDKN1C variants and their allelic status are reported. As already mentioned in Material and Method section, the methylation value represents the mean of the methylation percentage at each of the four analyzed CpG sites. In addition, reported methylation levels are the mean of at least two independent experiments
| Case Nr | Methylation analysis | CDKN1C sequencing | ||
|---|---|---|---|---|
| H19/IGF2: IG-DMR (%) | KCNQ1OT1: TSS-DMR (%) | Genomic Variants | Alleles | |
| 1 | 49.3 | 48.0 | c.555 T>C (p.A185A) | heterozygous |
| 2 | 48.5 | 38.6 a | c.555 T>C (p.A185A); c.600 A > G (p.P200P) | heterozygous |
| 3 | 51.0 | 47.0 | c.511_522delGCTCCGGTCGC (p.A171_A174del) | homozygous |
| 4 | 48.1 | 46.3 | wildtype | |
| 5 | 42.0 | 47.0 | c.555 T>C (p.A185A) | heterozygous |
| 6 | 47.5 | 47.0 | c.511_522delGCTCCGGTCGC (p.A171_A174del) | heterozygous |
| 7 | 47.0 | 49.4 | wildtype | |
| 8 | 47.0 | 45.3 | wildtype | |
| 9 | 49.6 | 48.0 | wildtype | |
| 10 | 49.3 | 47.1 | wildtype | |
| 11 | 49.1 | 47.9 | c.511_522delGCTCCGGTCGC (p.A171_A174del) | heterozygous |
| 12 | 44.3 | 45.3 | wildtype | |
| 13 | 50.0 | 50.0 | wildtype | |
| 14 | 44.0 | 45.0 | c.511_522delGCTCCGGTCGC (p.A171_A174del) | homozygous |
| 15 | 50.0 | 50.0 | c.-85 G>A, c.a5 + 24_a5 + 25insG | heterozygous |
| 16 | 48.0 | 48.0 | c.555 T>C (p.A185A); c.624_629delGGCCCC (p.A209_P210del) | homozygous |
| 17 | 49.0 | 47.0 | c.555 T>C (p.A185A) | homozygous |
| 18 | 45.0 | 49.0 | c.511_522delGCTCCGGTCGC (p.A171_A174del) | heterozygous |
| 19 | 41.0 | dysomogeneous pattern | c.a5 + 24_a5 + 25insG | heterozygous |
| 20 | 47.0 | 47.0 | c.555 T>C (p.A185A), c.624_629delGGCCCC (p.A209_P210del) | heterozygous |
| 21 | 48.0 | 49.0 | c.511_522delGCTCCGGTCGC (p.A171_A174del) | heterozygous |
aslightly below the normal range
Fig. 2.
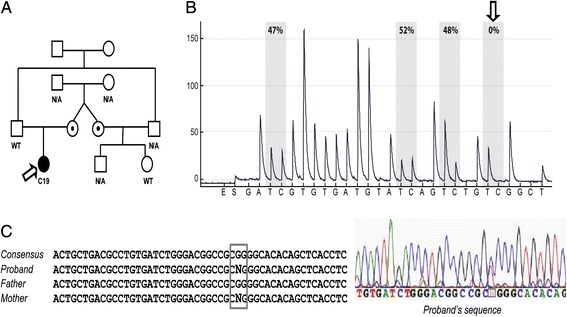
CDKN1C sequence variation in C19 case. In a, the pedigree of C19 is depicted and the proband is indicated by an arrow. As shown, the CDKN1C sequence variation (rs547284149) was inherited from the mother, was also present in the C19 maternal aunt but absent in her daughter who did not show omphalocele. As displayed in. b, this sequence variation determined a non-homogeneous methylation pattern, with a decrease in methylation at the fourth CpG site analyzed by pyrosequencing. The sequence variation and its maternal origin were confirmed by sanger sequencing of KCNQ1OT1:TSS-DMR: we found a heterozygous non-coding variant G > A at nucleotide 687 (NR_002728.3) (c)
CDKN1C mutation analysis
Among the 21 investigated subjects, seven did not show any sequence alteration in CDKN1C, while 14 presented with polymorphic variants. In two of these latter cases (cases 16 and 20), an additional variant, c.624-629delGGCCCC (p.A209_P210del), was detected (Fig. 1b). This variant is located in exon 1 of CDKN1C, in the region encoding the PAPA domain of the protein (Table 2). Patient 16 was homozygous for the deletion (the parents were first degree cousins). Case 20 inherited the deletion from the healthy mother and the same variant was also present in the twin who died in utero and did not showed omphalocele. The deletion is reported in the dbSNP database (rs760463569) without frequency information. We sequenced 100 CDKN1C alleles from 50 PBL samples from healthy individuals, and the variant was not identified (data not shown). To assess the consequence of the p.A209_P210del on p57 protein function, we took advantage of the in silico predictor sites MutationTaster and Provean, and we investigated the evolutionary conservation of the entire PAPA domain by a Pairwise Sequence Alignment. Mutation Taster (www.mutationtaster.org) analysis predicted that the variant should be classified as “polymorphic” with a high probability (0.99); Provean (www.provean.jvci.org) defined the effect of the deletion as “benign” with a score of 0.489. Moreover, pairwise alignment between the nucleotides encoding the human PAPA domain and the corresponding sequence of mouse CDKN1C cDNA revealed a percentage of identity lower than 50% (alignment score: 315).
Array CGH
No evidences of copy number variants with pathogenic significance were obtained (Additional file 2 A and B). The 1p31.3 duplication identified by Radhakrishna et al. [18] was not present in our cases.
Discussion
We report the analyses of the 11p15.5 locus and the array CGH results from 21 cases with isolated omphalocele diagnosed during pregnancy or at birth. In two patients additional mild signs were present, however other specific syndromes were not suspected. Among the 19 cases with isolated omphalocele confirmed by follow-up, we observed a VUS (rs760463569) in CDKN1C in two cases (cases 16 and 20) and a rare variant of KCNQ1OT1:TSS-DMR in one case (case 19). CDKN1C sequencing analysis led to the identification of the same deletion c.624-629delGGCCCC in two unrelated cases, mapping inside the highly polymorphic hexanucleotide tandem-repeats of exon 1, encoding the PAPA domain of the protein. This variant is annotated in the dbSNP database (rs760463569), however its allele frequency, its clinical significance, and the segregation in the dbSNP population are not reported. Moreover, the deletion was not observed in 100 CDKN1C alleles derived from the 50 genotyped healthy controls, therefore it can be classified as a rare variant with unknown clinical significance (VUS). Further studies would be necessary to clarify the functional impact of this variant on the pathogenesis of the omphalocele, although we know that the PAPA domain represents a hotspot for CDKN1C ins/del and an extremely unstable genomic region due to the presence of repeated stretches of hexanucleotides. Moreover, the same hexanucleotide lost in the c.624-629del has been found as an insertion in healthy controls. Truncating mutations mapping at the nucleotides encoding the PAPA motif have already been described [31]. In these cases, the huge effect on gene function is likely due to the introduction of a premature STOP codon inside CDKN1C gene sequence.
So far, in frame ins/del of tandem repeated sequences in the polymorphic region of PAPA domain have never been associated with altered phenotypes. Thus, at the moment we cannot draw a conclusion about the role of the c.624-629del, even though the in silico predictions and the arguments above suggest that it represent a neutral polymorphism.
We also found a rare heterozygous maternally inherited non-coding variant G > A at the KCNQ1OT1:TSS-DMR locus. Interestingly, this variant alters the methylation pattern of this DMR, resulting in its partial demethylation. This nucleotide alteration was also present in a healthy maternal aunt, but not in her healthy daughter, a pattern of transmission consistent with a possible influence on the phenotype.
As mentioned above, in BWS the presence of omphalocele is mainly related to KCNQ1OT1:TSS-DMR hypomethylation or loss-of-function mutations in the maternally derived allele of CDKN1C. Thus, the presence of a VUS in 2 of 19 individuals (10.5%) and of a rare variant in one case with the same rare phenotype might not be due to a casual association, even though data supporting their causative role are not currently available. In both cases, the variant allele was maternally inherited, according to the segregation pattern associated with the pathological phenotype, but the familial segregation of the variants could not be further investigated.
On the other hand, the presence of the G>A change in the stillborn twin without omphalocele may indicate that it is not clinically significant or that its penetrance is incomplete.
Conclusions
We can surmise that alterations of methylation levels at H19/IGF2:IG-DMR and KCNQ1OT1:TSS-DMR are not primarily associated to omphalocele, in our cohort; conversely, sequence variants at KCNQ1OT1:TSS-DMR and CDKN1C could represent susceptibility factors for the onset of isolated omphalocele. This hypothesis needs to be validated in a wider number of patients with the same clinical manifestation.
Additional files
Clinical data of pregnancies and exposition to known environmental risk factors. (XLS 23 kb)
A and B: Array CGH results indicating copy number variants and their pathogenic significance. (ZIP 30 kb)
Acknowledgements
The authors thank the patients and their parents for their participation.
Funding
This study was supported by Ministero della Salute Regione Lombardia (Ricerca Finalizzata 2011–2012, RF-2011-02347106) and by “5X1000” 2013 from Fondazione IRCCS Ca′ Granda, Ospedale Maggiore Policlinico, Milano to Monica Miozzo.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its additional files.
Author’s contributions
MFB, MC, LPa, LPe, SS, ST, MM: conception and design of the study and writing of the manuscript. LPa, LF, MB, SG: laboratory experiments, data interpretation, and writing of the manuscript. MFB, FL: genetic counseling. MFB, MC, FL, LCa, EL, LCo, FM: patients evaluation. All authors read and approved the manuscript.
Abbreviations
- ALG6
Alpha-1,3-Glucosyltransferase
- BWS
Beckwith-Wiedemann Syndrome
- CDKN1C
Cyclin Dependent Kinase Inhibitor 1C
- DLEU2L
Deleted In Lymphocytic Leukemia 2-Like
- DMR
Differentially Methylated Region
- FFPE
Formalin-Fixed Paraffin-Embedded
- FOXD3
Forkhead Box D3
- H19
Imprinted Maternally Expressed Transcript (Non-Protein Coding)
- IG
InterGenic
- IGF2
Insulin Like Growth Factor 2
- IRCCS
Istituto di Ricerca e Cura a Carattere Scientifico
- ITGB3BP
Integrin Subunit Beta 3 Binding Protein
- KCNQ1
Potassium Voltage-Gated Channel Subfamily Q Member 1
- KCNQ1OT1
KCNQ1 Opposite Strand/Antisense Transcript 1
- KIAA1799
EF-Hand Calcium Binding Domain 7
- MAALR
Minimum Absolute Average Log Ratio
- MAPK
Mitogen-Activated Protein Kinase
- OEIS
Omphalocele, Exstrophy, Imperforate anus, Spinal defects
- p21Cip1
cyclin-dependent kinase inhibitor 1A
- p27Kip1
Cyclin-dependent kinase inhibitor 1B
- PGM1
Phosphoglucomutase 1
- PHLDA
Pleckstrin Homology Like Domain Family A Member 2
- ROR1
Receptor Tyrosine Kinase Like Orphan Receptor 1
- SD
Standard Deviation
- SLC22A18
Solute Carrier Family 22 Member 18
- TSS
Transcription Start Site
- UPD
Uniparental disomy
- US
Ultrasound Screening
- VUS
Variants of Unknown Significance
- WOG
Weeks Of Gestation
Ethics approval and consent to participate
The Ethic Committee of Fondazione IRCCS Ca′ Granda-Ospedale Maggiore Policlinico of Milan approved on 15th July 2015 the project “EPI_GEN_DIAGNOSIS V. 1.0” (approval identification number: 526), including the experiments of this paper. After the ethics approval, the patients or their parents who agreed to a potential research program at the time of diagnosis were contacted again and were asked for an appropriate written informed consent to the current study. Consent was obtained from all of them. The study conformed to the principles of the Helsinki Declaration.
Consent for publication
The patients or their parents approved the use of their genetic data for scientific publications only by saving the condition of anonymity.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12881-017-0470-z) contains supplementary material, which is available to authorized users.
Contributor Information
Maria Francesca Bedeschi, Phone: +39 02 5503 4244, Email: mariafrancesca.bedeschi@policlinico.mi.it.
Mariarosaria Calvello, Email: Mariarosaria.Calvello@ieo.it.
Leda Paganini, Email: leda.paganini@unimi.it.
Lidia Pezzani, Email: lidiapezzani@yahoo.it.
Marco Baccarin, Email: m.baccarin@unicampus.it.
Laura Fontana, Email: laura.fontana@unimi.it.
Silvia M. Sirchia, Email: silvia.sirchia@unimi.it
Silvana Guerneri, Email: s.guerneri@policlinico.mi.it.
Lorena Canazza, Email: locanaoli@yahoo.it.
Ernesto Leva, Email: ernesto.leva@policlinico.mi.it.
Lorenzo Colombo, Email: lorenzo.colombo@mangiagalli.it.
Faustina Lalatta, Email: f.lalatta@policlinico.mi.it.
Fabio Mosca, Email: fabio.mosca@unimi.it.
Silvia Tabano, Email: silvia.tabano@unimi.it.
Monica Miozzo, Email: monica.miozzo@unimi.it.
References
- 1.Sadler TW. The embryologic origin of ventral body wall defects. Semin Pediatr Surg. 2010;19(3):209–214. doi: 10.1053/j.sempedsurg.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Cohen-Overbeek TE, Tong WH, Hatzmann TR, Wilms JF, Govaerts LC, Galjaard RJ, et al. Omphalocele: comparison of outcome following prenatal or postnatal diagnosis. Ultrasound Obstet Gynecol. 2010;36(6):687–692. doi: 10.1002/uog.7698. [DOI] [PubMed] [Google Scholar]
- 3.Hoyme HE, Higginbottom MC, Jones KL. The vascular pathogenesis of gastroschisis: intrauterine interruption of the omphalomesenteric artery. J Pediatr. 1981;98(2):228–231. doi: 10.1016/S0022-3476(81)80640-3. [DOI] [PubMed] [Google Scholar]
- 4.Brewer S, Williams T. Loss of AP-2a impacts multiple aspects of ventral body wall development and closure. Dev Biol. 2004;267(2):399–417. doi: 10.1016/j.ydbio.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 5.Brewer S, Williams T. Finally, a sense of closure? Animal models of human ventral body wall defects. BioEssays. 2004;26(12):1307–1321. doi: 10.1002/bies.20137. [DOI] [PubMed] [Google Scholar]
- 6.Prefumo F, Izzi C. Fetal abdominal wall defects. Best Pract Res Clin Obstet Gynaecol. 2014;28(3):391–402. doi: 10.1016/j.bpobgyn.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 7.Chen CP. Syndromes and disorders associated with omphalocele (I): Beckwith-Wiedemann syndrome. Taiwan J Obstet Gynecol. 2007;46(2):96–102. doi: 10.1016/S1028-4559(07)60002-3. [DOI] [PubMed] [Google Scholar]
- 8.Chen CP. Syndromes and disorders associated with omphalocele (II): OEIS complex and Pentalogy of Cantrell. Taiwan J Obstet Gynecol. 2007;46(2):103–110. doi: 10.1016/S1028-4559(07)60003-5. [DOI] [PubMed] [Google Scholar]
- 9.Chen CP. Syndromes and disorders associated with omphalocele (III): single gene disorders, neural tube defects, diaphragmatic defects and others. Taiwan J Obstet Gynecol. 2007;46(2):111–120. doi: 10.1016/S1028-4559(07)60004-7. [DOI] [PubMed] [Google Scholar]
- 10.Frolov P, Alali J, Klein MD. Clinical risk factors for gastroschisis and omphalocele in humans: a review of the literature. Pediatr Surg Int. 2010;26(12):1135–1148. doi: 10.1007/s00383-010-2701-7. [DOI] [PubMed] [Google Scholar]
- 11.Bliek J, Maas S, Alders M, Merks JH, Mannens M. Epigenotype, phenotype and tumors in patients with isolated hemihyperplasia. J Pediatr. 2008;153(1):95–100. doi: 10.1016/j.jpeds.2007.12.022. [DOI] [PubMed] [Google Scholar]
- 12.Martin RA, Grange DK, Zehnbauer B, Debaun MR. LIT1 and H19 methylation defects in isolated hemihyperplasia. Am J Med Genet. 2005;134:129–131. doi: 10.1002/ajmg.a.30578. [DOI] [PubMed] [Google Scholar]
- 13.Cerrato F, De Crescenzo A, Riccio A. Looking for CDKN1C enhancers. Eur J Hum Genet. 2014;22(4):442–443. doi: 10.1038/ejhg.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milani D, Pezzani L, Tabano S, Miozzo M. Beckwith-Wiedemann and IMAGe syndromes: two very different diseases caused by mutations on the same gene. Appl Clin Genet. 2014;7:169–175. doi: 10.2147/TACG.S35474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eggermann T, Binder G, Brioude F, Maher ER, Lapunzina P, Cubellis MV, et al. CDKN1C mutations: two sides of the same coin. Trend Mol Med. 2014;20(11):614–622. doi: 10.1016/j.molmed.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Romanelli V, Belinchón A, Benito-Sanz S, Martínez-Glez V, Gracia-Bouthelier R, Heath KE, et al. CDKN1C (p57(Kip2)) analysis in Beckwith-Wiedemann syndrome (BWS) patients: genotype-phenotype correlations, novel mutations, and polymorphisms. Am J Med Genet. 2010;152A:1390–1397. doi: 10.1002/ajmg.a.33453. [DOI] [PubMed] [Google Scholar]
- 17.Russo S, Calzari L, Mussa A, Mainini E, Cassina M, Di Candia S, et al. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann sindromes. Clinical. Epigenetics. 2016;8:23. doi: 10.1186/s13148-016-0183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mussa A, Russo S, Larizza L, Riccio A, Ferrero GB, et al. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2016;24:183–190. doi: 10.1038/ejhg.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li M, Squire J, Shuman C, Fei YL. Imprinting status of 11p15 genes in Beckwith Wiedemann syndrome patients with CDKN1C mutations. Genomics. 2001;74:3. doi: 10.1006/geno.2001.6549. [DOI] [PubMed] [Google Scholar]
- 20.Cooper WN, Luharia A, Evans GA, Raza H, Haire AC, Grundy R, et al. Molecular subtypes and phenotypic expression of Beckwith Wiedemann syndrome syndrome. Eur J Hum Genet. 2005;13:10. doi: 10.1038/sj.ejhg.5201463. [DOI] [PubMed] [Google Scholar]
- 21.Grati FR, Turolla L, D'Ajello P, Ruggeri A, Miozzo M, Bracalente G, et al. Chromosome 11 segmental paternal isodisomy in amniocytes from two fetuses with omphalocoele: new highlights on phenotype-genotype correlations in Beckwith-Wiedemann syndrome. J Med Genet. 2007;44(4):257–263. doi: 10.1136/jmg.2006.046854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radhakrishna U, Nath SK, McElreavey K, Ratnamala U, Sun C, Maiti AK, et al. Genome-wide linkage and copy number variation analysis reveals 710 kb duplication on chromosome 1p31.3 responsible for autosomal dominant omphalocele. J Med Genet. 2012;49(4):270–276. doi: 10.1136/jmedgenet-2012-100826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanagawa SL, Begleiter ML, Ostlie DJ, Holcomb G, Drake W, Butler MG. Omphalocele in three generations with autosomal dominant transmission. J Med Genet. 2002;39:184–185. doi: 10.1136/jmg.39.3.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feldkamp ML, Srisukhumbowornchai S, Romitti PA, Olney RS, Richardson SD, Botto LD. Self-reported maternal cigarette smoke exposure during the periconceptional period and the risk for omphalocoele. Paediatr Perinat Epidemiol. 2014;28(1):67–73. doi: 10.1111/ppe.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pryde PG, Greb A, Isada NB, Johnson MB, Klein M, Evans MI. Familial omphalocele: considerations in genetic counseling. Am J Med Genet. 1992;44:624–627. doi: 10.1002/ajmg.1320440519. [DOI] [PubMed] [Google Scholar]
- 26.Van Eijck FC, Hoogeveen YL, van Weel C, Rieu PN, Wijnen RM. Minor and giant omphalocele: long-term outcomes and quality of life. J Pediatr Surg. 2009;44(7):1355–1359. doi: 10.1016/j.jpedsurg.2008.11.034. [DOI] [PubMed] [Google Scholar]
- 27.Garne E. Atrial and ventricular septal defects - epidemiology and spontaneous closure. J Matern Fetal Neonatal Med. 2006;19(5):271–276. doi: 10.1080/14767050500433817. [DOI] [PubMed] [Google Scholar]
- 28.Tabano S, Colapietro P, Cetin I, Grati FR, Zanutto S, Mandò C, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics. 2010;5(4):313–324. doi: 10.4161/epi.5.4.11637. [DOI] [PubMed] [Google Scholar]
- 29.Calvello M, Tabano S, Colapietro P, Maitz S, Pansa A, Augello C, et al. Quantitative DNA methylation analysis improves epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. Epigenetics. 2013;8(10):1053–1060. doi: 10.4161/epi.25812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paganini L, Carlessi N, Fontana L, Silipigni R, Motta S, Fiori S, et al. Beckwith-Wiedemann syndrome prenatal diagnosis by methylation analysis in chorionic villi. Epigenetics. 2015;10(7):643–649. doi: 10.1080/15592294.2015.1057383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. 2012;44(7):788–792. doi: 10.1038/ng.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Clinical data of pregnancies and exposition to known environmental risk factors. (XLS 23 kb)
A and B: Array CGH results indicating copy number variants and their pathogenic significance. (ZIP 30 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its additional files.