

Abstract
Circulating levels of the pro-inflammatory cytokine C-C motif chemokine 11 (CCL11, also known as eotaxin-1) are increased in several animal models of neuroinflammation, including traumatic brain injury and Alzheimer’s disease. Increased levels of CCL11 have also been linked to decreased neurogenesis in mice. We hypothesized that circulating CCL11 levels would increase following ischemic stroke in mice and humans and that higher CCL11 levels would correlate with poor-long term recovery in patients. As predicted, circulating levels of CCL11 in both young and aged mice increased significantly 24 hours after experimental stroke. However, CCL11 levels were decreased in ischemic stroke patients compared to controls at 24 hours after stroke. Interestingly, lower post-stroke CCL11 levels were independently predictive of increased stroke severity and poorer functional outcomes in patients twelve months after ischemic stroke. These results illustrate the differences in the murine and human inflammatory response to ischemic stroke. In addition, it suggests CCL11 as a candidate biomarker for the prediction of acute and long-term functional outcomes in ischemic stroke patients.
Keywords: Stroke, eotaxin, CCLL1, inflammation, neuroinflammation, ischemia
Introduction
CCL11 (also known as eotaxin-1) is a chemokine that plays a role in a variety of pathologic conditions, including allergy, coronary heart disease and inflammatory bowel disease [1–3]. Although traditionally thought of as an eosinophil chemokine, CCL11 can be secreted by a variety of cell types, including lymphocytes, monocytes, endothelial cells and neurons [4, 5]. Levels of circulating CCL11 increase with normal aging in both human serum and plasma, and exogenous delivery of CCL11 was recently shown to decrease neurogenesis in rodents [6,7]. Circulating levels of CCL11 also increase significantly after traumatic brain injury in mice, suggesting that CCL11 may play a negative role in functional outcome after neurological injury by suppressing neurogenesis [8]. We hypothesized that peripheral CCL11 levels would increase significantly in mice following ischemic stroke, a pattern which would hold true in aged mice that better replicate the average biological age of a human ischemic stroke patient [9]. As such, the suppression of CCL11 release or inhibition of CCL11 with antibody therapies after ischemic stroke could represent an attractive therapeutic target to decrease age-related impairments in stroke recovery. However, the translation of successful experimental immunotherapies has failed in many clinical trials, due in part to profound differences in the immune system between species [10]. In order to determine the relevance of circulating CCL11 as a clinically relevant target, we examined circulating CCL11 levels in human stroke patients and controls. We hypothesized that CCL11 levels would be significantly higher in ischemic stroke patients compared to controls, and that this would be associated with poorer acute and long-term functional outcomes.
Methods
Animal Methods
Middle Cerebral Artery Occlusion (MCAO) Model of Ischemic Stroke: Male C57BL/6 mice were obtained from Envigo Laboratories. Young (10–12 wks) and aged (24 mos) male mice were subjected to focal transient cerebral ischemia by 60 min of reversible MCA occlusion under Isofluorane anesthesia as previously described [11]. In order to achieve equivalent levels of occlusion, 0.21 mm and 0.23 mm silicone coated sutures were used in young and aged mice, respectively [11]. Rectal temperatures were maintained at approximately 37° C during surgery and ischemia with an automated temperature control feedback system. Mice were sacrificed at 24 hrs after reperfusion for serum harvest. All animal procedures were performed in accordance with NIH guidelines for the care and use of laboratory animals and approved by the University of Texas Health Science Center at Houston.
Murine ELISA Cytokine Measurement: Serum CCL11 levels were determined by Multiplex ELISA (Bio-Rad, Hercules, CA). Briefly, mice were euthanized by avertin injection and blood was collected into syringes via cardiac puncture. Blood was allowed to clot for 30 minutes, followed by centrifugation at 2000g for 10 minutes at 4*C for serum separation. Serum was collected and stored at −80*C until use. Samples were thawed and 50ul of serum was loaded into each well in duplicate, with samples assayed according to manufacturer’s instructions using Luminex 200 magnetic bead array platform (Luminex Corporation, Austin, TX, USA). Data is presented as mean ± SEM and analyzed by Student T Test (GraphPad Prism, San Diego, CA). The p value <0.05 was considered statistically significant.
Human Subject Methods
Human Sample Collection: Collection was conducted at an 868-bed community based teaching hospital certified as a JCO Comprehensive Stroke Center (CSC). Blood was drawn from patients at 24 +/− 6 hrs after stroke onset. Ischemic stroke was defined as an acute-onset focal neurological deficit with corresponding evidence of cerebral infarction on radiographic imaging (CT or MRI). Exclusion criteria included active cancer, autoimmune disease, iatrogenic stroke and active immunosuppressive therapy. Over the five-year period from 2011–2015, approximately 3300 patients admitted to Hartford Hospital for stroke-like symptoms were screened as potential biobank candidates. Patients used in this specific study were those identified as having available serum samples collected at 24 hours +/− 6 hours after ischemic stroke onset, with imaging-confirmed, non-dissection ischemic stroke. In addition, eligible patients showed no evidence of active infection, no exclusion criteria and had detailed in-hospital outcomes, demographic and co-morbidity information available (n=138). For detailed study selection methods, see Supplementary Figure 1. CCL11 levels were measured in 138 serum samples, with 5 returning undetectable results. Therefore, the final number of patients included in this study was n=133.
Blood was drawn from 17 asymptomatic vascular-risk factor controls recruited from the cardiac clinic at the University of Connecticut Health Center. Cytokine levels were measured by multiplex ELISA (BioRad, Hercules, CA). All human sample collection was approved by the Institutional Review Board at Hartford Hospital and the University of Connecticut Health Center.
Measurements: The primary functional outcomes were defined as modified Rankin score (mRS) and modified Barthel Index (mBI) in hospital (n=119), 3 months (n=57) and 12 months (n=33). 3 and 12 month functional outcomes were assessed via telephone interview by a trained nurse. Secondary outcomes were defined as NIH severity (as determined by NIH Stroke Scale) at admission, NIH severity at discharge and mortality (in-hospital, 3 months and 12 months). The modified Rankin score (mRS) is used to measure overall disability and independence, while the modified Barthel Index (MBI) measures physical disability and the performance of activities of daily living.
Statistical analysis: As the distribution of CCL11 levels was found to be non-normally distributed, several statistical approaches were taken. To determine the effect of ischemic stroke on CC11 levels, CCL11 was log transformed and analysis of covariance analysis (ANCOVA) was performed on serum CCL11 levels from ischemic stroke patients, with asymptomatic control patients enrolled at the University of Connecticut Health Center serving as risk-factor matched controls. The ANCOVA analysis was designed to control for variables determined to be significantly different between ischemic stroke and control groups via Fisher’s Exact Test (age, hyperlipidemia and hypertension).
For outcome analysis, outcome measures were dichotomized into groups and the Wilcoxon ranked sum test was used to compare CCL11 levels. Patients were assessed for functional status in-hospital, and at 3 and 12 months by mBI (poor = ≤14) and mRS (poor = 3–5). Note that for mRS, 6 (deceased), was considered separately in mortality analysis. The univariate relationship between CCL11 and NIHSS on admission was analyzed as a continuous variable using non-parametric correlation analysis.
Significant findings identified in the univariate analyses of outcome measures were included in a multivariate logistic regression to control for significant confounders at a level of p=.05. In order to account for the non-normality of CCL11 in the multivariate analysis, CCL11 was logarithmically transformed in order to normalize the distribution. For multivariate analysis of the association between CCL11 and stroke severity, NIHSS was dichotomized into severe (NIHSS ≥11) and mild/moderate (NIHSS ≤10). For all other multivariate analysis, where NIHSS was examined as a potential confounding variable, NIHSS was considered as a continuous variable.
The criterion of statistical significance was set at 0.05. All analysis were performed using Statistical Package for the Social Science v21. Data was illustrated using GraphPad Prism (San Diego, CA).
Results
Peripheral CCL11 levels increase 24 hours after experimental ischemic stroke in mice
In order to determine the effect of ischemic stroke on acute circulating CCL11 levels in mice, we measured serum levels of CCL11 in both young (3 month old) and aged (24 month old) animals 24 hours after stroke. The aged mice used in our experimental studies were 24 months of age, which closely approximates the biological age of a 70-year old human (12). Mice of this age were selected to best mirror our clinical patient population, which had an average age of 70.42 years (Table 1). In agreement with our hypothesis, levels of circulating CCL11 significantly increased 24 hrs after ischemic stroke in an experimental mouse model (Figure 1). Stroke mice demonstrated significantly higher levels of serum CCL11 than sham mice at 24 hours, regardless of age (p=.0026).
Table 1.
Demographic characteristics for stroke patients included in this study
| Category | Total (n=133) | |
|---|---|---|
|
| ||
| Age, mean (standard deviation) | 70.42 (13.87) | |
|
| ||
| Sex (male) | 57.1% (n=76) | |
|
| ||
| Race | ||
| Caucasian, % | 84.0% (n=110) | |
| African American, % | 5.3% (n=7) | |
| Latino, % | 9.9% (n=13) | |
| Other, % | 0.8% (n=4) | |
|
| ||
| Stroke risk factor history | ||
| Hypertension, % | 81.8% (n=108) | |
| Coronary Artery Disease, % | 32.6% (n=43) | |
| Diabetes, % | 34.8% (n=46) | |
| Smoking, % | 18.2% (n= 24) | |
| Hyperlipidemia, % | 68.9% (n= 91) | |
|
| ||
| Pre-stroke condition | ||
| Baseline Modified Barthel, Median, IQR | 20 (20, 20) | |
| Baseline Modified Rankin, Median, IQR | 0 (0, 0) | |
|
| ||
| Acute Function | ||
| Admission NIHSS, median (IQR) | 5 (2, 15) | |
| In-Hospital Modified Barthel, Median, IQR | 13 (7, 20) | |
| In-Hospital Modified Rankin, Median, IQR | 3 (1, 4) | |
|
| ||
| Stroke Subtype | ||
| Cardioembolic | 49.6% (n=66) | |
| Large Vessel | 16.5% (n=21) | |
| Small Vessel | 15.7% (n=22) | |
| Undetermined | 18.0% (n=24) | |
Figure 1.
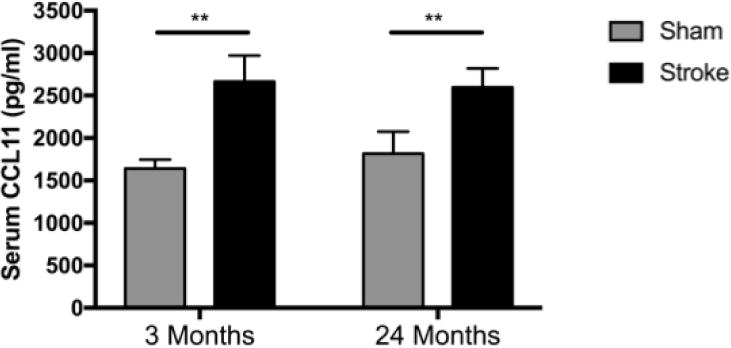
Peripheral CCL11 levels 24 hrs in serum after sham or stroke MCAO surgery in young and aged mice. N=3–4/group. Error bars show Standard Error of the Mean (SEM). **=p<0.01.
CCL11 levels do not increase 24 hours after ischemic stroke in human patients
We next sought to determine whether serum CCL11 levels also increase acutely after ischemic stroke in human patients. Serum levels of CCL11 were measured in patients 24 +/− 6 hours after ischemic stroke and compared to levels from vascular risk factor control patients By univariate analysis, there was no significant difference in serum CCL11 levels in control patients (47.81 pg/ml) when compared to ischemic controls (41.07 pg/ml). As several important biological variables differed between control and ischemic stroke patients, including age, hyperlipidemia and hypertension (see Supplementary Table 1), we then generated a multivariate linear model to adjust for these potential confounders. CCL11 levels were log-transformed to normalize the distribution. Following adjustment for the above confounding factors, CCL11 levels were found to be significantly lower in ischemic stroke patients 24 hours after stroke onset than in healthy controls (p=.028, see Figure 2 and Supplementary Table 2).
Fig 2.
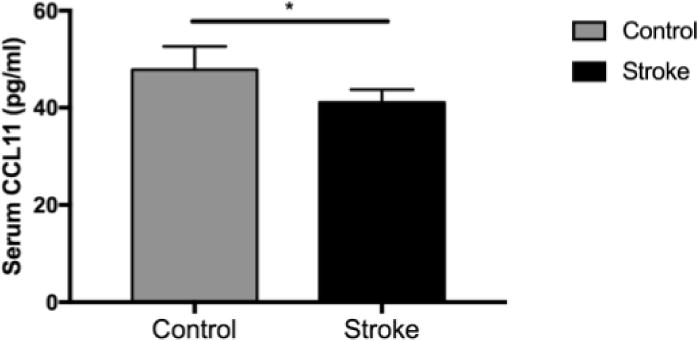
Peripheral CCL11 levels 24 hours after the onset of ischemic stroke (n=133) in human patients compared to levels in asymptomatic control serum (n=17). Error bars show SEM. * = p<.05.
CCL11 levels correlate with stroke severity and etiology in human patients
Evidence suggests that age and initial stroke severity significantly impact the post-stroke inflammatory response. As we included patients with a wide range of stroke severities (from NIH 0–29) and ages (age 50–95) in our study, we then determined whether patient variability could potentially mask an effect between ischemic stroke and acute CCL11 levels. To this end, we then examined the association between these variables and acute CCL11 levels in our stroke cohort.
Importantly, no significant association was found between CCL11 and poor pre-stroke neurological function on the modified Barthel index or modified Rankin scale. However, we observed a trend towards an inverse relationship between admission stroke severity (as measured by NIHSS) and CCL11 levels (p=.098, Figure 3A). In addition, lower CCL11 levels at 24 hours after ischemic stroke were found to be significantly correlated with poorer NIHSS at discharge from the hospital (p=.007, Figure 3B). No significant association between CCL11 levels and in-hospital mortality was found.
Figure 3.
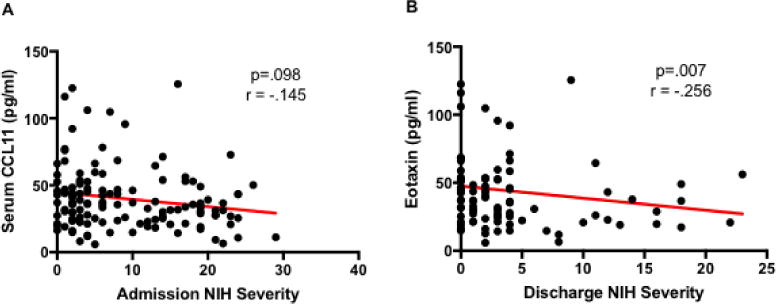
A. Correlation between serum concentrations of CCL11 at 24 hours post-stroke and admission NIH stroke severity from ischemic stroke patients as measured by Multiplex Assay. NIHSS was assessed as a continuous variable by non-parametric correlation analysis. B. Correlation between serum concentrations of CCL11 at 24 hours post-stroke and discharge NIH stroke severity in ischemic stroke patients.
Additionally, we examined whether CCL11 levels at 24 hours after stroke correlated with stroke subtype. As stroke subtype may influence on serum CCL11 levels 24 hours after stroke. (Supplemental Figure 2). However, even after correction for stroke severity (NIH on admission), age, coronary artery disease and hyperlipidemia, CCL11 levels at 24 hours were not significantly associated with stroke subtype.
Low CCL11 levels 24 hours after ischemic stroke are associated with poor outcome at 3 and 12 months post-stroke
Long-term outcome analysis demonstrated that lower levels of CCL11 at 24 hrs after ischemic stroke was significantly associated with poor outcome at 3 months as measured by modified Barthel Index (p=.024). Acute CCL11 levels were also found to be significantly lower in living patients with poor functional outcomes at 12 months as measured by modified Rankin Score (poor mRS = 3–5) (p=.029). A trend towards a similar pattern in function as measured by modified Barthel Index was also seen (Table 2).
Table 2.
Univariate analysis of CCL11 serum levels and long-term functional outcomes
| Modified Rankin Score | Good (0–2) | Poor (3–5) | Sig. |
|---|---|---|---|
|
| |||
| mRS, In-Hospital (n=116) | 37.08 pg/ml | 34.08 pg/ml | p = .352 |
| mRS, 3 Months (n=57) | 37.4 pg/ml | 32.33 pg/ml | p = .320 |
| mRS, 12 Months (n=33) | 38.82 pg/ml | 27.35 pg/ml | p = .029 |
|
| |||
| Modified Barthel Index | Good (≥ 15) | Poor (≤ 14) | Sig. |
|
| |||
| mBI, In-Hospital (n=116) | 37.02 pg/ml | 35.76 pg/ml | p = .365 |
| mBI, 3 Months (n=58) | 39.2 pg/ml | 23.33 pg/ml | p = .024 |
| mBI, 12 Months (n=35) | 36.17 pg/ml | 23.50 pg/ml | p = .134 |
Low CCL11 is Associated with Increased NIH Severity and Poor 3 and 12 Month Outcome in a Multivariate Model
To control for potential confounders, we constructed a multivariate model to assess the relationship between peripheral CCL11 levels at 24 hrs post-stroke and acute stroke severity. After controlling for patient age, sex and common co-morbidities (coronary artery disease, hyperlipidemia), low CCL11 levels at 24 hrs remained a significant predictor of increased NIH stroke severity at admission (Table 3).
Table 3.
Association of CCL11 with long-term functional outcomes after ischemic stroke
| Category | Significance |
|---|---|
|
Correlation with NIH Severity Adjusted for age, sex and co-morbidities |
p = .002 |
|
Negative Outcome at 3 Months (mBI ≤ 14) Adjusted for age, NIH severity, gender, co-morbidities |
p = .014 |
|
Negative Outcome at 3 Months (mRS 3–5) Adjusted for age, NIH severity, gender, co-morbidities |
p= .371 |
|
Negative Outcome at 12 Months (mBI ≤ 14) Adjusted for age, NIH severity, gender, co-morbidities |
p = .066 |
|
Negative Outcome at 12 Months (mRS 3–5) Adjusted for age, NIH severity, gender, co-morbidities |
p = .029 |
We next sought to determine the utility of CCL11 as a biomarker of long-term post-stroke functional outcome. As initial stroke severity is an important confounding factor in evaluating long-term functional outcome, we controlled for initial stroke severity in our multivariate assessment of long-term outcome, in addition to age and common co-morbidities. Low levels of CCL11 24 hrs after stroke were significantly predictive of poor functional outcome by mBI at 3 months (p=.014) and mRS at 12 months after stroke (p=.029).
Discussion
CCL11 (also known as eotaxin-1) is a chemokine that has been identified as a contributor to neurodegeneration in the central nervous system [7,13]. CCL11 is produced by epithelial cells, fibroblasts, smooth muscle cells and macrophages and is important for the recruitment of basophils, eosinophils, mast cells, macrophages and TH2-type T Cells in humans [14–16]. Our results demonstrate that serum CCL11 concentrations increase significantly in mice 24 hours after ischemic stroke. However, an inverse relationship was seen in humans, where ischemic stroke patients had significantly lower levels of circulating CCL11 than control patients 24 hours after stroke.
Previous pre-clinical experiments have shown that circulating CCL11 is capable of crossing the blood brain barrier and results in decreased neurogenesis in mice [6,17]. In primary cell lines obtained from mice, treatment with exogenous CCL11 increases microglial migration and induces the production of microglial reactive oxygen species, directly contributing to neuronal death in co-culture systems [18]. Previous studies have shown that serum CCL11 levels rise acutely in a mouse model of traumatic brain injury [8]. In line with these results, our studies found that CCL11 levels increase significantly in mice 24 hours after ischemic stroke, regardless of age.
Importantly, in disagreement with our animal data, we found that serum CCL11 levels were significantly lower in ischemic stroke patients relative to risk-factor matched asymptomatic controls after adjustment for potential confounding variables. Moreover, serum CCL11 was inversely associated with stroke severity at discharge, as measured by NIH Stroke Scale. This association remained significant after multivariate adjustment for potential confounders. Similar findings were recently reported in a human traumatic brain injury (TBI) cohort, where circulating CCL11 levels were lower in TBI patients than in healthy controls in a severity-dependent manner [19]. These results suggest that that peripheral CCL11 may have divergent responses after brain injury in mice and humans, supported by the fact that murine and human CCL11 share only 59% protein homology [20]. Interestingly, our data also demonstrated that mice have 10-fold higher levels of serum CCL11 compared to humans at baseline, confirming species differences in circulating CCL11 levels reported by other groups [7,19].
Although the role of CCL11 in neurodegeneration and impaired neurogenesis has been clearly demonstrated in mouse models, the role CCL11 plays in acute neurological injury in humans remains unclear. CCR3, a receptor for CCL11, is expressed on microglia in the human brain, but immunoreactivity for CCL11 itself in human brain samples was not detected in brains from either control or Alzheimer’s disease patients [21]. Our studies suggest that CCL11 may serve divergent function in neuroinflammation in humans vs. mouse models. Importantly, these factors call into question the utility of using CCL11-related findings in murine models to identify and develop therapeutic targets for patients suffering from acute neurological injury. Early use of human samples to validate early pre-clinical findings is important in translational and clinical trial design.
There are several potential reasons for the lower levels of CCL11 seen in patients with severe ischemic strokes. CCL11 is a pro-inflammatory cytokine released in response to TNF-α and IL-1β [22]. Once released, CCL11 serves as a chemoattractant for eosinophils and TH2 T-helper cells, contributing to subsequent inflammation [20]. However, in both ischemic stroke and traumatic brain injury, the pro-inflammatory response is accompanied by a massive systemic immunosuppression and the release of high levels of IL-10, which plays a role in stroke-induced immunosuppression and has been shown to directly suppress the release of CCL11 [23]. In addition, TH1-type cytokines like IFN-γ have been shown to inhibit the release of CCL11 from human cells [24, 25]. Enhanced TH1 pro-inflammatory cytokine release secondary to greater tissue damage in larger strokes could suppress the release of CCL11, resulting in the lower CCL11 levels seen in severe stroke patients.
After controlling for well-known predictors of stroke outcome, including age and co-morbidities, low CCL11 was significantly predictive of worse stroke severity (NIHSS) at discharge from the hospital. More importantly, after controlling for initial stroke severity, low CCL11 levels were predictive of negative functional outcomes at both 3 and 12 months after stroke. Importantly, the biology behind the association of low acute CCL11 levels and long-term functional outcomes remains unknown. In light of the significant differences between protein structure and peripheral levels of CCL11 between mice and humans, future studies of CCL11 in acute neurological injury may be better suited to an alternative model organism or human cell lines. However, as our human studies were limited to the analysis of peripheral CCL11 levels, we cannot rule out the potential impact of local CCL11 within the brain acutely after stroke.
The primary limitation of this study was the small sample size, particularly in the control group and in patients available for follow-up functional measurements at 3 and 12 months. In addition, this study only investigates one acute timepoint (24 hours). A larger cohort of ischemic stroke patients and controls, with samples taken at multiple timepoints, is necessary to confirm the relationship between CCL11 and stroke outcomes, as well as the relationship between circulating CCL11 and stroke etiology. Understanding the influence of age, co-morbidities and inflammation on stroke pathology is critical for the development of sensitive, specific biomarkers for clinical prognostic use. This study also underscores the importance of validating murine findings in human patients, as the association between CCL11 and ischemic stroke appears to vary significantly between species and may have significant effects on the translational utility of manipulation of CCL11 levels based on murine studies.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health NS094543 and NS076293 (to LDM) the Hartford Hospital Research Department, and the Russell and Diana Hawkins Family Foundation Discovery Fellowships to the Graduate School at UTHealth (to MAR).
Funding: This study was funded by the National Institutes of Health NS094543 and NS076293 (to LDM) and Hartford Hospital Research Department.
Footnotes
Compliance with Ethical Standards:
Ethical Approval: “All sample collection performed in studies involving human participants were conducted in accordance with the ethical standards of the Institutional Review Board at Hartford Hospital and the University of Connecticut Health Center and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.”
“All applicable international, national and institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. All animal procedures were performed in accordance with NIH guidelines for the care and use of laboratory animals and approved by the Animal Care Committee of the University of Connecticut Health Center. “
Informed Consent: “Informed consent was obtained from all individual participants included in the study.”
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Rothenberg ME, Maclean JA, Pearlman E, Luster AD, Leder Pa. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J Exp Med. 1997;185(4):785–90. doi: 10.1084/jem.185.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emanuele E, Falcone C, D’angelo A. Association of plasma eotaxin levels with the presence and extent of angiographic coronary artery disease. Atherosclerosis. 2006;186(1):140–5. doi: 10.1016/j.atherosclerosis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Mir A, Minguez M, Tatay J, et al. Elevated serum eotaxin levels in patients with inflammatory bowel disease. Am J Gastroenterol. 2002;97(6):1452–7. doi: 10.1111/j.1572-0241.2002.05687.x. [DOI] [PubMed] [Google Scholar]
- 4.Fryer AD, Stein LH, Nie Z, et al. Neuronal eotaxin and the effects of CCR3 antagonist on airway hyperreactivity and M2 receptor dysfunction. J Clin Invest. 2006;116(1):228–36. doi: 10.1172/JCI25423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adar T, Shteingart S, Ben ya’acov A, Bar-gil shitrit A, Goldin E. From airway inflammation to inflammatory bowel disease: eotaxin-1, a key regulator of intestinal inflammation. Clin Immunol. 2014;153(1):199–208. doi: 10.1016/j.clim.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 6.Targowski T, Jahnz-rózyk K, Plusa T, Glodzinska-wyszogrodzka E. Influence of age and gender on serum eotaxin concentration in healthy and allergic people. J Investig Allergol Clin Immunol. 2005;15(4):277–82. [PubMed] [Google Scholar]
- 7.Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477(7362):90–4. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shein SL, Shellington DK, Exo JL, et al. Hemorrhagic shock shifts the serum cytokine profile from pro- to anti-inflammatory after experimental traumatic brain injury in mice. J Neurotrauma. 2014;31(16):1386–95. doi: 10.1089/neu.2013.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manwani B, Liu F, Xu Y, Persky R, Li J, Mccullough LD. Functional recovery in aging mice after experimental stroke. Brain Behav Immun. 2011;25(8):1689–700. doi: 10.1016/j.bbi.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zschaler J, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol. 2014;34(5):433–54. [PubMed] [Google Scholar]
- 11.Manwani B, Friedler B, Verma R, Venna VR, Mccullough LD, Liu F. Perfusion of ischemic brain in young and aged animals: a laser speckle flowmetry study. Stroke. 2014;45(2):571–8. doi: 10.1161/STROKEAHA.113.002944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flurkey K, Currer JM, Harrison DE. JG Fox The Mouse in Biomedical Research 2nd Edition Burlington. MA, American: College Laboratory Animal Medicine (Elsevier); 2007. The Mouse in Aging Research; pp. 637–672. [Google Scholar]
- 13.Huber AK, Giles DA, Segal BM, Irani DN. An emerging role for eotaxins in neurodegenerative disease. Clin Immunol. 2016 doi: 10.1016/j.clim.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 14.Menzies-gow A, Ying S, Sabroe I, et al. Eotaxin (CCL11) and eotaxin-2 (CCL24) induce recruitment of eosinophils, basophils, neutrophils, and macrophages as well as features of early- and late-phase allergic reactions following cutaneous injection in human atopic and nonatopic volunteers. J Immunol. 2002;169(5):2712–8. doi: 10.4049/jimmunol.169.5.2712. [DOI] [PubMed] [Google Scholar]
- 15.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277(5334):2005–7. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 16.De paulis A, Annunziato F, Di gioia L, et al. Expression of the chemokine receptor CCR3 on human mast cells. Int Arch Allergy Immunol. 2001;124(1–3):146–50. doi: 10.1159/000053694. [DOI] [PubMed] [Google Scholar]
- 17.Erickson MA, Morofuji Y, Owen JB, Banks WA. Rapid transport of CCL11 across the blood-brain barrier: regional variation and importance of blood cells. J Pharmacol Exp Ther. 2014;349(3):497–507. doi: 10.1124/jpet.114.213074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parajuli B, Horiuchi H, Mizuno T, Takeuchi H, Suzumura A. CCL11 enhances excitotoxic neuronal death by producing reactive oxygen species in microglia. Glia. 2015;63(12):2274–84. doi: 10.1002/glia.22892. [DOI] [PubMed] [Google Scholar]
- 19.Di battista AP, Rhind SG, Hutchison MG, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflammation. 2016;13(1):40. doi: 10.1186/s12974-016-0500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitaura M, Nakajima T, Imai T, et al. Molecular cloning of human eotaxin, an eosinophil-selective CC chemokine, and identification of a specific eosinophil eotaxin receptor, CC chemokine receptor 3. J Biol Chem. 1996;271(13):7725–30. doi: 10.1074/jbc.271.13.7725. [DOI] [PubMed] [Google Scholar]
- 21.Xia MQ, Qin SX, Wu LJ, Mackay CR, Hyman BT. Immunohistochemical study of the beta-chemokine receptors CCR3 and CCR5 and their ligands in normal and Alzheimer’s disease brains. Am J Pathol. 1998;153(1):31–7. doi: 10.1016/s0002-9440(10)65542-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung KF, Patel HJ, Fadlon EJ, et al. Induction of eotaxin expression and release from human airway smooth muscle cells by IL-1beta and TNFalpha: effects of IL-10 and corticosteroids. Br J Pharmacol. 1999;127(5):1145–50. doi: 10.1038/sj.bjp.0702660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garcia-zepeda EA, Rothenberg ME, Ownbey RT, Celestin J, Leder P, Luster AD. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat Med. 1996;2(4):449–56. doi: 10.1038/nm0496-449. [DOI] [PubMed] [Google Scholar]
- 24.Gerber BO, Zanni MP, Uguccioni M, et al. Functional expression of the eotaxin receptor CCR3 in T lymphocytes co-localizing with eosinophils. Curr Biol. 1997;7(11):836–43. doi: 10.1016/s0960-9822(06)00371-x. [DOI] [PubMed] [Google Scholar]
- 25.Miyamasu M, Misaki Y, Yamaguchi M, et al. Regulation of human eotaxin generation by Th1-/Th2-derived cytokines. Int Arch Allergy Immunol. 2000;122(Suppl 1):54–8. doi: 10.1159/000053634. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.