

Abstract
During epithelial ovarian cancer (EOC) progression, intraperitoneally disseminating tumor cells and multi-cellular aggregates (MCAs) present in ascites fluid adhere to the peritoneum and induce retraction of the peritoneal mesothelial monolayer prior to invasion of the collagen-rich sub-mesothelial matrix and proliferation into macro-metastases. Clinical studies have shown heterogeneity among EOC metastatic units with respect to cadherin expression profiles and invasive behavior, however the impact of distinct cadherin profiles on peritoneal anchoring of metastatic lesions remains poorly understood. In the current study, we demonstrate that metastasis-associated behaviors of ovarian cancer cells and MCAs are influenced by cellular cadherin composition. Our results show that mesenchymal N-cadherin expressing (Ncad+) cells and MCAs invade much more efficiently than E-cadherin expressing (Ecad+) cells. Ncad+ MCAs exhibit rapid lateral dispersal prior to penetration of three-dimensional collagen matrices. When seeded as individual cells, lateral migration and cell-cell junction formation precede matrix invasion. Neutralizing the Ncad extracellular domain with the monoclonal antibody GC-4 suppresses lateral dispersal and cell penetration of collagen gels. In contrast, use of a broad spectrum matrix metalloproteinase (MMP) inhibitor (GM6001) to block endogenous membrane type 1 matrix metalloproteinase (MT1-MMP) activity does not fully inhibit cell invasion. Using intact tissue explants, Ncad+ MCAs were also shown to efficiently rupture peritoneal mesothelial cells, exposing the sub-mesothelial collagen matrix. Acquisition of Ncad by E-cadherin expressing cells (Ecad+) increased mesothelial clearance activity, but was not sufficient to induce matrix invasion. Furthermore, co-culture of Ncad+ with Ecad+ cells did not promote a “leader-follower” mode of collective cell invasion, demonstrating that matrix remodeling and creation of invasive micro-tracks are not sufficient for cell penetration of collagen matrices in the absence of Ncad. Collectively, our data emphasize the role of Ncad in intraperitoneal seeding of EOC and provide the rationale for future studies targeting Ncad+ in pre-clinical models of EOC metastasis.
Keywords: ovarian cancer, metastasis, cadherins, extracellular matrix, invasion, mesothelium
INTRODUCTION
Epithelial ovarian carcinoma (EOC) is the leading cause of death among all female reproductive cancers, with a predicted 14 240 deaths estimated in the US in 2016 (1). In the overwhelming majority of cases, tumors are detected at a late stage when metastatic disease is already present, resulting in poor prognosis (2). Moreover, most patients with good initial response to chemotherapy eventually exhibit drug resistance and relapse (3). Therefore a detailed molecular-level understanding of the mechanisms involved in the acquisition of aggressive metastatic disease is needed to develop novel therapeutic approaches to prevent metastatic dissemination and improve long-term survival of women with ovarian cancer.
Ovarian tumors are thought to arise from the ovarian surface epithelium as well as the distal fimbriae of the fallopian tubes, from which shed cells implant on the ovarian surface (4–7). Unlike most solid tumors, EOC metastasis occurs by shedding of cancer cells from the ovarian tumor directly into the peritoneal cavity as both single cells and multicellular aggregates (MCAs) that disseminate with the peritoneal fluid flow (5,7). Hematogenous metastasis with homing to the peritoneal cavity has also been described (8,9). These anchorage-independent cancer cells subsequently adhere to peritoneal mesothelial cells, induce rapid mesothelial cell retraction (10,11), and invade the sub-mesothelial interstitial collagen-rich extracellular matrix (ECM) wherein they proliferate to form widely disseminated metastatic lesions on the peritoneum, omentum, and bowel (7). The MCA as a metastatic unit is poorly understood. It has been demonstrated that individual cells as well as MCAs from malignant primary human ovarian cancer ascites adhere to and dissociate upon both collagen and human mesothelial cell monolayers in vitro (12). However, the majority of these MCAs fail to invade, suggesting heterogeneity in the MCA population that may influence metastatic success (13).
Most experimental studies of matrix invasion utilize dissociated suspensions of cells and have implicated multiple molecular events in the invasive process. For example, the ability to overcome steric hindrance imposed by ‘pores’ within matrices that are smaller than invading cells is related to a complex combination of cell adhesiveness, nuclear volume, cell contractility and matrix stiffness (14). Invasion is also facilitated by expression of matrix degrading metalloproteinases that remove physical constraints on cellular migration (15–20). However the role of cadherin-mediated cell-cell adhesion, particularly in MCA invasion, remains unclear. Cadherins are calcium-dependent cell-cell adhesion molecules that function to maintain epithelial integrity. While fallopian tube epithelia express epithelial cadherin (Ecad) (21,22), the mesodermally derived ovarian surface epithelium expresses neural cadherin (Ncad) (4,23). We have previously shown that approximately 30% of human ovarian tumors exhibit simultaneous positive immunoreactivity for both Ecad and Ncad with two predominant expression patterns (5,24). Tumors exhibit ‘mixed’ cadherin expression, wherein distinct cells within one tumor express either Ecad or Ncad, as well as ‘hybrid’ cadherin expression, characterized by Ecad and Ncad expression in the same cell (24). Furthermore, in a paired analysis of primary and metastatic ovarian tumors from the same patient, Ncad immunoreactivity was maintained in ~70% of metastases while only 25% retained Ecad expression (5). Therefore, to evaluate cellular activities associated with metastatic success in the context of cellular ultrastructure (individual cells vs MCAs) and cadherin expression profile, we used live imaging and electron microscopy to quantify interaction with intact peritoneal explants, organotypic meso-mimetic cultures and three-dimensional (3D) interstitial collagen gels to model sub-mesothelial anchoring of EOC cells in the peritoneum.
RESULTS
The peritoneum, which lines the peritoneal cavity and covers all the abdominal organs, is the initial point of contact for disseminating EOC cells and MCAs during metastatic progression. Underlying the mesothelial monolayer, the sub-mesothelial matrix displays highly organized loosely woven collagen fibers in tumor-free animals, as observed using second harmonic generation (SHG) imaging of intact peritoneal explants [Fig. 1A]. Using a murine allograft model of EOC metastasis, we observe widely disseminated intraperitoneal seeding by cells and cell clusters, with subsequent penetration of the collagen-rich sub-mesothelial matrix [Fig. 1B–E]. Cellular invasion is accompanied by significant deformation of the surrounding collagen matrix, resulting in loss of the highly organized parallel collagen fiber orientation seen in tumor-free animals, as well as the appearance of collagen-free areas [Fig. 1B] continuing into micro-tracks as observed by combined fluorescence/SHG imaging of peritoneal explants [Fig. 1C–E; Suppl. Movie 1–2].
Figure 1. Murine allograft model of ovarian cancer metastasis demonstrates peritoneal seeding by cancer cells/MCAs with subsequent penetration and remodeling of sub-mesothelial collagen.
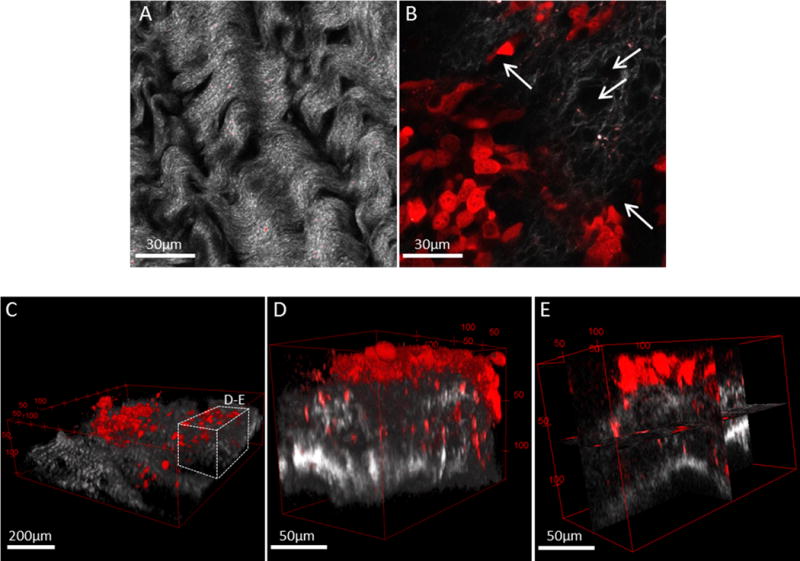
C57Bl/6 female mice were injected intraperitoneally with ID8-RFP murine EOC cells and sacrificed at 8–10 weeks post injection. The parietal peritoneum was dissected and prepared for combined fluorescence/SHG microscopy as described in Methods. Shown are examples of (A) tumor-free mouse peritoneal explant (collagen, grey) and (B) peritoneal explant (collagen, grey) containing a metastatic lesion (cancer cells, red) exhibiting collagen reorganization and peri-cellular collagen clearance areas (arrows). Scale bars: as indicated. Murine metastatic lesions depict (C) seeding of cancer cells and cell clusters (red) atop of peritoneal collagen layer (grey), (D) 3D volume view and (E) orthoslice view of cancer cells penetrating the sub-mesothelial collagen layer. Scale bars: as indicated.
Penetration of 3D collagen gels by EOC cells and MCAs is cadherin-dependent
To model EOC sub-mesothelial collagen invasion and anchoring and to address the role of the cadherin expression repertoire in these processes [Suppl. Fig. 1], we performed continuous live imaging of Ecad+ and Ncad+ EOC cells and MCAs. OvCa433 cells (Ecad+) tagged with red fluorescent protein (RFP) and DOV13 cells (Ncad+) expressing green fluorescent protein were seeded as individual cells or MCAs onto 3D collagen gels and continuously traced with a laser scanning confocal microscope in reflectance mode for matrix imaging and fluorescence mode for cell visualization [Fig. 2A]. Quantitation of the depth of penetration of individual cells revealed that both Ncad+ and Ecad+ cells remain relatively superficially localized, penetrating the collagen gel to a depth equivalent to one cell diameter (12–15 μm for DOV13, and 20-30μm for OvCa433) [Fig. 2B, C–D]. Relative to individual cells, MCAs showed distinctly different patterns of dispersal on collagen gels. Ecad+ MCAs exhibited moderate aggregate dissociation on the collagen gel surface and invasion of the lower cell layer to a depth comparable to that observed with individual cells [Fig. 2B, E]. In contrast, Ncad+ MCAs underwent rapid lateral dispersal by 24h, followed by significant penetration of the collagen gel [Fig. 2B, F]. Ncad+ MCAs also displayed considerable lateral motility (8.8±1.8 μm/h), rapidly contacting neighboring aggregates and fusing into super-aggregates within 4–8h [Fig. 3]. Ecad+ MCAs were less motile (7.0±1.4 μm/h) and aggregate fusion was not visible prior to ~30h [Fig. 3]. Peri-cellular collagen clearance was also enhanced in MCAs relative to individual cells [Suppl. Fig. 2A, B]. This is not due to mechanical deformation of the matrix, as control experiments using fluorescent beads (31μm diameter) showed no signs of lateral motility, collagen penetration or collagen clearance [Fig. 2B; Suppl. Fig. 3]. Interestingly, at longer incubation times, individual Ncad+ cells establish a cell-cell contact network that then initiates collagen gel penetration while retaining cellular contacts [Fig. 4A, B]. This is consistent with the multicellular streaming invasion phenotype characteristic of mesenchymal cells, wherein cells retain tip-like cell:cell contacts [Fig. 4B, arrows; (25–27)]. Similar tip-like junctions were observed between ID8 cells (Ncad+; [Suppl. Fig. 1A]) invading intra-peritoneal collagen in vivo [Suppl. Fig. 4, arrows]. This behavior was not observed in Ecad+ cells and MCAs, even when incubated for >7d (data not shown).
Figure 2. Cadherin composition impacts matrix invasion by EOC cells and MCAs.
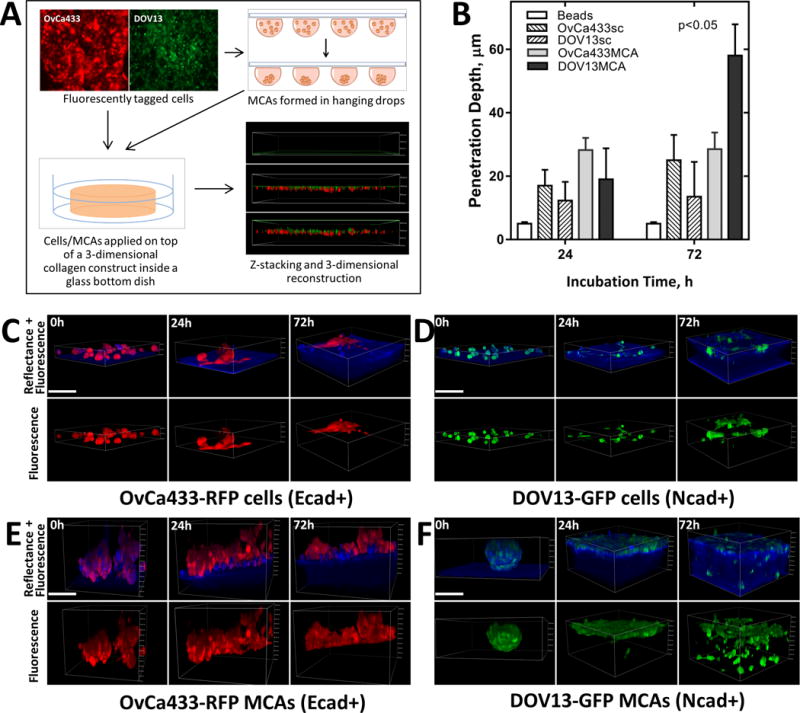
A) Overview of collagen invasion live imaging assay. EOC cells are fluorescently tagged with RFP or GFP via lentiviral transduction and applied as either individual cells or pre-formed MCAs on top of a 3D collagen gel (1.5mg/ml collagen concentration in complete medium) inside a glass-bottom dish. Continuous z-stack imaging of cell/MCA dynamics (green or red) and collagen (blue) was performed using confocal fluorescence and reflectance modes, respectively. B–F) Imaging and analysis of collagen invasion. Multiple representative z-stack snapshots of (C) OvCa433-RFP (Ecad+) cells, (D) DOV13-GFP (Ncad+) cells, (E) OvCa433-RFP (Ecad+) MCAs, (F) DOV13-GFP MCAs, and (Suppl. fig. 2) control fluorescent beads were obtained for up to 72h, and depth of penetration depth quantified (Mean±SD, N=100 in three assays);scale bar: 100μm. Statistical significance shown between the penetration depth of each cell line relative to fluorescent beads; p<0.05, Mann-Whitney U test.
Figure 3. EOC individual cells and MCAs exhibit distinct rates of lateral motility.
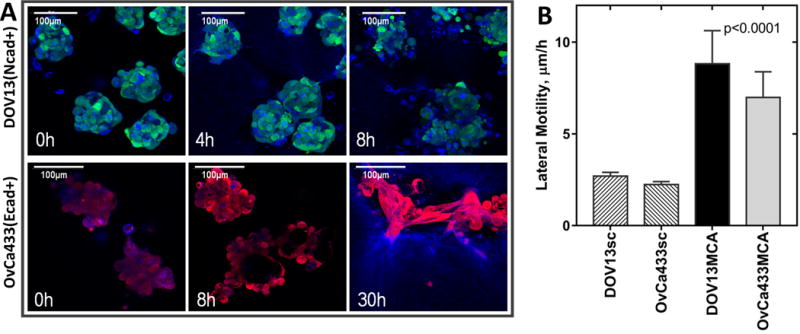
DOV13-GFP (Ncad+) and OvCa433-RFP (Ecad+) individual cells or MCAs were applied on top of a 3D collagen gel construct (1.5mg/ml) inside a glass-bottom dish; 8-hour time-lapse confocal imaging of cell/MCAs (green or red) and collagen matrix (blue) was performed in fluorescent and reflectance modes, respectively. A) Representative images of DOV13-GFP and OvCa433-RFP MCA dynamics are shown at stated time points (scale bars as indicated). B) Evaluation of lateral motility for different cell types (Mean±SD); all assays were repeated in triplicate; statistical significance shown between the lateral motilities of each cell line and fluorescent beads; p<0.05, Mann-Whitney U test.
Figure 4. Lateral motility of mesenchymal-type Ncad+ cells creates an invasion-permissive cell:cell network.
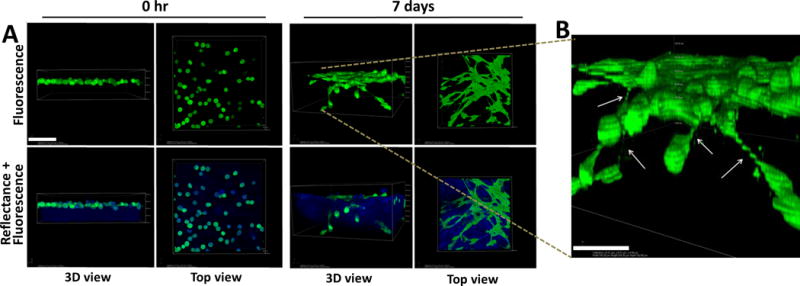
Individual DOV13 (Ncad+) cells were applied atop 3D collagen gels (1.5mg/ml) inside a glass-bottom dish, and series of z-stack confocal microscopy images were acquired using fluorescence and reflectance modes to visualize cells (green) and collagen (blue), respectively, during the course of incubation. A) Representative images demonstrate cellular network formation via tip-like cell:cell junctions (top view) and matrix invasion (3D view) by cancer cells after 7 days of incubation. Scale bar: 100μm. B) A magnified 3D volume view depicts cellular junctions between invading cells and adjacent superficially located cells (indicated by arrows). Scale bar: 50 μm.
Contribution of Ncad expression and MMP activity to collagen invasion
Matrix proteolysis can facilitate matrix remodeling and cell migration through tissue barriers. The membrane-anchored interstitial collagenase membrane type 1 matrix metalloproteinase (MT1-MMP) is overexpressed in EOC (19,28,29) and has been shown to process collagen fibers, creating tube-like micro-tracks for invading cells (15,16). To differentiate the functional effect of a gain of MT1-MMP or Ncad expression on collagen invasion, we evaluated two genetically modified subclones of OvCa433 (Ecad+) cells: OvCa433Ncad+ cells, that express Ncad in addition to endogenous Ecad (24) and OvCa433-MT cells, that express catalytically active MT1-MMP (30). Examination of cells and MCAs showed that neither OvCa433-MT [Fig. 5A–B, E] nor OvCa433Ncad+ [Fig. 5C–D, E] gained the ability to invade collagen gels. Of note, both clones had a smaller spherical cell diameter (18.9±3.2μm and 18.0±3.0 μm for OvCa433-MT and OvCa433Ncad+ vs 22.9±4.3μm for OvCa433), resulting in an overall lesser penetration depth relative to parental OvCa433 cells [Fig. 5E]. At 72 hours of incubation an occasional cell migrated from OvCa433Ncad+ MCAs, but did not penetrate deeply [Fig. 5F], possibly due to polarity cues from Ecad (31). In this context, it is interesting to note that the murine EOC cell line ID8 expresses Ncad and lacks MT1-MMP, yet penetrates intra-peritoneal collagen in vivo [Fig. 1; Suppl. Fig. 1A; Suppl. Fig. 4].
Figure 5. Acquisition of MT1-MMP or Ncad by Ecad+ EOC cells is not sufficient to induce matrix invasion.
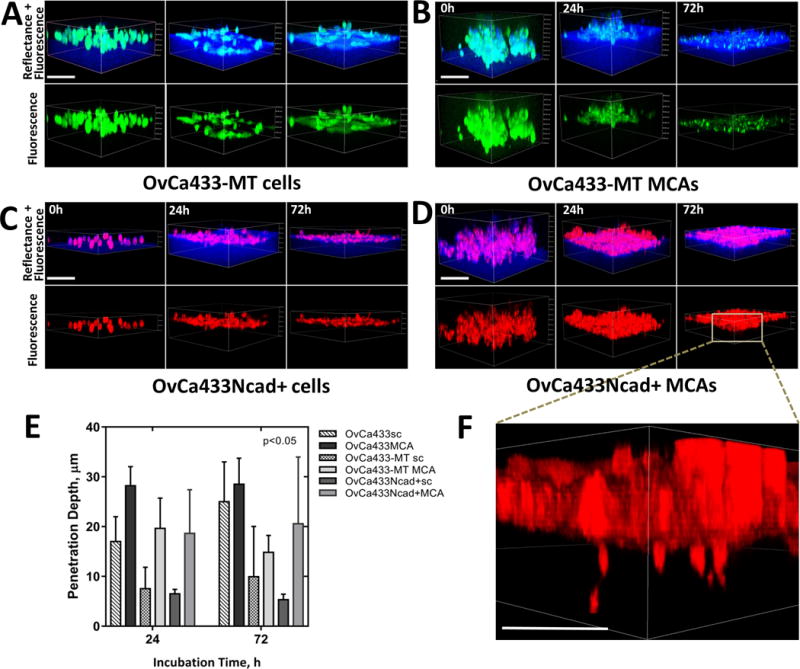
Ecad+ OvCa433MT1 and OvCa433Ncad+ cells were transiently stained with green CMFDA or red CMTPX CellTracker dyes, respectively, as described in Methods, applied atop 3D collagen gels (1.5mg/ml) inside a glass-bottom dish as individual cells (A and C) or MCAs (B and D), and imaged in confocal fluorescence (cancer cells, green or red) and reflectance (collagen, blue) modes over the course of incubation. Scale bars: 100μm. E) Matrix invasion by cells evaluated in terms of cell penetration depth changes with incubation time (M±SD, N=100 single cells and 30 MCAs of three assays). Statistical significance shown between the penetration depths of each cell type and passive beads; p<0.05, Mann-Whitney U test. F) Enlarged 3D view of OvCa433Ncad+ MCA dispersed on collagen after 72h of incubation is shown. Scale bar: 50μm.
To further examine the role of Ncad-mediated cell-cell communication in collagen invasion, function blocking antibodies (GC-4) or isotype control IgG were used to inhibit Ncad junctional activity. Blocking Ncad function with GC-4 resulted in a significant delay in lateral dispersal atop the gel and retarded de novo cell-cell junction formation compared to controls [Fig. 6A]. Subsequently, GC-4-treated cells exhibited a 3.4-fold reduction in depth of penetration. Occasional cell invasion was observed, but only after 7d of incubation [Fig. 6B]. MT1-MMP levels were not altered by GC-4 treatment [Suppl. Fig. 5]. Interestingly, previous studies have highlighted the importance of blocking Ncad to inhibit collective migration of cell clusters in 3D matrices, and predicted a lack of impact against single cell invasion [38]. However, data in the current study show that individual cells engage in lateral motility and adherens junction formation prior to collagen invasion, and Ncad-neutralizing antibody successfully blocked EOC network formation and subsequent cell invasion through 3D collagen matrices.
Figure 6. Blocking Ncad suppresses matrix invasion.
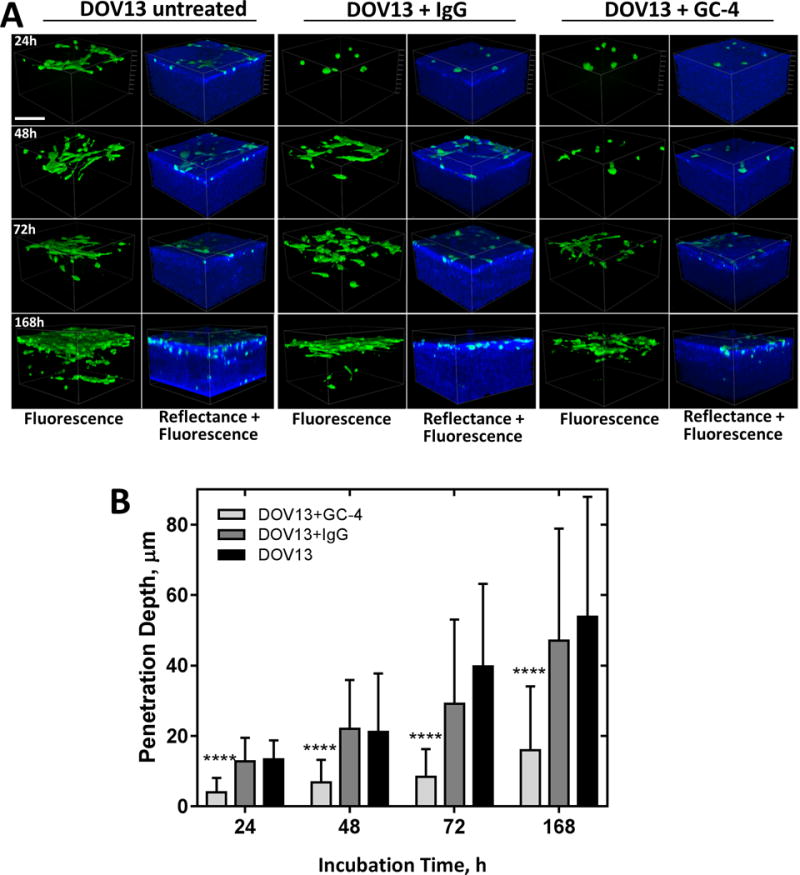
A) Individual Ncad+ DOV13 cells were applied on top of a 3D collagen gel (1.5mg/ml) inside a glass-bottom dish, and incubated in the presence of Ncad-blocking antibody clone GC-4 (200μg/ml), isotype control IgG (200μg/ml), or no drug, as detailed in Methods. Representative reconstructed 3D images of cancer cells (green, fluorescence mode) and collagen matrix (blue, reflectance mode) acquired after 0h, 24h, 72h, and 7 days of incubation. Scale bar: 100μm. B) Evaluation of matrix invasion by EOC cells at different time points of incubation in terms of penetration depth changes (M±SD, N=100). Statistical significance shown between the penetration depths of each cell type and passive beads; ****p<0.0001, Mann-Whitney U test.
To elucidate the contribution of MT1-MMP-driven matrix remodeling to the invasive activity observed in DOV13 cells, the broad-spectrum MMP inhibitor GM6001 was used. DOV13 cells or MCAs were pre-incubated with GM6001 and inhibitor was replenished every 24h throughout the experiment (72h). Although inhibition of MT1-MMP activity significantly blocked collagen remodeling by both cells and MCAs [Fig. 7A, B], this treatment mitigated, but did not completely block, matrix invasion by cancer cells [Fig. 7A, C; Suppl. Fig. 5].
Figure 7. Inhibition of MT1-MMP modulates matrix invasion and collagen remodeling by EOC cells.
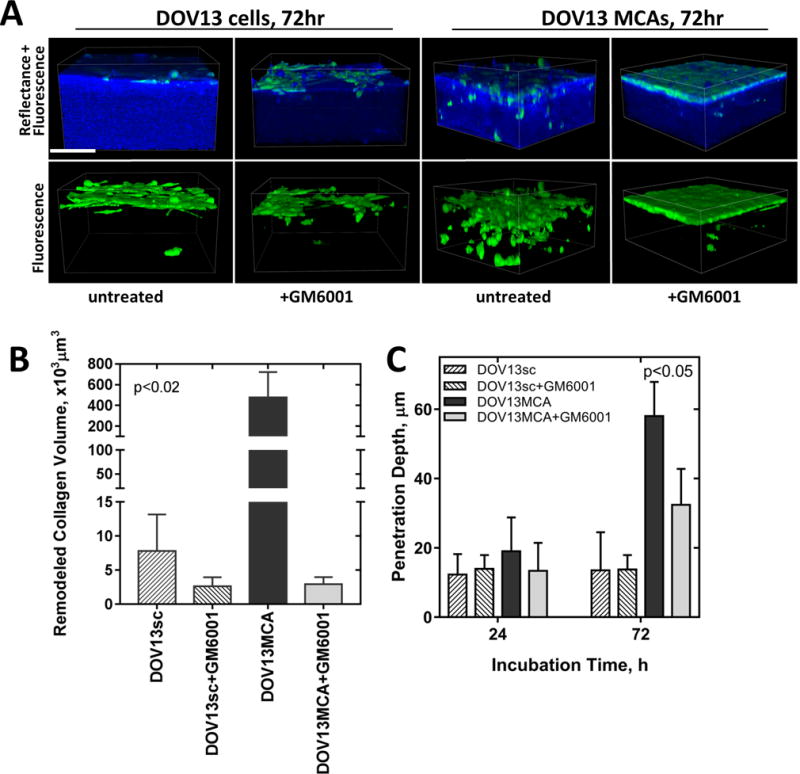
A) Individual Ncad+ DOV13 cells (that endogenously express MT1-MMP) were applied atop 3D collagen gels (1.5mg/ml) inside a glass-bottom dish, and incubated with the broad spectrum MMP inhibitor GM6001 (25μM) or with no inhibitor, as detailed in Methods. Nikon A1R-MP confocal microscope was used for continuous z-stack imaging of cancer cells (green, fluorescence mode) and collagen (blue, reflectance mode). Representative images at 72h incubation time point are shown. Scale bar: 100μm. Evaluation of (B) peri-cellular collagen remodeling after 72hr of incubation, and (C) cell penetration depth at 24 and 72h. All assays were repeated in triplicate and statistical analysis was performed using Mann-Whitney U test. Statistical significance is shown for Ovca433MT1-MMP and OvCa433Ncad+ with respect to OvCa433 invasiveness.
Lack of collective cell behavior in co-cultured epithelial and mesenchymal cells
Recent reports describe the occurrence of collective cell behavior in co-cultured cells, wherein invasive mesenchymal (Ncad+) cells (“invading leaders”) generate micro-tracks that facilitate the movement of non-invasive Ecad-expressing cells (“followers”) through the existing tracks (32,33). For example, formation of Ecad-Ncad junctions between highly invasive Ncad+ breast or prostate cancer cells and non-invasive Ecad-expressing cells derived from normal mammary or normal prostate epithelium, respectively, was reported with subsequent “leader-follower” invasive behavior (33). A related study demonstrated collective invasion initiated by invasive fibroblasts and followed by non-invasive squamous cell carcinoma cells (32). As heterogeneous cadherin expression patterns are observed in ovarian tumors, with many human EOC tumors containing both Ecad- and Ncad-expressing cells, (5,24), we sought to determine whether invasive mesenchymal cells create micro-tracks in collagen that may serve as a conduit for less invasive cells. Mixtures of invasive Ncad+ DOV13 and non-invasive Ecad+ OvCa433 cells were seeded onto 3D collagen gels and observed by live imaging [Fig. 8]. At earlier time points (24–48h), cells sorted laterally into distinct superficial networks based on cadherin composition [Fig. 8A]. Subsequent collagen invasion was observed only by Ncad+ DOV13 cells, while Ecad+ OvCa433 cells remained superficially localized and did not invade through pre-formed micro-tracks [Fig. 8A,B; Suppl. Movie 3]. After 14d of incubation, DOV13 cells completely penetrated and degraded most of the matrix, while non-invasive OvCa433 cells have dissociated from one another and undergone cell death [Fig. 8C; Suppl. Movie 4].
Figure 8. Cadherin-dependent sorting of EOC cells does not promote collective migration.
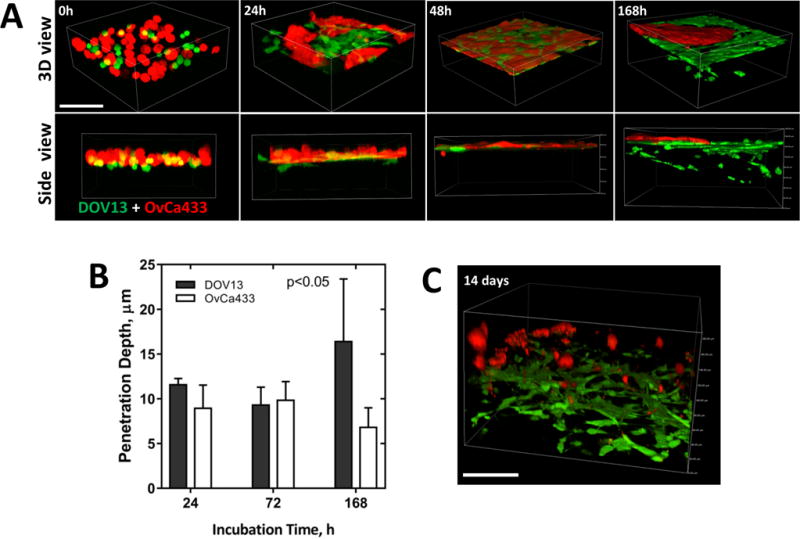
Ncad+ DOV13 and Ecad+ OvCa433 cells were applied atop 3D collagen gels (1.5mg/ml) inside a glass-bottom dish, and series of z-stack confocal images were acquired to observe cell-cell and cell-collagen interactions up to 14 days of incubation. Fluorescent and reflectance confocal modes were utilized to image cells (red, green) and collagen matrix (blue), respectively. A) Representative images of sorting and homotypic network formation (top view) and collagen invasion (3D view) at stated time points. Scale bar: 100μm. B) Evaluation of penetration depth of cell populations (M±SD, N=100 of three assays). Statistical significance is shown between the penetration depths of each cell line type and passive beads; p<0.05, Mann-Whitney U test. C) A representative 3D volume view of cellular dynamics after 14 days of incubation. Scale bar: 100μm.
Cadherin composition impacts mesothelial clearance in vitro and ex vivo
To access the sub-mesothelial collagen matrix and effectively establish secondary lesions, EOC cells and MCAs initially contact the peritoneal mesothelium, a monolayer of cells lining the peritoneal cavity which serves as barrier for tumor cell penetration. Using intact ex vivo peritoneal tissue explants, we observed that individual Ncad+ DOV13 cells avidly adhere to mesothelial cells and extrude cellular processes between mesothelial junctions within 2h of cell seeding [Fig. 9A], but did not disrupt the mesothelial monolayer. In striking contrast, lateral dissociation of DOV13 MCAs [Fig. 9B] induces substantial disruption to and clearance of the mesothelial cell monolayer, accompanied by mesothelial cell rupture within 4h of MCA seeding, as evidenced by disintegration of the mesothelial cell membrane and exposure of the cytoskeleton [Fig. 9Bd]. To further examine the role of Ncad in mesothelial cell clearance activity, a quantitative in vitro assay was used (34) wherein cells were seeded atop a fluorescently tagged confluent mesothelial cell layer in meso-mimetic cultures (35). Similar to results observed with ex vivo intact tissue explants, DOV13 MCAs seeded atop a mesothelial cell monolayer quickly attached, migrated through the mesothelium within the first 24h and dispersed on the culture dish underneath mesothelial cells by 72h of incubation [Fig. 9C, F; Suppl. Fig. 6]. Ecad+ OvCa433 MCAs adhered to the mesothelium, but demonstrated minimal mesothelial clearance by 72h [Fig. 9D, F]. However acquisition of Ncad expression (OvCa433Ncad+) significantly increased the rate of mesothelial clearance after 24–48h [Fig. 9E, F].
Figure 9. Cadherin composition impacts mesothelial clearance in intact tissue explants and organotypic meso-mimetic cultures.
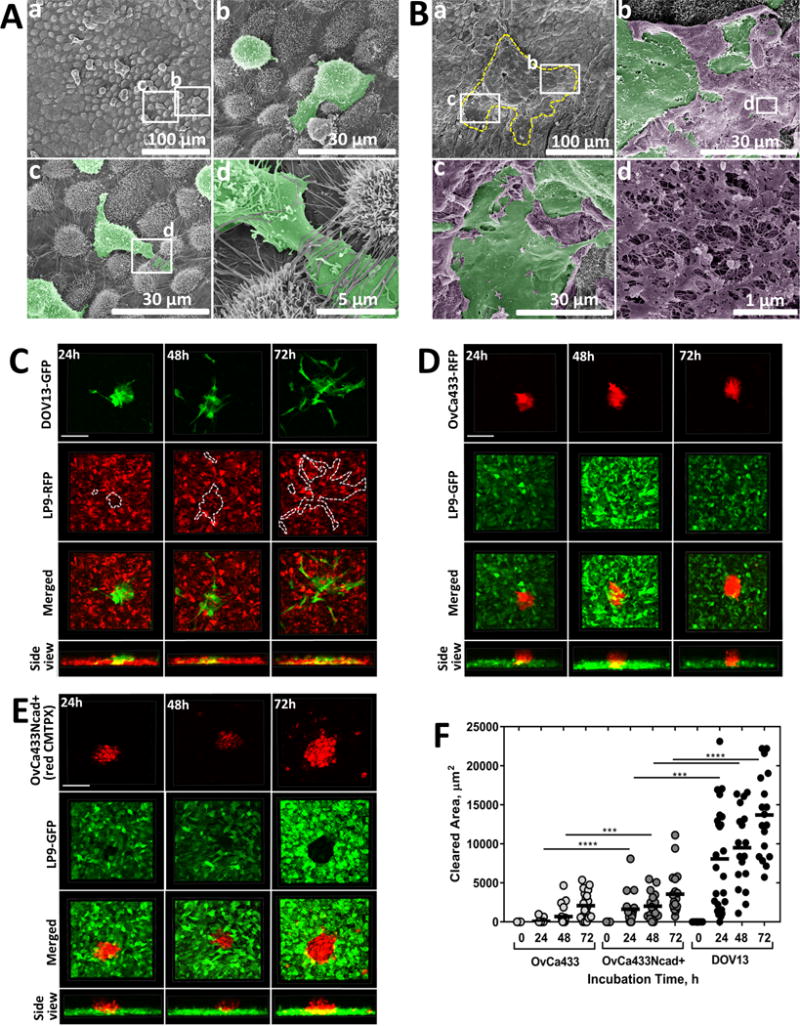
(A–B) Ex vivo peritoneal adhesion and mesothelial clearance. Murine peritoneal tissue explants were dissected and pinned ‘mesothelium-side-up’ on optically clear silastic resin as described in Methods and incubated with (A) DOV13 cells (2h) or (B) DOV13 MCAs (4h). Explants were rinsed with ice-cold PBS 3 × 3 min, subjected to SEM processing and imaged with FEI-Magellan 400 field emission microscope (scale bars as indicated). For clarity of visualization, DOV13 cells (Ab–d) and MCAs (Bb–c) are pseudo-colored green while ruptured mesothelial cells are pseudo-colored purple (Bb–d). The yellow dashed line in (Ba) depicts the borders of the dispersed MCA in the lower magnification image while the white rectangles identify areas magnified in (Bb) and (Bc), respectively. The white rectangle in panel (Bb) identifies the area of ruptured mesothelium magnified in (Bd). (C–E) Imaging of MCA mesothelial clearance using in vitro meso-mimetic cultures. (C) DOV13-GFP, (D) OvCa433-RFP or (E) CMTPX-stained OvCa433Ncad+ MCAs were applied on top of (C) RFP-tagged or (D–F) GFP-tagged LP9 mesothelial cell layers grown in 35mm glass-bottom dishes to 100% confluence, and subsequent MCA dispersal and mesothelial clearance activity (indicated by dotted line for DOV13 MCAs, visually detectable for OvCa433 and OvCa433Ncad+ MCAs) were observed using confocal microscopy during the course of incubation. (F) Mesothelial clearance was quantified in terms of the LP9 cell area cleared by the individual EOC MCAs for different incubation time points (M±SD, N=16); ***p<0.001, ****p<0.0001, Mann-Whitney U test; statistical significance is shown between different cell types.
DISCUSSION
Metastatic dissemination of EOC is largely mediated by direct extension of cells into the peritoneal cavity, wherein they survive suspended in ascites fluid as individual cells and MCAs that subsequently adhere to peritoneal mesothelial cells, induce mesothelial cell retraction and penetrate the underlying 3D collagen matrix to establish secondary lesions (5,7,10,11). Human ascites-derived cells and MCAs exhibit a range of adhesive behaviors, suggesting that specific cellular phenotypes dictate metastatic propensity. Using multiple in vitro, ex vivo and organotypic cultures to model events in EOC metastasis, results from the current study show that both Ncad expression and MMP-mediated proteolysis are key determinants driving mesothelial clearance and matrix invasion by EOC cells, thus characterizing the aggressive phenotype. Importantly, blocking Ncad adhesion complexes and MMP-mediated matrix proteolysis significantly reduced mesothelial clearance ability and collagen invasion.
Experiments using live cell fluorescence microscopy to visualize movement through 3D collagen matrices by individual cells and MCAs with differentially expressed cadherins showed that only Ncad+ EOC cells and clusters effectively penetrate the collagen matrix and that this process is dependent upon Ncad-mediated junctional integrity. These results support the concept of collective cell behavior, which simultaneously integrates motility with matrix remodeling, and has been observed in normal morphogenesis as well as in cancer progression (25,26,36–41). Even when seeded at low density, Ncad+ cells migrate laterally prior to penetration of the collagen gel, wherein they retain loose ‘tip-like’ cell-cell junctions that involve only a fraction of the cell surface. Our in vivo results with ID8 cells (Ncad+) show that cells invading collagen-rich intra-peritoneal tissue also retain tip-like junctions. Similar morphological variants of collective migration were found in mesenchymal type tumors with conserved Ncad adherens junctions (26,38,40) and were histologically detected in in vivo studies of invasive epithelial cancer (42,43) and melanoma (44,45). Consistent with the current data, retention of Ecad expression is not associated with multicellular streaming (25,38). It is interesting to note that this is observed even in cells with the hybrid (dual cadherin) phenotype (24), suggesting that even low level Ecad expression provides morphological cues that modulate the collagen invasive phenotype. Based on these data, Ncad+ MCAs in ascites are predicted to more efficiently seed metastatic lesions due to the presence of pre-formed Ncad cell-cell junctions. This prediction is supported in pre-clinical murine models of EOC, wherein Ecad+ cells such as OvCa433 grow subcutaneously but do not seed intraperitoneal xenografts (data not shown and (46,47)) while Ncad+ lines including human DOV13, SKOV3ip, and OVCAR8 as well as murine ID8 cells readily anchor and proliferate, forming widely disseminated peritoneal implants (48–50). Furthermore, this observation is consistent with clinical data comparing primary tumors to metastatic lesions obtained from the same patient, showing prevalence of Ncad+ metastases (5).
In addition to Ncad, invasion of collagen matrices is also impacted by MMP expression. EOCs express the interstitial collagenase MT1-MMP (15–19), and expression is enhanced in peritoneal metastases relative to paired primary tumors (19). In this study, Ncad+ DOV13 cells also endogenously express MT1-MMP, which catalyzes collagenolysis as well as matrix reorganization to facilitate movement of migrating cancer cells (15,17,51). It is interesting to note that MT1-MMP mRNA and protein levels are upregulated in MCA cultures (30), consistent with our current results that show enhanced collagen remodeling by MCAs. Nevertheless, inhibition of MMP activity with the broad spectrum small molecule GM6001 significantly decreased collagen peri-cellular collagen clearance, but only partially suppressed collagen matrix invasion by both individual cells and MCAs. It was recently shown that MT1-MMP-mediated proteolysis is critical for “leader-follower” behavior in heterotypic co-cultures of non-invasive normal breast epithelial MCF-10A cells and malignant MDA-MB-231 breast cancer cells. MCF-10A cells were shown to “follow” the MDA-MB-231 “leader” cells through invasive strands in collagen gels, and addition of GM6001 abolished this behavior (33). This is in contrast to our co-culture experiments, wherein non-invasive Ecad+ OvCa433 cells segregated away from invasive Ncad+ DOV13 cells and did not “follow” tracks formed by DOV13 cells through the collagen gel. Together these data suggest that MT1-MMP dependent proteolysis and collagen reorganization are not wholly sufficient for metastatic anchoring in sub-mesothelial collagen in peritoneal metastases. This is consistent with our in vivo data using ID8 (Ncad+) cells that invade intra-peritoneally but do not express MT1-MMP, and suggest that factors in addition to MMP activity contribute to invasion.
Prior to engaging collagen, metastasizing EOC cells first encounter the mesothelial monolayer that lines the peritoneal cavity. Consistent with previous findings (52), our results show that Ncad+ MCAs exhibit enhanced mesothelial clearance relative to Ecad+ aggregates. Furthermore, acquisition of Ncad augments this process, even in hybrid Ecad/Ncad-expressing cells. Contrary to previous studies, however, our results using intact peritoneal tissue explants reveal a distinct fate for peritoneal mesothelial cells. While in vitro studies of mesothelial clearance suggest temporary disassembly of mesothelial layers (11), our ex vivo data indicate that EOC MCAs physically rupture the underlying mesothelial cells, leading to a permanently compromised mesothelial monolayer with exposed sub-mesothelial collagen that provides an adhesive substratum to which EOC cells avidly adhere (19,53).
Our results using Ncad function blocking antibodies show delayed lateral dispersal and reduced collagen gel penetration relative to IgG-treated controls. While the underlying mechanism requires further investigation, it is interesting to speculate that the GC-4 antibody may alter cell-matrix mechanosensing through the HAVDI adhesive site in the Ncad EC1 ectodomain. HAVDI ligation alters signaling via the YAP/TAZ complex, known to be an important factor in the transduction of mechanical signals (54,55). Furthermore, HAVDI ligation attenuates YAP/TAZ cellular mechanosensing, reducing myosin IIA accumulation in focal adhesions and thereby decreasing traction force generation, cell contraction, motility and invasion (56,57). Additionally, collective cancer cell invasion may involve mechanotransduction at adherens junctions, triggering β-catenin recruitment and adhesion reinforcement, which may be particularly important when heterotypic junctions are formed (58). Alternatively, binding of GC-4 to Ncad may inhibit the cis interaction between Ncad and the fibroblast growth factor (FGF) receptor. This interaction has been shown to regulate FGF receptor dimerization and activation of signal transduction pathways, thereby promoting cell motility (59–62). Further studies are needed to probe the precise contribution of Ncad and its binding partners to mechanisms of EOC invasion.
Pre-clinical studies have been reported using several distinct Ncad antagonists including linear and cyclic peptides that harbor the HAV cell adhesion recognition motif (63,64) as well as function-blocking antibodies. For example, the pentapeptide ADH-1 (Exherin™, Adherex Technologies Inc., Durham, NC) is currently under active investigation in preclinical and clinical trials based on its ability to induce tumor cell apoptosis and disrupt vasculature (63–68). However, the very short half-life (~2.2h for Exherin (69)) is a drawback of this class of therapeutic agents for usage in vivo as it would require continuous intraperitoneal delivery of the drug. A recently reported linear peptide antagonist of Ncad, harboring a Trp residue in the second position (65,70), has been used in vitro to block aggregation of beads coated with the Ncad ectodomain and to inhibit endothelial cell tube formation; however its effect on tumor cell interaction with tissues and matrices has not been evaluated. Monoclonal antibodies against Ncad have been shown to inhibit heterotypic adhesion between Ncad+ invasive breast cancer cells and mammary stromal cells (71), to repress adhesion, invasion and proliferation of prostate cancer cells in vitro, and to mitigate prostate cancer xenograft growth and metastasis in vivo (72). Collectively, our findings delineate the importance of Ncad expression for effective peritoneal seeding by metastasizing EOC cells and provide the rationale to support future pre-clinical studies using intraperitoneal delivery of Ncad blocking molecules as a therapeutic strategy to suppress EOC metastatic anchoring.
MATERIALS AND METHODS
Cell Lines
The EOC cell lines OvCa433 and DOV13 were kindly provided by Dr. Robert Bast (M.D. Anderson Cancer Center, Houston, TX). Cells were maintained in Minimal Essential Medium (MEM; Gibco, Big Cabin, OK) containing 10% Fetal Bovine Serum (FBS; Gibco), 1% Non-Essential Amino Acids (Corning Cellgro, Manassas, VA), 1% Penicillin/Streptomycin (Lonza, Allendale, NJ), 1% Sodium Pyruvate (Corning Cellgro), 0.1% Amphotericin B (Cellgro), and supplemented with 10μg/ml Insulin (Gibco) for DOV13 cell medium only. The human peritoneal mesothelial cell line LP9 was obtained from Coriell NIA Aging Cell Repository (Camden, NJ) and maintained in a 1:1 ratio of M199 and Ham F12 media (Gibco), supplemented with 15% FBS, 1% Penicillin/Streptomycin, 1% HEPES (Gibco), 1% GlutaMAX (Gibco), 10ng/ml epidermal growth factor (EGF; Sigma, St Louis, MO), 400ng/ml hydrocortisone (Sigma). ID8 murine EOC cells were obtained from Dr. Katherine Roby (University of Kansas Medical Center, Kansas City, KS). Cells were maintained in Dulbecco’s Modification of Eagle’s Medium (DMEM; Corning Cellgro) containing 4% FBS, 1% Penicillin/Streptomycin, and supplemented by 5 μg/mL Insulin, 5 μg/mL Transferrin, and 5 ng/mL Sodium Selenite (1% ITS; Sigma). Cell lines were tested and authenticated by Genetica DNA Laboratories using short tandem repeat (STR) DNA profiling and were found to be >95% concordant. Cells tested negative for mycoplasma in 2015. Generation of stable OvCa433Ncad+ (OvCa433 that express N-cadherin in addition to endogenous E-cadherin) and OvCa433MT (OvCa433 that express catalytically active MT1-MMP) cell lines has been described previously (24,30). Transfected cells were maintained in medium supplemented with Geneticin (600μg/ml; Enzo Life Sciences, Farmingdale, NY) and routinely sorted every five passages on a BD FACSAria III cell sorter to ensure continuous protein expression.
RFP lentiviral vector (GenTarget, San Diego, CA) was used to generate fluorescently tagged OvCa433-RFP and LP9-RFP cell lines. GFP lentiviral vector (AddGene, Cambridge, MA) was utilized to create DOV13-GFP and LP9-GFP stable cell lines. Lentiviral transductions were performed according to manufacturers’ protocols and successfully tagged cells were further selected via BD FACSAria III cell sorter. Generation of fluorescently labeled ID8-RFP cell line has been previously described (49). Short-term fluorescent labeling of cells was performed as described (24,30).
Fluorescence/SHG Imaging of Murine Metastatic Lesions
Female C57Bl/6 mice (Mus musculus) of varying ages (6–22mo) (n=12, Jackson Laboratories, Bar Harbor, ME) were injected intraperitoneally with 1–10×106 ID8-RFP murine EOC cells as previously described (49). Normal collagen was also examined in tumor free mice (n=30) as a component of a larger study. No randomization method was used. Animal procedures were carried out according to the regulations of the University of Notre Dame Animal Care and Use Committee. Mice were sacrificed by CO2 inhalation followed by cervical dislocation at 8–10 weeks post-injection. The parietal peritoneum was dissected, rinsed with PBS and placed between coverslips for imaging with the mesothelium side facing the objective (25X XLPlanN, 1.05na WATER) of the 2-Photon confocal microscope (Olympus FV1000, software FLUOVIEW FV1000). Using a Mai Tai DeepSee titanium-sapphire 690–1040nm laser, ID8 metastatic implants and peritoneal collagen were visualized by the RFP and SHG signals, respectively. The investigator was not blinded during animal study. Areas of peritoneum with collagen-tumor interactions present were specifically imaged. At 12% laser power, the 2-photon laser was set to 860 nm and emission was simultaneously collected at 425–465 nm and 575–625 nm for SHG and RFP, respectively.
MCA Formation via Hanging Drop Method
EOC cells were harvested, centrifuged and re-suspended in fresh medium at 5,000 cells/ml. Droplets (20μl) were seeded on inner surface of a 150 × 25 mm tissue culture dish lid (100 cells/droplet). PBS was added to the lower dish and the lid was gently inverted atop the dish. Cells were incubated at 37°C for 48 hours and MCA formation was confirmed by light microscopy.
Invasion of 3D collagen gels
3D collagen gels were constructed by diluting Rat Tail Collagen Type I (RTCI; BD Biosciences, San Jose, CA) with the corresponding complete cell medium to a 1.5mg/ml final concentration. This mixture (200ul) was placed into 20 mm glass wells of glass-bottom 35mm dishes (#D35-20-1-N, In Vitro Scientific, Mountain View, CA) and incubated at 37°C × 20–30 min until gel polymerization. For individual cell collagen invasion experiments, harvested cells were diluted to 50,000 cell/ml. For MCA collagen invasion experiments, MCAs (100cell/MCA) were collected from the dish lid after 48 hours of incubation into 15 ml falcon tubes and centrifuged at 2,200 rpm × 2 min at 22°C and MCA pellets were diluted in 200 μl of fresh full medium. Single cell or MCA suspensions (200 μl) were applied to solidified 3D collagen gels and continuously monitored using Nikon A1R-MP confocal microscope in reflectance mode for collagen imaging and fluorescence mode for cell imaging as detailed below. In a control experiment 31 μm beads (Green Fluorescent Polymer Microspheres, 10% CV, Duke Scientific Corp., Palo Alto, CA) were used to exclude potential effects of mechanical stresses due to gravity on invasion and lateral motion. In some experiments, DOV13 cells were pre-incubated with mouse monoclonal anti-Ncad antibody GC-4 (Sigma-Aldrich, St Louis, MO) or mouse isotype control IgG (ThermoFisher Scientific, Waltham, MA) (200 μg/ml) for 1h at 37°C prior to the start of collagen invasion experiments and replenished every 12h. Similarly, DOV13 cells were incubated with the broad spectrum MMP inhibitor GM6001 (EMD Millipore, Billerica, MA) (25μM) for 24h prior to invasion experiments or during hanging drop MCA formation. Fresh inhibitor was added every 24 hours.
Imaging of cellular dynamics in collagen gels was performed using a Nikon A1R-MP laser confocal microscope with ApoLWD 40× WI lambda-S DIC N2 water immersion objective lens to obtain a series of high resolution 100 μm thick z-stacks taken at time zero, continuously for 8h and then every 24h for 7–14 days at multiple (3–5) sites. An Argonne laser with a 488-nm wavelength and helium-neon laser beam with 594-nm wavelength were utilized for fluorescence confocal and reflectance imaging of tagged cells and unlabeled collagen, respectively. The voxel size of the obtained z-stacks was 0.2076×0.2076×0.6896 μm3. All cell invasion assays were performed using the microscope environmental chamber to maintain 37°C and 5% CO2.
Cell/MCA Peritoneal Adhesion and Ex Vivo Mesothelial Clearance
Cell/MCA adhesion to murine peritoneum and mesothelial clearance were assessed using ex vivo explants of intact peritoneal tissue (35,73). C57Bl/6 mice (Jackson Laboratories, Bar Harbor, ME) were dissected using a ventral midline incision; 4 peritoneal tissue pieces were removed and pinned to the bottom of 24-well dishes pre-coated with optically transparent silicone using Sylgard 184 Silicone Elastomer Kit (Fisher, Waltham, MA). Cells (200,000 cell/ml) or pre-formed MCAs (100cell/MCA) were applied atop murine peritoneal explants and incubated for 2 or 4h, as indicated. The assay was stopped with 3 × 3min ice-cold PBS washes and tissue explants were subjected to further SEM processing and imaging.
Scanning Electron Microscopy (SEM)
Murine peritoneal explants were fixed in a 2% Glutaraldehyde, 2% Paraformaldehyde in 0.1M Cacodylate buffer pH 7.35 primary fixative solution overnight at 4°C, followed by 3 × 20min washes in 0.1 Cacodylate buffer. Secondary processing was performed with 2% Osmium tetroxide in 0.1 Cacodylate buffer using a PELCO® EM Pro Microwave vacuum chamber. Samples were then washed with ultrapure water 3 × 5min, dehydrated in a series of increasing Ethanol concentrations (20%, 50%, 70%, 90%, 3 × 100%), followed by critical point drying using Tousimis-931 dryer, placed on carbon stubs, sputter coated with iridium, and examined under FEI-Magellan 400 field emission SEM. Electron micrograph false colorization was applied using Adobe Photoshop CC 2014 software.
MCA In Vitro Mesothelial Clearance
LP9-RFP or LP9-GFP human peritoneal mesothelial cells were grown on collagen coated (20ng/ml) glass surfaces to 100% confluence. DOV13-GFP, OvCa433-RFP or CMTPX-stained OvCa433Ncad+ MCAs (100cell/MCA) were applied atop the mesothelial cell layers. Subsequent dispersal of cancer cells/MCAs and mesothelial clearance activity were monitored using Nikon A1R-MP confocal microscope at 0, 24, 48 and 72 hour time points.
Image Analysis
Nikon software (NIS-Elements AR Analysis) software was used to analyze cell-collagen invasion and mesothelial clearance by a) quantifying the penetration depth of cells/MCAs, their lateral motility, the degree of deformation of collagen matrices in the collected z-stacks and b) the mesothelial clearance area, respectively. The penetration depth of cells or MCAs was defined as the distance from the lowest point on the bottom portion of cell/MCA surface to the top of collagen matrix. Cell lateral motility was quantified in terms of the cell/MCA average displacement per unit time in a horizontal plane from 8-hour time lapse by tracking the centers of each cell. For this, the coordinates of the center of each cell/MCA rc (xc, yc) were first determined from the collected z-stack images for each time point and the average speed was calculated as <dc/δt>, where dc is the displacement during the time interval δt. The degree of collagen remodeling was calculated in terms of the volume of voids surrounding the cell/MCAs inside the collagen matrix using confocal z-stack images acquired over the incubation period of 72 hours. The voids volume per cell or cluster was calculated as the sum of the collagen excluding areas measured for each z-slice, multiplied by the distance between slices, and divided by the number of cells/MCAs in the analyzed z-stack. Mesothelial clearance analysis in the confocal z-stack images in GFP and RFP channels was performed using NIS-Elements software by assessing the area cleared by EOC cells at the bottom of the LP9 layer. Fluorescence/SHG images of murine metastatic lesions were reconstructed and visualized using Fiji open source software.
Statistical Analysis
For each type of assay, statistical analysis using 3–6 z-stacks of confocal/reflectance microscopy images for a single sample in parallel multiple sample measurements was conducted. All data are obtained from at least 3 independent measurements and are expressed in terms of mean ± S.D. Statistical significance (defined as p<0.05) was calculated using a two-sided Mann-Whitney U test.
Supplementary Material
Acknowledgments
This work was supported in part by Research Grants RO1CA109545 (MSS) and RO1CA086984 (MSS) from the National Institutes of Health/National Cancer Institute; from the Leo and Anne Albert Charitable Trust (MSS); the Research Like a Champion grant (YK, RL); the Walther Cancer Foundation Seeding Research in Cancer grant (OK); NSF DGE1313583 (EL); U01-HL116330 (OK, MA), and University of Notre Dame Integrated Imaging Facility. We wish to thank Dr. Charles Tessier, Indiana University School of Medicine – South Bend, for assistance with second harmonic generation imaging microscopy.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
SUPPLEMENTARY INFORMATION
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015;65(1):5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Howlader N, Noone A, Krapcho M, Neyman N, Aminou R, Waldron W, et al. SEER cancer statistics review 1975–2008. Bethesda, MD: National Cancer Institute; 2011. p. 19. [Google Scholar]
- 3.Marcus CS, Maxwell GL, Darcy KM, Hamilton CA, McGuire WP. Current approaches and challenges in managing and monitoring treatment response in ovarian cancer. Journal of Cancer. 2014;5(1):25. doi: 10.7150/jca.7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auersperg N, Wong AST, Choi K, Kang SK, Leung PCK. Ovarian Surface Epithelium: Biology, Endocrinology, and Pathology. Endocr Rev. 2001;22(2):255–288. doi: 10.1210/edrv.22.2.0422. 04/01; 2015/03. [DOI] [PubMed] [Google Scholar]
- 5.Hudson LG, Zeineldin R, Stack MS. Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clin Exp Metastasis. 2008;25(6):643–655. doi: 10.1007/s10585-008-9171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levanon K, Crum C, Drapkin R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J Clin Oncol. 2008 Nov 10;26(32):5284–5293. doi: 10.1200/JCO.2008.18.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lengyel E. Ovarian cancer development and metastasis. The American journal of pathology. 2010;177(3):1053–1064. doi: 10.2353/ajpath.2010.100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pradeep S, Kim SW, Wu SY, Nishimura M, Chaluvally-Raghavan P, Miyake T, et al. Hematogenous metastasis of ovarian cancer: rethinking mode of spread. Cancer Cell. 2014;26(1):77–91. doi: 10.1016/j.ccr.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coffman LG, Burgos-Ojeda D, Wu R, Cho K, Bai S, Buckanovich RJ. New models of hematogenous ovarian cancer metastasis demonstrate preferential spread to the ovary and a requirement for the ovary for abdominal dissemination. Translational Research. 2016 doi: 10.1016/j.trsl.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niedbala MJ, Crickard K, Bernacki RJ. Interactions of human ovarian tumor cells with human mesothelial cells grown on extracellular matrix: an in vitro model system for studying tumor cell adhesion and invasion. Exp Cell Res. 1985;160(2):499–513. doi: 10.1016/0014-4827(85)90197-1. [DOI] [PubMed] [Google Scholar]
- 11.Iwanicki MP, Davidowitz RA, Ng MR, Besser A, Muranen T, Merritt M, et al. Ovarian cancer spheroids use myosin-generated force to clear the mesothelium. Cancer Discov. 2011 Jul;1(2):144–157. doi: 10.1158/2159-8274.CD-11-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burleson KM, Casey RC, Skubitz KM, Pambuccian SE, Oegema TR, Jr, Skubitz APN. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol Oncol. 2004 Apr;93(1):170–181. doi: 10.1016/j.ygyno.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 13.Burleson KM, Boente MP, Pambuccian SE, Skubitz AP. Disaggregation and invasion of ovarian carcinoma ascites spheroids. J Transl Med. 2006 Jan 24;4:6. doi: 10.1186/1479-5876-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lautscham LA, Kämmerer C, Lange JR, Kolb T, Mark C, Schilling A, et al. Migration in Confined 3D Environments Is Determined by a Combination of Adhesiveness, Nuclear Volume, Contractility, and Cell Stiffness. Biophys J. 2015;109(5):900–913. doi: 10.1016/j.bpj.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9(8):893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 16.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res. 2008 Sep 15;68(18):7247–7249. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 17.Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three-dimensional extracellular matrix. Cell. 2003;114(1):33–45. doi: 10.1016/s0092-8674(03)00513-0. [DOI] [PubMed] [Google Scholar]
- 18.Ellerbroek SM, Wu YI, Overall CM, Stack MS. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J Biol Chem. 2001 Jul 6;276(27):24833–24842. doi: 10.1074/jbc.M005631200. [DOI] [PubMed] [Google Scholar]
- 19.Barbolina MV, Adley BP, Ariztia EV, Liu Y, Stack MS. Microenvironmental regulation of membrane type 1 matrix metalloproteinase activity in ovarian carcinoma cells via collagen-induced EGR1 expression. J Biol Chem. 2007 Feb 16;282(7):4924–4931. doi: 10.1074/jbc.M608428200. [DOI] [PubMed] [Google Scholar]
- 20.Sodek K, Ringuette M, Brown T. MT1-MMP is the critical determinant of matrix degradation and invasion by ovarian cancer cells. Br J Cancer. 2007;97(3):358–367. doi: 10.1038/sj.bjc.6603863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.King SM, Hilliard TS, Wu LY, Jaffe RC, Fazleabas AT, Burdette JE. The impact of ovulation on fallopian tube epithelial cells: evaluating three hypotheses connecting ovulation and serous ovarian cancer. Endocr Relat Cancer. 2011 Sep 20;18(5):627–642. doi: 10.1530/ERC-11-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poncelet C, Cornelis F, Tepper M, Sauce E, Magan N, Wolf JP, et al. Expression of E-and N-cadherin and CD44 in endometrium and hydrosalpinges from infertile women. Fertil Steril. 2010;94(7):2909–2912. doi: 10.1016/j.fertnstert.2010.04.055. [DOI] [PubMed] [Google Scholar]
- 23.Ahmed N, Thompson EW, Quinn MA. Epithelial-mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J Cell Physiol. 2007;213(3):581–588. doi: 10.1002/jcp.21240. [DOI] [PubMed] [Google Scholar]
- 24.Klymenko Y, Johnson J, Bos B, Lombard R, Campbell L, Loughran E, et al. Heterogeneous cadherin expression and multi-cellular aggregate dynamics in ovarian cancer dissemination. Neoplasia. 2017 doi: 10.1016/j.neo.2017.04.002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14(8):777–783. doi: 10.1038/ncb2548. [DOI] [PubMed] [Google Scholar]
- 26.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nature reviews Molecular cell biology. 2009;10(7):445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 27.Roussos ET, Balsamo M, Alford SK, Wyckoff JB, Gligorijevic B, Wang Y, et al. Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J Cell Sci. 2011 Jul 1;124(Pt 13):2120–2131. doi: 10.1242/jcs.086231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davidson B, Goldberg I, Berner A, Nesland JM, Givant-Horwitz V, Bryne M, et al. Expression of membrane-type 1, 2, and 3 matrix metalloproteinases messenger RNA in ovarian carcinoma cells in serous effusions. Am J Clin Pathol. 2001 Apr;115(4):517–524. doi: 10.1309/B1YX-L8DB-TGY1-7905. [DOI] [PubMed] [Google Scholar]
- 29.Davidson B, Goldberg I, Gotlieb WH, Kopolovic J, Ben-Baruch G, Nesland JM, et al. The prognostic value of metalloproteinases and angiogenic factors in ovarian carcinoma. Mol Cell Endocrinol. 2002;187(1):39–45. doi: 10.1016/s0303-7207(01)00709-2. [DOI] [PubMed] [Google Scholar]
- 30.Moss NM, Barbolina MV, Liu Y, Sun L, Munshi HG, Stack MS. Ovarian cancer cell detachment and multicellular aggregate formation are regulated by membrane type 1 matrix metalloproteinase: a potential role in I.p. metastatic dissemination. Cancer Res. 2009 Sep 1;69(17):7121–7129. doi: 10.1158/0008-5472.CAN-08-4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Desai RA, Gao L, Raghavan S, Liu WF, Chen CS. Cell polarity triggered by cell-cell adhesion via E-cadherin. J Cell Sci. 2009 Apr 1;122(Pt 7):905–911. doi: 10.1242/jcs.028183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392–1400. doi: 10.1038/ncb1658. [DOI] [PubMed] [Google Scholar]
- 33.Carey SP, Starchenko A, McGregor AL, Reinhart-King CA. Leading malignant cells initiate collective epithelial cell invasion in a three-dimensional heterotypic tumor spheroid model. Clin Exp Metastasis. 2013;30(5):615–630. doi: 10.1007/s10585-013-9565-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davidowitz RA, Iwanicki MP, Brugge JS. In vitro mesothelial clearance assay that models the early steps of ovarian cancer metastasis. J Vis Exp. 2012 Feb 17;(60) doi: 10.3791/3888. pii: 3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lengyel E, Burdette J, Kenny H, Matei D, Pilrose J, Haluska P, et al. Epithelial ovarian cancer experimental models. Oncogene. 2014;33(28):3619–3633. doi: 10.1038/onc.2013.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedl P, Hegerfeldt Y, Tusch M. Collective cell migration in morphogenesis and cancer. Int J Dev Biol. 2004;48:441–450. doi: 10.1387/ijdb.041821pf. [DOI] [PubMed] [Google Scholar]
- 37.Friedl P. Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol. 2004;16(1):14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 38.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 39.Hegerfeldt Y, Tusch M, Brocker EB, Friedl P. Collective cell movement in primary melanoma explants: plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 2002 Apr 1;62(7):2125–2130. [PubMed] [Google Scholar]
- 40.Ilina O, Friedl P. Mechanisms of collective cell migration at a glance. J Cell Sci. 2009 Sep 15;122(Pt 18):3203–3208. doi: 10.1242/jcs.036525. [DOI] [PubMed] [Google Scholar]
- 41.Wolf K, Friedl P. Molecular mechanisms of cancer cell invasion and plasticity. Br J Dermatol. 2006;154(s1):11–15. doi: 10.1111/j.1365-2133.2006.07231.x. [DOI] [PubMed] [Google Scholar]
- 42.Bell C, Stadler J, Waizbard E, Chaitchik S, Greif F. Tumor differentiation and histological type in human breast cancer. J Surg Oncol. 1986;31(1):39–43. doi: 10.1002/jso.2930310109. [DOI] [PubMed] [Google Scholar]
- 43.Page DL. Diagnostic histopathology of the breast. 1987 [Google Scholar]
- 44.Ackerman AB, Godomski J. Neurotropic malignant melanoma and other neurotropic neoplasms in the skin. Am J Dermatopathol. 1984 Summer;6(Suppl):63–80. [PubMed] [Google Scholar]
- 45.Day CL, Jr, Harrist TJ, Gorstein F, Sober AJ, Lew RA, Friedman RJ, et al. Malignant melanoma. Prognostic significance of “microscopic satellites” in the reticular dermis and subcutaneous fat. Ann Surg. 1981 Jul;194(1):108–112. doi: 10.1097/00000658-198107000-00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takai N, Jain A, Kawamata N, Popoviciu LM, Said JW, Whittaker S, et al. 2C4, a monoclonal antibody against HER2, disrupts the HER kinase signaling pathway and inhibits ovarian carcinoma cell growth. Cancer. 2005;104(12):2701–2708. doi: 10.1002/cncr.21533. [DOI] [PubMed] [Google Scholar]
- 47.Shaw TJ, Senterman MK, Dawson K, Crane CA, Vanderhyden BC. Characterization of intraperitoneal, orthotopic, and metastatic xenograft models of human ovarian cancer. Molecular Therapy. 2004;10(6):1032–1042. doi: 10.1016/j.ymthe.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Afzal S, Lalani E, Poulsom R, Stubbs A, Rowlinson G, Sato H, et al. MT1-MMP and MMP-2 mRNA expression in human ovarian tumors: Possible implications for the role of desmoplastic fibroblasts. Hum Pathol 1998. 1998 Feb;29(2):155–165. doi: 10.1016/s0046-8177(98)90226-x. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Metzinger MN, Lewellen KA, Cripps SN, Carey KD, Harper EI, et al. Obesity Contributes to Ovarian Cancer Metastatic Success through Increased Lipogenesis, Enhanced Vascularity, and Decreased Infiltration of M1 Macrophages. Cancer Res. 2015 Dec 1;75(23):5046–5057. doi: 10.1158/0008-5472.CAN-15-0706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitra AK, Davis DA, Tomar S, Roy L, Gurler H, Xie J, et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol Oncol. 2015 Aug;138(2):372–377. doi: 10.1016/j.ygyno.2015.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fraley SI, Wu PH, He L, Feng Y, Krisnamurthy R, Longmore GD, et al. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci Rep. 2015 Oct 1;5:14580. doi: 10.1038/srep14580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Davidowitz RA, Selfors LM, Iwanicki MP, Elias KM, Karst A, Piao H, et al. Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J Clin Invest. 2014 Jun;124(6):2611–2625. doi: 10.1172/JCI69815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moser TL, Pizzo SV, Bafetti LM, Fishman DA, Stack MS. Evidence for preferential adhesion of ovarian epithelial carcinoma cells to type I collagen mediated by the αA2β1 integrin. International Journal of Cancer. 1996;67(5):695–701. doi: 10.1002/(SICI)1097-0215(19960904)67:5<695::AID-IJC18>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 54.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154(5):1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 55.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 56.Cosgrove BD, Mui KL, Driscoll TP, Caliari SR, Mehta KD, Assoian RK, et al. N-cadherin adhesive interactions modulate matrix mechanosensing and fate commitment of mesenchymal stem cells. Nature Materials. 2016 doi: 10.1038/nmat4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pasapera AM, Plotnikov SV, Fischer RS, Case LB, Egelhoff TT, Waterman CM. Rac1-dependent phosphorylation and focal adhesion recruitment of myosin IIA regulates migration and mechanosensing. Current Biology. 2015;25(2):175–186. doi: 10.1016/j.cub.2014.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Labernadie A, Kato T, Brugués A, Serra-Picamal X, Derzsi S, Arwert E, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19(3):224–237. doi: 10.1038/ncb3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Utton MA, Eickholt B, Howell FV, Wallis J, Doherty P. Soluble N‐cadherin stimulates fibroblast growth factor receptor dependent neurite outgrowth and N‐cadherin and the fibroblast growth factor receptor co‐cluster in cells. J Neurochem. 2001;76(5):1421–1430. doi: 10.1046/j.1471-4159.2001.00140.x. [DOI] [PubMed] [Google Scholar]
- 60.Takeda H, Shimoyama Y, Nagafuchi A, Hirohashi S. E-cadherin functions as a cis-dimer at the cell–cell adhesive interface in vivo. Nature Structural & Molecular Biology. 1999;6(4):310–312. doi: 10.1038/7542. [DOI] [PubMed] [Google Scholar]
- 61.Nieman MT, Prudoff RS, Johnson KR, Wheelock MJ. N-cadherin promotes motility in human breast cancer cells regardless of their E-cadherin expression. J Cell Biol. 1999 Nov 1;147(3):631–644. doi: 10.1083/jcb.147.3.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Doherty P, Walsh FS. CAM-FGF receptor interactions: a model for axonal growth. Molecular and Cellular Neuroscience. 1996;8(2):99–111. doi: 10.1006/mcne.1996.0049. [DOI] [PubMed] [Google Scholar]
- 63.Mariotti A, Perotti A, Sessa C, Rüegg C. N-cadherin as a therapeutic target in cancer. Expert Opin Investig Drugs. 2007;16(4):451–465. doi: 10.1517/13543784.16.4.451. [DOI] [PubMed] [Google Scholar]
- 64.Blaschuk OW, Devemy E. Cadherins as novel targets for anti-cancer therapy. Eur J Pharmacol. 2009 Dec 25;625(1–3):195–198. doi: 10.1016/j.ejphar.2009.05.033. [DOI] [PubMed] [Google Scholar]
- 65.Blaschuk OW. N-cadherin antagonists as oncology therapeutics. Philos Trans R Soc Lond B Biol Sci. 2015 Feb 5;370(1661):20140039. doi: 10.1098/rstb.2014.0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shintani Y, Fukumoto Y, Chaika N, Grandgenett PM, Hollingsworth MA, Wheelock MJ, et al. ADH‐1 suppresses N‐cadherin‐dependent pancreatic cancer progression. International journal of cancer. 2008;122(1):71–77. doi: 10.1002/ijc.23027. [DOI] [PubMed] [Google Scholar]
- 67.Sadler NM, Harris BR, Metzger BA, Kirshner J. N-cadherin impedes proliferation of the multiple myeloma cancer stem cells. Am J Blood Res. 2013;3(4):271–285. [PMC free article] [PubMed] [Google Scholar]
- 68.Augustine CK, Yoshimoto Y, Gupta M, Zipfel PA, Selim MA, Febbo P, et al. Targeting N-cadherin enhances antitumor activity of cytotoxic therapies in melanoma treatment. Cancer Res. 2008 May 15;68(10):3777–3784. doi: 10.1158/0008-5472.CAN-07-5949. [DOI] [PubMed] [Google Scholar]
- 69.Final clinical and pharmacokinetic (PK) results from a phase 1 study of the novel N-cadherin (N-cad) antagonist, Exherin (ADH-1), in patients with refractory solid tumors stratified according to N-cad expression. ASCO Annual Meeting Proceedings. 2006 [Google Scholar]
- 70.Devemy E, Blaschuk OW. Identification of a novel N-cadherin antagonist. Peptides. 2008;29(11):1853–1861. doi: 10.1016/j.peptides.2008.06.025. [DOI] [PubMed] [Google Scholar]
- 71.Hazan RB, Kang L, Whooley BP, Borgen PI. N-Cadherin Promotes Adhesion Between Invasive Breast Cancer Cells and the Stroma. Cell Communication & Adhesion. 1997 Jan 01;4(6):399–411. doi: 10.3109/15419069709004457. [DOI] [PubMed] [Google Scholar]
- 72.Tanaka H, Kono E, Tran CP, Miyazaki H, Yamashiro J, Shimomura T, et al. Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat Med. 2010;16(12):1414–1420. doi: 10.1038/nm.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bruney L, Conley KC, Moss NM, Liu Y, Stack MS. Membrane-type I matrix metalloproteinase-dependent ectodomain shedding of mucin16/CA-125 on ovarian cancer cells modulates adhesion and invasion of peritoneal mesothelium. Biol Chem. 2014;395(10):1221–1231. doi: 10.1515/hsz-2014-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.