

Abstract
Background
Most studies on Enterocytozoon bieneusi are conducted based on the internal transcribed spacer (ITS) region of the rRNA gene, whereas some have examined E. bieneusi population structures. Currently, the population genetics of this pathogen in giant panda remains unknown. The objective of this study was to determine the E. bieneusi population in captive giant pandas in China.
Results
We examined 69 E. bieneusi-positive specimens from captive giant pandas in China using five loci (ITS, MS1, MS3, MS4 and MS7) to infer E. bieneusi population genetics. For multilocus genotype (MLG) analysis of E. bieneusi-positive isolates, the MS1, MS3, MS4, and MS7 microsatellite and minisatellite loci were amplified and sequenced in 48, 45, 50 and 47 specimens, respectively, generating ten, eight, nine and five types. We successfully amplified 36 specimens and sequenced all five loci, forming 24 MLGs. Multilocus sequence analysis revealed a strong and significant linkage disequilibrium (LD), indicating a clonal population. This result was further supported by measurements of pairwise intergenic LD and a standardized index of association (I S A) from allelic profile data. The analysis in STRUCTURE suggested three subpopulations in E. bieneusi, further confirmed using right’s fixation index (F ST). Subpopulations 1 and 2 exhibited an epidemic structure, whereas subpopulation 3 had a clonal structure.
Conclusions
Our results describe E. bieneusi population genetics in giant pandas for the first time, improving the current understanding E. bieneusi epidemiology in the studied region. These data also benefit future studies exploring potential transmission risks from pandas to other animals, including humans.
Keywords: E. bieneusi, Multilocus sequence typing, Gene diversity, Population genetics, Population structure, Linkage disequilibrium
Background
Diarrheal disease is the second leading cause of morbidity and mortality in children under 5 years old (including infants) in low-income countries [1, 2]. The illness can often be traced to the unicellular fungus microsporidian, recognized as a category B biodefense agent on the National Institutes of Health list [3]. Enterocytozoon bieneusi is among the most common microsporidia; in immunocompetent individuals, the anthropozoonotic pathogen may be asymptomatic, but it has been implicated in self-limiting diarrhoea and malnutrition. In immunosuppressed individuals, however, persistent diarrhoea, abdominal pain, and weight loss are common symptoms [3–5]. Enterocytozoon bieneusi can also infect various invertebrates and vertebrates, including domestic animals, through a wide variety of transmission routes: water-borne, food-borne, anthroponotic and zoonotic [6, 7]. Currently, effective commercial treatments for E. bieneusi infection do not exist, although some reports have suggested that the fungicide fumagillin is effective. However, this drug is toxic to mammals [6].
Enterocytozoon bieneusi cannot be identified based on morphology because its spores are too small and do not have characteristic staining properties, making them indistinguishable. Moreover, in vitro cultivation of the fungus has been unsuccessful thus far [8, 9]. Thus, molecular screening is currently the only option. ITS gene analysis is widely used in molecular epidemiological studies of genetically diverse E. bieneusi, identifying over 200 genotypes [10, 11]. ITS analysis is adequate for genotyping and epidemiological studies of E. bieneusi in the absence of recombination. However, ITS analysis has major limitations if genetic recombination occurs at any reproduction stage [12]. As a result, multilocus genotyping (MLST) has been developed, targeting three microsatellites (MS1, MS3 and MS7) and a minisatellite (MS4) to provide high-resolution E. bieneusi genotyping based on length polymorphisms and single nucleotide polymorphisms (SNP) [13]. Together with ITS analysis, multilocus genotyping has been gradually applied to E. bieneusi genetic characterization. The results contribute to the establishment of effective control measures by enabling elucidation of transmission routes, characterization of the public health consequences of animal-derived E. bieneusi isolates, and identification of the sources of human E. bieneusi infections.
For E. bieneusi, MLST has been applied to humans, non-human primates, swine, Asiatic black bears, red kangaroos, dairy and native beef, foxes, and red-bellied tree squirrels [14–21]. However, no population genetic analysis is available on E. bieneusi in giant pandas. Therefore, this study aimed to amplify the 12 ITS sequences in E. bieneusi-positive isolates from captive giant pandas of China using micro/minisatellite markers and characterize them using MLST. The resultant data on population genetic structure will help to prevent or reduce microsporidiosis occurrence in giant pandas through the development of efficient control strategies. These data can also be used to compare E. bieneusi population genetics across multiple hosts, improving risk assessment of potential human and livestock infection through infected giant pandas.
Methods
Enterocytozoon bieneusi isolates
In total, 69 ITS-positive E. bieneusi isolates from captive giant pandas were previously genotyped in our laboratory. Samples were collected from captive giant pandas at conservation bases and zoological gardens in china. Genotypes SC02 (n = 50), D (n = 3), SC06 (n = 2), CHB1 (n = 2), F (n = 2), EbpC (n = 2), SC01 (n = 2), SC04 (n = 2), SC05 (n = 1), SC07 (n = 1), SC08 (n = 1) and Peru 6 (n = 1) were used for MLST analysis.
MLST PCR and sequencing
Three microsatellites (MS1, MS3 and MS7) and one minisatellite (MS4) were further amplified in all 69 samples to determine their multilocus genotypes (MLGs). Primers and amplification conditions were described previously [13]. Secondary PCR products were visualized by Golden View staining and 1% agarose gel electrophoresis. Amplicons of the expected size (MS1, 676 bp; MS3, 537 bp; MS4, 885 bp; MS7, 471 bp) were bi-directionally sequenced by Invitrogen (Shanghai, China) to ensure accuracy.
Genetic analysis
We analyzed E. bieneusi genetic diversity at each marker (ITS, MS1, MS3, MS4 and MS7) in DnaSP version 5.10.1 (http://www.ub.edu/dnasp/). Detailed methods for calculating genotype frequency, genetic diversity (Hd), and intragenic linkage disequilibrium (LD) followed a previous publication [14]. We then concatenated sequences of E. bieneusi isolates for all five loci separately. Sequence alignment determined MLGs. DnasP and Arlequin version 3.5.2.2 (http://cmpg.unibe.ch/software/arlequin35/) were used to calculate nucleotide diversity (Pi), genotype frequency, Hd, LD, and recombination rates. To determine the nature of mutations in the population, Fu’s neutrality tests (Fs) were conducted in both programs based on segregating sites (DnaSP), as well as on segregating sites and indels (Arlequin). Significant differences were assessed with coalescence simulations in a parameter test. To confirm the occurrence of recombination, rates were estimated using GENECONV, MaxChi, and SiScan in RDP (Recombination Detection Program) version 4.92 (http://web.cbio.uct.ac.za/~darren/rdp.html).
A population genetic analysis of allelic profiles was also performed. In Arlequin, we applied exact tests with Markov chain parameters to allelic profile data to assess pairwise intergenic LD. Standardized index of association (I S A) values were calculated using LIAN 3.7 (http://guanine.evolbio.mpg.de/cgi-bin/lian/lian.cgi.pl) on five-loci haplotypes. I S A equal to zero or with a negative value indicates randomly mating populations and alleles in linkage equilibrium (LE). An I S A value greater than zero indicates a non-panmictic population structure is exhibiting LD. The variance of pairwise differences (VD) was also calculated as another test of LD; when less than L (95% critical value for VD relative to the null hypothesis of panmixia), the population is panmictic and is in LE. When VD > L, the population is non-panmictic and has some LD [22]. Intra-/intergenic LD, I S A, neutrality, and recombination events were combined to assess E. bieneusi population structure.
Substructure analysis
Allelic profile data were analyzed to determine subpopulations of E. bieneusi genotypes, using K-means partitional clustering and the admixture model in the Bayesian analysis tool STRUCTURE version 2.3.4 (http://web.stanford.edu/group/pritchardlab/structure.html) [23]. Twenty simulation runs were conducted for each of K value = 2–8, with 104 and 105 replicates of the Markov chain Monte Carlo simulation discarded as burn-in. Wright’s fixation index (F ST), a measure of population divergence, was calculated in Arlequin; F ST = 0 indicates similar polymorphisms across all markers and F ST = 1 indicates high levels of between-population divergence. We also performed a median-joining analysis implemented in the software Network version 5.0 (http://www.fluxus-engineering.com/sharenet.htm) to estimate the potential for geographic segregation.
Results and discussion
Among the 69 ITS-positive E. bieneusi isolates, the MS1, MS3, MS4 and MS7 loci were successfully amplified in 48, 45, 50 and 47 specimens, respectively, yielding ten, eight, nine, and five types (Table 1). MS1 exhibited the highest sequence polymorphism and MS7 the lowest. A previous study of E. bieneusi population genetics in humans reported the highest resolution for MS1 and the lowest for MS3 and MS4 [14]. Another study conducted in non-human primates also suggested that MS1 had the highest resolution and MS3 the lowest [16]. In contrast, research on E. bieneusi populations in swine revealed that MS3 and MS4 possessed higher resolution than the other tested loci [15]. These inter-study differences may result from variation in hosts and sample size. The Hd and intragenic linkage disequilibrium shown in Table 2 indicate that all five markers were in significant LD. Assessment of intragenic recombination revealed that ITS and MS4 exhibited one and two recombination events (Rms), respectively, and LD was incomplete in these two loci, whereas the remaining three markers (with complete LD) did not have Rms (Table 2).
Table 1.
Samples used in this study and their genotype distribution
| Category | No. of samples used | No. of types at each micro and mini satellite locia | No. MLGs | |||
|---|---|---|---|---|---|---|
| MS1 | MS3 | MS4 | MS7 | |||
| CRBGPB | 25 | 7 (15) | 5 (14) | 4 (15) | 4 (14) | 10 (11) |
| DGPB | 16 | 5 (14) | 2 (12) | 5 (14) | 2 (14) | 7 (11) |
| WGGPB | 7 | 1 (6) | 4 (5) | 2 (6) | 1 (6) | 3 (4) |
| WHGPB | 3 | 1 (1) | 2 (2) | 2 (2) | 0 (0) | 0 (0) |
| YBGPB | 4 | 2 (3) | 3 (3) | 3 (3) | 1 (3) | 3 (3) |
| ZGs | 14 | 4 (9) | 2 (9) | 6 (10) | 1 (10) | 6 (7) |
| Total | 69 | 10 (48) | 8 (45) | 9 (50) | 5 (47) | 24 (36) |
aNumber in the parenthesis indicates the number of samples
Abbreviations: CRBGPB Chengdu Research Base of Giant Panda Breeding, DGPB Dujiangyan Giant Panda Base, WGGPB Wolong Gengda Giant Panda Base, WHGPB Wolong Hetaoping Giant Panda Base, YBGPB Yaan Bifengxia Giant Panda Base, ZGs zoological gardens
Table 2.
Hd, polymorphic sites, intragenic linkage disequilibrium and recombination events in individual genetic markers among 36 E. bieneusi specimens
| Marker | Hd | Polymorphic sites | LD (|D’|) | Rm |
|---|---|---|---|---|
| ITS | 1 | 43 | Y = 0.9994 + 0.0000X | 1 |
| MS1 | 0.53 | 6 | Y = 1.0000 + 0.0000X | 0 |
| MS3 | 0.86 | 7 | Y = 1.0000 + 0.0000X | 0 |
| MS4 | 0.97 | 28 | Y = 0.9866 + 0.0163X | 2 |
| MS7 | 1 | 14 | Y = 1.0000 + 0.0000X | 0 |
Hd gene diversity, LD (|D’|) linkage disequilibrium per site, Rm minimum number of recombination events
All five loci were amplified and sequenced for 36 specimens, forming 24 MLGs with an Hd of 0.77 (Table 1). The 36 contigs had 49 polymorphic sites, with nucleotide diversity Pi = 0.00439 and an average number of nucleotide differences k = 8.379. In this study, the 36 isolates yielded 24 MLGs with a Hd value of 0.994 or 0.768, depending on whether the analysis used polymorphic or segregating sites. The results indicate that E. bieneusi genotypes are relatively abundant in our giant panda population. However, nucleotide diversity was very low (0.033 based on polymorphic sites, 0.004 based on segregating sites). This pattern of high Hd but low nucleotide diversity suggests the presence of a bottleneck followed by population expansion in E. bieneusi infecting giant pandas.
The observed ZnS, a measure of LD, was 0.254. Fisher’s exact test found 339 significant pairwise correlations out of 1176, with 111 remaining significant after a Bonferroni correction. In contrast, chi-squared tests found 605 significant correlations, with 300 remaining significant after Bonferroni corrections. Coalescence simulations with 1000 replicates showed that average ZnS was 0.120, and its 95% confidence interval ranged from 0.066 to 0.462. The probability for expected ZnS ≤ 0.254 was 0.200 (i.e. the observed ZnS had a 0.200 probability of being higher than the expected ZnS if the former was within the 95% confidence interval). Linkage disequilibrium was strong and significant (|D’|: Y = 0.9652 - 0.0204X), but a negative slope in the |D’| graph (Fig. 1) revealed a decline in an LD with greater nucleotide distance, leading to the possibility of genetic recombination. Accordingly, we also detected a minimum of four Rms, but we note that they could be due to genetic changes. To further confirm the occurrence of recombination, we utilized GENECONV, SiScan, and MaxChi analyses implemented in RDP 4 software. SiScan analysis did not detect any Rm, whereas both GENECONV and MaxChi detected two Rms (all three tests ignored indels).
Fig. 1.
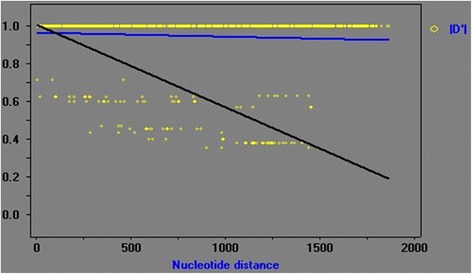
Linkage disequilibrium (LD) in 36 isolates determined by DnaSP software after end-to-end linking of the sequences of 5 loci. Y coordinate represents the LD value, and X coordinate represents the nucleotide distance in kb
The results of Fu’s neutrality test shown in Table 3 indicate that the population experienced molecular selection. Moreover, the observation of Fs = -6.680 (the infinite model of mutation) suggested that a great number of alleles were present that probably resulted from a recent population expansion.
Table 3.
Genetic diversity in 36 E. bieneusi specimens based on the analysis of concatenated multilocus gene sequences
| Test model | Variability of multilocus gene sequences | ||||||
|---|---|---|---|---|---|---|---|
| No. of MLGs | Hd | k | Pi | Theta (k) | Fs (Obs) | P (Fs ≤ Obs) | |
| Polymorphic sites | 662 | 0.994 | 83.173 | 0.0330 | 83.173 | -6.680 | 0.010 |
| Segregating sites | 49 | 0.768 | 8.379 | 0.004 | 8.379 | 1.144 | 0.133 |
Abbreviations: Hd gene diversity, k mean number of pairwise differences, Pi nucleotide diversity (average over loci), theta (k) gene variance based on the mean number of pair-wise differences, Fs Fu’s statistic testing selective neutrality based on genotype frequency, Obs observed value, P (Fs ≤ Obs) probability of obtaining Fs values equal or lower than the observed
We found an I S A of 0.0903 and a VD (0.9725) greater than L (0.7652) (Table 4), indicating a non-panmictic population exhibiting LD. Furthermore, our Monte Carlo simulations revealed a PMC (significance for obtaining this value in 1000 permutations) below the threshold for retaining the null hypothesis of panmixia (Table 4). Thus, all three tests indicated a non-panmictic population is exhibiting LD. To exclude the possibility that the LD was due to clonal expansion of one or more MLGs, we also calculated I S A and VD for MLGs another way, considering isolates with the same MLG as a single individual. The I A S value was 0.0408 and VD value was still higher than L (Table 4). Thus, the population of giant pandas in our study was clonal with strong LD and limited recombination, which suggests that MLGs are relatively stable across time and location. Thus, they should be used for tracking the transmission of microsporidiosis.
Table 4.
Results of linkage disequilibrium analysis based on allelic profile data
| Population | n | H ± SD | I S A | PMC | VD | L | VD > L |
|---|---|---|---|---|---|---|---|
| All | 36 | 0.5151 ± 0.1396 | 0.0903 | 1.10 × 10−02 | 0.9725 | 0.7652 | Y |
| Alla | 23 | 0.6107 ± 0.1358 | 0.0408 | 2.11 × 10−01 | 0.8191 | 0.7297 | Y |
| Population 1 | 14 | 0.2637 ± 0.1539 | 0.0482 | 1.97 × 10−01 | 0.5636 | 0.4925 | Y |
| Population 2 | 10 | 0.2833 ± 0.1641 | -0.0358 | – | 0.4364 | 0.4889 | N |
| Population 3 | 12 | 0.6856 ± 0.0908 | 0.0739 | 1.76 × 10−01 | 0.9326 | 0.7633 | Y |
| Population 1a | 7 | 0.5833 ± 0.1463 | -0.0331 | 7.75 × 10−01 | 0.6443 | 0.7153 | N |
| Population 2a | 6 | 0.3167 ± 0.1873 | -0.0690 | – | 0.3524 | 0.4444 | N |
| Population 3a | 12 | 0.6856 ± 0.0908 | 0.0739 | 1.76 × 10−01 | 0.9326 | 0.7633 | Y |
aConsidering each group of isolates with the same MLG as one individual
Abbreviations: n number of isolates, H mean genetic diversity, SD standard deviation, I S A standardized index of association, P MC significance for obtaining this value in 1000 simulations using the Monte Carlo method, V D variance of pair-wise differences, L 95% critical value for VD, VD > L indicates linkage disequilibrium, Y, V D > L N, VD ≤ L
A STRUCTURE analysis was used to further assess the formation of subpopulations. As shown in Fig. 2, when K = 2 (Fig. 2a) and 3 (Fig. 2b), the subpopulations generated were clear, whereas a staggered pattern was formed when K = 4 (Fig. 2c). α values remained stable at < 0.2 when K = 2 (α = 0.0886), 3 (α = 0.0432) and 4 (α = 0.0424), which demonstrates the presence of subpopulations [14]. The presence of three subpopulations was also confirmed with 20 Monte Carlo simulation runs, which showed a peak value of Delta K at K = 3 (Fig. 3). Furthermore, two subpopulations (K = 2) exhibited F ST = 0.230, suggesting extensive genetic differences. Subpopulations were further confirmed with comparisons of F ST across any two subpopulations (K = 3), which yielded F ST values (0.066, 0.168, and 0.127) consistently over 0.05.
Fig. 2.
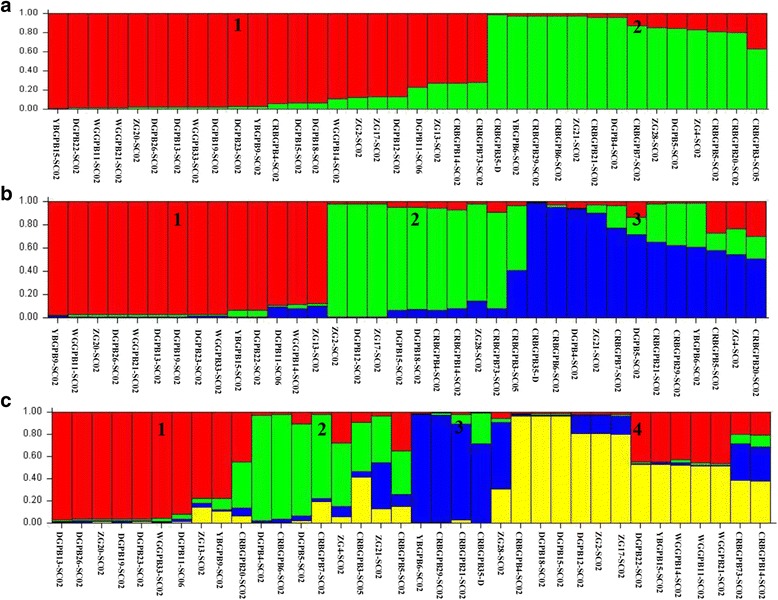
Subpopulation structure of 36 E. bieneusi isolates. Different subpopulation patterns are shown depending on the setting of K values. a K = 2. b K = 3. c K = 4
Fig. 3.
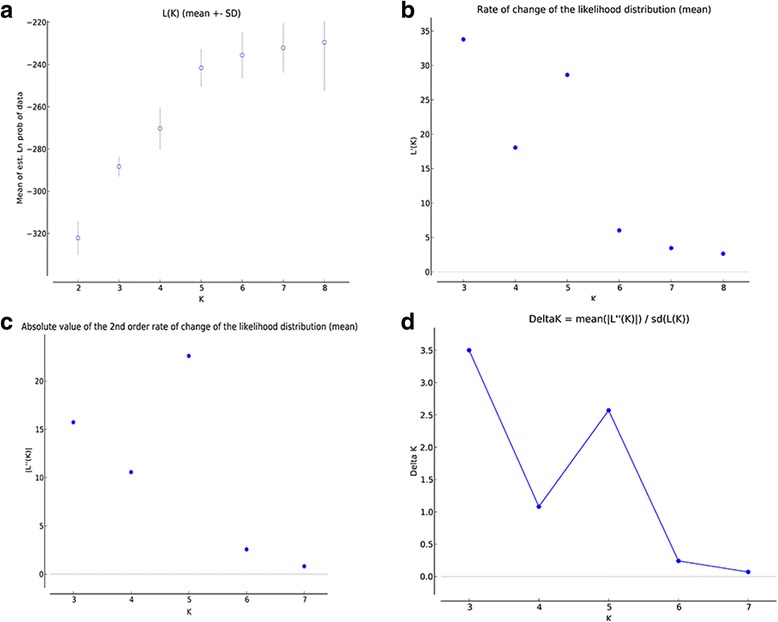
K value calculation for population substructuring after burn-in length of 104 and 105 replicates of 20 Markov chain Monte Carlo simulation runs. a Plot of mean likelihood L (K) and variance per K value from STRUCTURE on a dataset containing 36 specimens for five loci. b-d Plots for detecting the number of K groups that best fit the data, where (d) represents the peak delta K value of 3
The LD analysis of the allelic profiles from subpopulations 1, 2 and 3 revealed positive I S A for subpopulation 1 and 3, along with and VD > L with insignificant PMC. However, subpopulation 2 had negative I S A and VD < L (Table 4). To confirm the existence of epidemic population structure, we then recalculated I S A for MLGs (isolates with the same MLG were treated as one individual) of the three subpopulations. Subpopulations 1 and 2 had negative I S A and VD < L with insignificant PMC, whereas subpopulation 3 had positive I S A and VD > L. Thus, both subpopulations 1 and 2 were in LE and had epidemic population structure, whereas subpopulation 3 remained in LD (despite non-significant PMC), exhibiting a clonal population structure. This outcome was similar to a previous study in AIDS patients, showing one E. bieneusi subpopulation with a clonal structure and another with an epidemic population structure [24]. A clonal population structure of E. bieneusi was also found in non-human primates and swine [15, 16].
All the clusters formed mainly came from Sichuan province in the substructure analysis (Fig. 2b). Thus, no clear geographically segregated subpopulation was observed. While, subpopulation structure analysis showed that subpopulation 1 consisted of isolates only from adults (> 5.5 years); subpopulation 2 had admixture of isolates from adults and yearlings (< 1.5 years), and adults isolates dominated; subpopulation 3 mainly consisted of isolates from juveniles (1.5–5.5 years) which may have contributed to the significance for LD and I S A analyses of each population. In the median-joining network analysis, all the clusters were all mainly from Sichuan province (Fig. 4). We still observed no clear geographical segregation of E. bieneusi isolates from different areas, which is similar to the report of Li et al. in 64 isolates from AIDS patients and captive baboons [24].
Fig. 4.
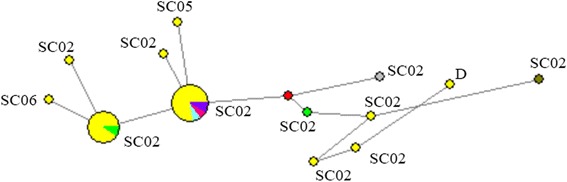
Median-joining analysis of the multilocus sequence contigs from 36 E. bieneusi isolates in this study. The size of the circles is proportional to the frequency of each of the 24 multilocus genotypes obtained based on segregating sites. The yellow, green, pink, purple, blue, grey and brown colours in circles represent the isolates from Sichuan, Zhejiang, Guangdong, Shandong, Fujian, Hunan provinces, and Shanghai city, respectively
Conclusions
We used ITS and micro/minisatellite markers to analyze 69 E. bieneusi isolates from captive giant pandas of China. We found an overall clonal population of E. bieneusi with strong LD and limited recombination. Within the larger population, three subpopulations were present, with two exhibiting epidemic structure (subpopulations 1 and 2), whereas the third exhibited clonal structure (subpopulation 3). Additional large-scale MLST studies on E. bieneusi in other hosts should be conducted to characterize the pathogen’s population genetics better. Such data should prove highly beneficial for characterizing E. bieneusi risk factors, including transmission, host specificity, and exact clinical presentation.
Acknowledgments
We thank Huai-Yi Su for collecting samples.
Funding
This study was supported by Chengdu Giant Panda Breeding Research Foundation (CPF2014-10; CPF2015-4).
Availability of data and materials
The data sets supporting the conclusions of this article are included in the article. Representative sequences are submitted to the GenBank database under the accession numbers: KY950545–KY950576.
Abbreviations
- Fs
Fu’s neutrality tests
- FST
Right’s fixation index
- Hd
Genetic diversity
- ISA
A standardized index of association
- ITS
Internal transcribed spacer
- LD
Linkage disequilibrium
- LE
Linkage equilibrium
- MLG
Multilocus genotype
- MLGs
Multilocus genotypes
- MLST
Multilocus sequence typing
- Pi
Nucleotide diversity
- RDP
Recombination detection program
- SNPs
Single nucleotide polymorphisms
- VD
Variance of pairwise differences; Rms: recombination events
Authors’ contributions
GP, WL and ZZ conceived and designed the research. XH, HL, ZR and CW collected samples. WL, JL, KW and YS performed the experiments. WL, CL and HY analyzed the data. WL wrote the paper. All authors read and approved the final manuscript.
Ethics approval
This study was performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Health, China. Before experiments, the protocol of the current study was reviewed and approved by the Institutional Animal Care and Use Committee of the Sichuan Agricultural University under permit number DYY-S20153801. No animals were harmed during the sampling process. Permission was obtained from the relative institutions of the giant pandas before the collection of faecal specimens.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wei Li, Email: castalli@163.com.
Yuan Song, Email: 601127698@qq.com.
Zhijun Zhong, Email: zhongzhijun488@126.com.
Xiangming Huang, Email: 542291569@qq.com.
Chengdong Wang, Email: 28593401@qq.com.
Caiwu Li, Email: 83330019@qq.com.
Haidi Yang, Email: 4909454@qq.com.
Haifeng Liu, Email: 276760202@qq.com.
Zhihua Ren, Email: 112283458@qq.com.
Jingchao Lan, Email: 327617708@qq.com.
Kongju Wu, Email: 646401864@qq.com.
Guangneng Peng, Email: pgn.sicau@163.com.
References
- 1.Wang T, Fan Y, Koehler AV, Ma G, Li T, Hu M, et al. First survey of Cryptosporidium, Giardia and Enterocytozoon in diarrhoeic children from Wuhan, China. Infect Genet Evol. 2017;51:127–131. doi: 10.1016/j.meegid.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 2.Zhang SX, Zhou YM, Xu W, Tian LG, Chen JX, Chen SH, et al. Impact of co-infections with enteric pathogens on children suffering from acute diarrhea in southwest China. Infect Dis Poverty. 2016;5(1):64. doi: 10.1186/s40249-016-0157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graczyk TK, Majewska AC, Schwab KJ. The role of birds in dissemination of human waterborne enteropathogens. Trends Parasitol. 2008;24(2):55–59. doi: 10.1016/j.pt.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Yu F, Wu Y, Li T, Cao J, Wang J, Hu S, et al. High prevalence of Enterocytozoon bieneusi zoonotic genotype D in captive golden snub-nosed monkey (Rhinopithecus roxellanae) in zoos in China. BMC Vet Res. 2017;13(1):158. doi: 10.1186/s12917-017-1084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li W, Li Y, Song M, Lu Y, Yang J, Tao W, et al. Prevalence and genetic characteristics of Cryptosporidium, Enterocytozoon bieneusi and Giardia duodenalis in cats and dogs in Heilongjiang province, China. Vet Parasitol. 2015;208(3-4):125–134. doi: 10.1016/j.vetpar.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 6.Didier ES. Microsporidiosis: an emerging and opportunistic infection in humans and animals. Acta Trop. 2005;94(1):61–76. doi: 10.1016/j.actatropica.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 7.Thellier M, Breton J. Enterocytozoon bieneusi in human and animals, focus on laboratory identification and molecular epidemiology. Parasite. 2008;15(3):349–358. doi: 10.1051/parasite/2008153349. [DOI] [PubMed] [Google Scholar]
- 8.Dengjel B, Zahler M, Hermanns W, Heinritzi K, Spillmann T, Thomschke A, et al. Zoonotic potential of Enterocytozoon bieneusi. J Clin Microbiol. 2001;39(12):4495. doi: 10.1128/JCM.39.12.4495-4499.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santin M, Fayer R. Microsporidiosis: Enterocytozoon bieneusi in domesticated and wild animals. Res Vet Sci. 2011;90(3):363–371. doi: 10.1016/j.rvsc.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Yue DM, Ma JG, Li FC, Hou JL, Zheng WB, Zhao Q, et al. Occurrence of Enterocytozoon bieneusi in donkeys (Equus asinus) in China: a public health concern. Front Microbiol. 2017;8:565. doi: 10.3389/fmicb.2017.00565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng L, Li W, Zhong Z, Gong C, Liu X, Huang X, et al. Molecular characterization and multilocus genotypes of Enterocytozoon bieneusi among horses in southwestern China. Parasit Vectors. 2016;9(1):561. doi: 10.1186/s13071-016-1844-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Widmer G, Akiyoshi DE. Host-specific segregation of ribosomal nucleotide sequence diversity in the microsporidian Enterocytozoon bieneusi. Infect Genet Evol. 2010;10(1):122–128. doi: 10.1016/j.meegid.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Y, Li N, Dearen T, Lobo ML, Matos O, Cama V, et al. Development of a multilocus sequence typing tool for high-resolution genotyping of Enterocytozoon bieneusi. Appl Environ Microbiol. 2011;77(14):4822–4828. doi: 10.1128/AEM.02803-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Cama V, Feng Y, Gilman RH, Bern C, Zhang X, et al. Population genetic analysis of Enterocytozoon bieneusi in humans. Int J Parasitol. 2012;42(3):287–293. doi: 10.1016/j.ijpara.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Wan Q, Xiao L, Zhang X, Li Y, Lu Y, Song M, et al. Clonal evolution of Enterocytozoon bieneusi populations in swine and genetic differentiation in subpopulations between isolates from swine and humans. PLoS Negl Trop Dis. 2016;10(8):e0004966. doi: 10.1371/journal.pntd.0004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karim MR, Wang R, He X, Zhang L, Li J, Rume FI, et al. Multilocus sequence typing of Enterocytozoon bieneusi in non-human primates in China. Vet Parasitol. 2014;200(1-2):13–23. [DOI] [PubMed]
- 17.Deng L, Li W, Zhong Z, Gong C, Cao X, Song Y, et al. Multi-locus genotypes of Enterocytozoon bieneusi in captive Asiatic black bears in southwestern China: high genetic diversity, broad host range, and zoonotic potential. PLoS One. 2017;12(2):e0171772. doi: 10.1371/journal.pone.0171772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhong Z, Tian Y, Song Y, Deng L, Li J, Ren Z, et al. Molecular characterization and multi-locus genotypes of Enterocytozoon bieneusi from captive red kangaroos (Macropus rufus) in Jiangsu province, China. PLoS One. 2017;12(8):e0183249. [DOI] [PMC free article] [PubMed]
- 19.Wang XT, Wang RJ, Ren GJ, Yu ZQ, Zhang LX, Zhang SY, et al. Multilocus genotyping of Giardia duodenalis and Enterocytozoon bieneusi in dairy and native beef (Qinchuan) calves in Shaanxi province, northwestern China. Parasitol Res. 2016;115(3):1355–1361. doi: 10.1007/s00436-016-4908-6. [DOI] [PubMed] [Google Scholar]
- 20.Zhang XX, Cong W, Lou ZL, Ma JG, Zheng WB, Yao QX, et al. Prevalence, risk factors and multilocus genotyping of Enterocytozoon bieneusi in farmed foxes (Vulpes lagopus), northern China. Parasit Vectors. 2016;9:72. doi: 10.1186/s13071-016-1356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng L, Li W, Yu X, Gong C, Liu X, Zhong Z, et al. First report of the human-pathogenic Enterocytozoon bieneusi from red-bellied tree squirrels (Callosciurus erythraeus) in Sichuan, China. PLoS One. 2016;11(9):e0163605. doi: 10.1371/journal.pone.0163605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haubold B, Travisano M, Rainey PB, Hudson RR. Detecting linkage disequilibrium in bacterial populations. Genetics. 1998;150(4):1341–1348. doi: 10.1093/genetics/150.4.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falush D, Stephens M, Pritchard JK. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164(4):1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Cama V, Akinbo FO, Ganguly S, Kiulia NM, Zhang X, et al. Multilocus sequence typing of Enterocytozoon bieneusi: lack of geographic segregation and existence of genetically isolated subpopulations. Infect Genet Evol. 2013;14:111–119. doi: 10.1016/j.meegid.2012.11.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data sets supporting the conclusions of this article are included in the article. Representative sequences are submitted to the GenBank database under the accession numbers: KY950545–KY950576.