

Abstract
NAD+ is a dinucleotide cofactor with the potential to accept electrons in a variety of cellular reduction-oxidation (redox) reactions. In its reduced form, NADH is a ubiquitous cellular electron donor. NAD+, NADH, and the NAD+/NADH ratio have long been known to control the activity of several oxidoreductase enzymes. More recently, enzymes outside those participating directly in redox control have been identified that sense these dinucleotides, including the sirtuin family of NAD+-dependent protein deacylases. In this review, we highlight examples of non-redox enzymes that are controlled by NAD+, NADH, or NAD+/NADH. In particular, we focus on the sirtuin family and assess the current evidence that the sirtuin enzymes sense these dinucleotides and discuss the biological conditions under which this might occur; we conclude that sirtuins sense NAD+, but neither NADH nor the ratio. Finally, we identify future studies that might be informative to further interrogate physiological and pathophysiological changes in NAD+ and NADH, as well as enzymes like sirtuins that sense and respond to redox changes in the cell.
Keywords: Sirtuins, Nicotinamide adenine dinucleotide, Metabolism, Mitochondria, Redox
Introduction
Nicotinamide adenine dinucleotide (NAD+) is a biological hydride acceptor that forms the reduced dinucleotide NADH. The NAD+/NADH dinucleotide pair is crucial for driving a wide range of reduction–oxidation (redox) reactions in cellular bioenergetics. Additionally, NAD+ is the precursor for the phosphorylated dinucleotides NADP+ and NADPH, which play a key role in protecting cells from reactive oxygen species (ROS) and in several cellular biosynthetic pathways.
In mammals, NAD+ is synthesized by two discrete routes: the deamidated and amidated pathways (Figure 1), which has been reviewed recently [1, 2]. Briefly, the deamidated pathway generates NAD+ via de novo synthesis from tryptophan generates quinolinic acid which is converted to nicotinic acid mononucleotide (NAMN). Alternatively, the Preiss-Handler pathway uses nicotinic acid salvaged from the diet that is then phosphoribosylated to generate NAMN. In either case, NAMN is condensed with ATP by nicotinamide mononucleotide adenylyltransferase (NMNAT) enzymes to generate nicotinic acid dinucleotide (NAAD+). In the final step, conversion of the nicotinic acid moiety to nicotinamide by glutamine-dependent NAD+ synthetase (NADS) generates NAD+. In the amidated pathway – so called because the pyridine moiety is amidated, as opposed to carboxylated) – precursors such as vitamin B3 compounds nicotinamide (NAM) or nicotinamide riboside (NR), generate nicotinamide mononucleotide (NMN) by nicotinamide phosphoribosyltransferase (NAMPT) or nicotinamide riboside kinase (NRK), respectively. In the final step, NMN is again condensed with ATP by NMNAT enzymes to synthesize cellular NAD+ (Figure 1).
Figure 1.
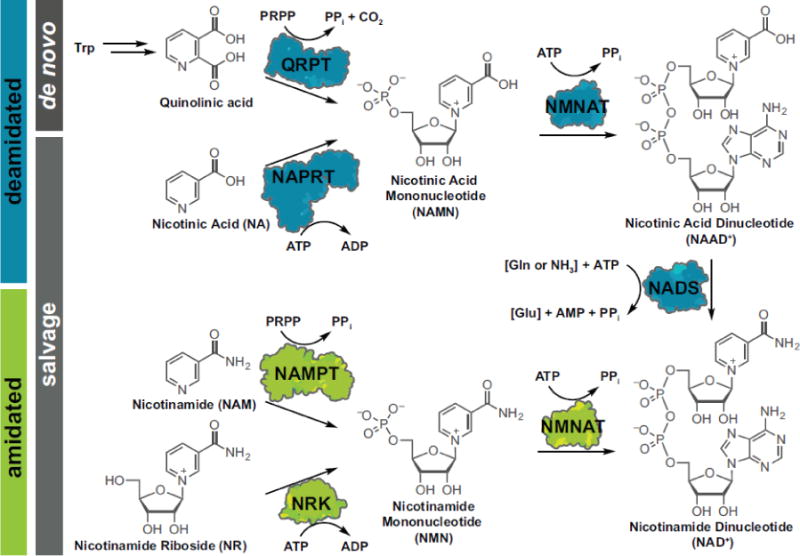
Overview of NAD+ metabolism. The deamidated (blue) and amidated (green) pathways are two discrete routes to synthesize intracellular NAD+. QRPT: Quinolinate Phosphoribosyltransferase; NAPRT: Nicotinate Phosphoribosyltransferase; NAMPT: Nicotinamide Phosphoribosyltransferase; NRK: Nicotinamide Riboside Kinases; NMNAT: Nicotinamide (Mono)nucleotide Adenylyltransferase; NADS: glutamine-dependent NAD+ synthetase.
The crucial role of cellular NAD+ is highlighted by the findings that ablation of the NAD+ biosynthetic enzyme NMNAT1 or NAMPT causes embryonic lethality [3, 4]. Indeed, NAD+ and/or NADP+ [NAD(P)+], are key metabolic cofactors. For example, oxidoreductases, such as lactate dehydrogenase and glutamate dehydrogenase, use NAD+ and NADH as substrates and are inherently sensitive to the redox state of the cell. More recently, the roles of NAD+ beyond redox are being studied. Here, we review the current state of knowledge of measuring intracellular NAD+ and NADH levels. We highlight key proteins and metabolic processes that are known to sense NAD+, NADH, or their ratio. In particular, we focus on the NAD+-dependent sirtuin family of protein deacylases, and consider the evidence for NAD+, NADH, or NAD+/NADH sensing. Finally, we identify outstanding questions and future directions to study physiological and pathophysiological changes in NAD+ and NADH, and the enzymes that sense them.
Physiological concentrations and states of NAD+ in vivo
Before assessing whether a protein senses the intracellular concentration of NAD+, NADH, or its ratio, knowing the concentrations of these dinucleotides is required. These dinucleotides can be either free or bound to protein, with three major pools compartmentalized in the nucleus, cytosol, and mitochondria. The total intracellular dinucleotide concentrations (free and bound) have been reported to be 1–3 mM [5], with an [NAD+]total/[NADH]total ratio of 2–10/1 (depending on species, cell type, and metabolic state) [5–8].
The first estimates of the free NAD+/NADH ratio were determined indirectly in the 1960s by Krebs and coworkers who measured the concentrations of the oxidized and reduced substrates of lactate dehydrogenase (LDH) and glutamate dehydrogenase (GDH) [9]. The substrates of these highly active dehydrogenases were considered in equilibrium with free NAD+ and NADH, so their ratios, together with the equilibrium constants, were used to calculate the free NAD+/NADH [9]. The NAD+/NADH ratio in liver cytoplasm and mitochondria from fed rats was found to be 725 and 8, respectively [9]. The ratios changed to 208 and 10 in liver cytoplasm and mitochondria, respectively, in diabetic rats. These findings were among the first to describe the existence of subcellular NAD+ pools that differ drastically and which do not change in the same direction in response to alterations in the metabolic state. These findings were an early indication that subcellular NAD+ and NADH pools are maintained at distinct equilibria.
Since that time, other methods have been used to investigate various parameters of the redox state. Using the same principle of indirect measurement as Krebs, hyperpolarized 13C-labelled glucose, which is converted to pyruvate and then lactate, was used in an NMR-based method to determine cytosolic [NAD+]free/[NADH]free ratios of 700–2500/1 in intact human cancer cells [10].
By taking advantage of the fluorescent properties of NAD(P)H, combined levels of these reduced dinucleotides can be directly detected in cellular compartments. Furthermore, the fraction of dinucleotide unbound to protein can be determined by fluorescence lifetime measurements or two-photon fluorescence microscopy. For example, fluorescence intensity, lifetime, and anisotropy determined by two-photon excitation microscopy allowed measurement of changes in NAD(P)H levels in pancreatic islet beta-cells under varying glucose concentrations [11], and estimated [NADH]free in the nucleus to be ~130 nM [12]; interestingly, increased protein binding of NADH was observed upon hypoxia [13]. Recently, fluorescence lifetime imaging was extended to deconvolute NADH and NADPH fluorescence signals to determine concentrations of each individual reduced dinucleotide [14].
In the past few years, the use of circular permuted fluorescent proteins has emerged as a promising strategy for measuring free dinucleotide concentrations and ratios [15], and at least four such systems have been reported. In two parallel publications, the bacterial Rex proteins were used as dinucleotide binding domains in the “Peredox” and “Frex” systems, respectively [16, 17]. Peredox is a fusion between two T-Rex molecules and the green fluorescent protein T-Sapphire, that allows the measurement of NAD+/NADH ratios and this system reported a uniform signal for cytosolic and nuclear NAD+/NADH ratios [16]. Frex, which has no apparent affinity for NAD+, has been used to report relative amounts of NADH in the cytosolic, nuclear, and mitochondrial pools although mitochondrial and cytosolic versions of the sensor differ from each other, complicating the comparison of NADH in these compartments [17]. In a third study, a smaller sized biosensor called RexYFP was generated by fusing YFP to a single molecule of T-rex. RexYFP reports the NAD+/NADH ratio in live cells in the cytosolic and mitochondrial compartments [18]. Using this same strategy, a genetically encoded fluorescent biosensor was developed for directly monitoring free NAD+ concentrations in subcellular compartments [19]. Using this sensor, systematic depletion of the NMNAT enzymes that catalyze the final step of NAD+ biosynthesis revealed specific mechanisms for maintaining mitochondrial, but not cytosolic, NAD+ concentrations. Together, these tools allowed selective monitoring of total cellular and compartment-specific responses of NAD+ and NADH, and revealed free NAD+ concentrations in cytosolic, nuclear, and mitochondrial compartments of 106 μM, 109 μM, and 230 μM, respectively.
One major challenge of measuring total NAD+ and NADH concentrations directly in subcellular compartments is the technical requirement of purifying subcellular compartments by fractionating cell or tissue lysates. During the purification procedure, alterations in NAD+ and NADH concentrations are expected. This limitation was recently overcome, at least in part, by leveraging affinity-tag-based purification of mitochondrial organelles, prior to metabolite extraction and analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS) [20]. In line with earlier findings, this study reported a total mitochondrial NAD+ concentration of 818 μM and an NAD+/NADH ratio of 111.
When integrated (Figure 2), these studies estimate cytosolic, nuclear, and mitochondrial [NAD+]free to be 106 μM, 109 μM, and 230 μM, respectively [19]; cytosolic and mitochondrial [NADH]free to be ~130 nM and ~30 μM, respectively [12, 17]; and a cytosolic ratio of [NAD+]free/[NADH]free 70–2500/1 (depending on cell type and growth media) [10, 16]. One major caveat to these measurements is the ambiguity surrounding free versus bound dinucleotides. Some studies have estimated 40–90% of the total NADH to be bound to proteins [11, 12, 21, 22]. However, direct biochemical measurement of total tissue NAD+ and NADH content often does not differentiate between the free and bound forms of the nucleotides. Since the majority of cellular NAD+ and NADH appears to be bound to protein, and therefore potentially unavailable to influence redox state, methods used to determine free NAD+ and NADH levels and free NAD+/NADH could give varying results.
Figure 2.
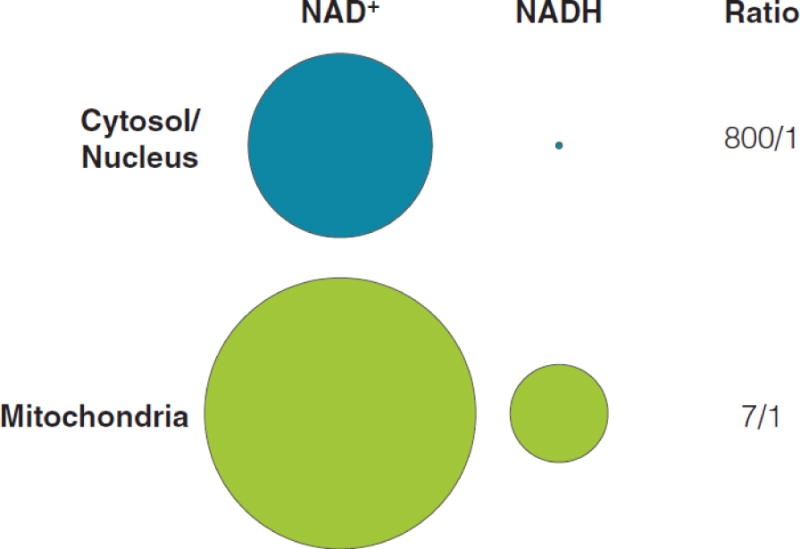
Summary sub-cellular dinucleotide pools and ratios. Based on published values, concentrations of NAD+ or NADH were converted to circle diagrams where the area of each shape accurately reflect the relative size of the nucleotide pool and the relationship between the sizes.
Despite this challenge, emerging data support the notion that cells partition discrete dinucleotide pools. Because NAD+ and NADH pass unassisted through nuclear pores, the free concentrations of each, as well as the NAD+/NADH ratio, is predicted to be similar in the nuclear and cytosolic cellular compartments; the data summarized above support this prediction. In contrast, the mitochondrial pool is discrete. Although the outer mitochondrial membrane is porous and freely permeable to small molecules, the inner membrane permeability is strictly regulated by the mitochondrial permeability transition pore (mtPTP) and a family of specific membrane transporters called solute carriers (SLC); neither NAD+ nor NADH freely traverses the inner membrane. The mitochondrial NAD+ pool also responds differently from that of the nuclear and cytosolic pools. Mitochondria maintain NAD+ levels during genotoxic stress and promote cell survival even when NAD+ in the nucleus and cytosol have fallen well below normal physiological levels [23]. Additionally, chronic treatment of FK866 – a potent inhibitor of NAMPT – was found to deplete cytosolic but not mitochondrial NAD+ [24]. Thus, partitioning discrete dinucleotide pools allows cells to maintain NAD+ levels under environmental, pharmacological, or genetic stressors.
Regulation by NAD+, NADH, or their ratio
Almost 30,000 proteins in the UniProtKB/Swiss-Prot database [25] are annotated as having NAD(P)+ binding sites [26], highlighting an essential and ubiquitous role for this metabolite. Beyond hydride transfer in redox reactions, proteins involved in transcription or cellular signaling, for example, have been identified that can sense NAD+ or NADH levels. Below, we highlight a few salient, well-studied examples.
C-terminal Binding Protein (CtBP)
CtBP is a transcription corepressor that interacts with a several cellular and viral transcriptional repressors, with the overall effect of silencing the expression of specific genes involved in development, cell cycle regulation, and cellular transformation. CtBP contains a domain resembling a Rossman fold [27] that binds NADH and NAD+ with affinities of 66 nM and ~10 μM, respectively [28], both within the estimated range of nuclear dinucleotide concentrations. Crystal structures of CtBP in complex with NAD(H), together with mutagenesis, binding, and modeling data, indicate that NAD(H) binding promotes stabilization of the dimeric form of CtBP [29]. The dimeric form is thought to be critical for corepressor activity, as it provides a stable scaffold for bridging repressors and their targets. In line with this notion, CtBP binding to target transcriptional repressors is enhanced by NAD+ and NADH, with NADH being three orders of magnitude more effective, and conditions that increase NADH levels potentiate CtBP-mediated repression in vivo [12]. The E-cadherin gene, one well-studied target of CtBP repression, suppresses tumorigenesis by restricting tumor cell motility and invasion. CtBP-mediated repression of E-cadherin is enhanced by hypoxia [12]. Since tumor cells are often hypoxic, with increased NADH/NAD+ ratios, the parallel stimulation of CtBP activity by NADH is thought to contribute to tumor invasion.
Clock
CLOCK and its functional analog NPAS2 (Neuronal PAS domain protein 2) both form heterodimers with BMAL1, are part of the transcriptional feedback system controlling circadian rhythms, and have been described as NAD(P)+/NAD(P)H redox sensors [30, 31]. Both NPAS2:BMAL1 and BMAL1:BMAL1 dimers can bind DNA. At NAD(P)H concentrations below 1 mM, the transcription factor complex binding to DNA is primarily the BMAL1 homodimer, whereas incubation with NAD(P)H induces NPAS2:BMAL1 heterodimer DNA binding. Furthermore, NAD(P)+ prevents NPAS2:BMAL1 binding to DNA by modulating the Kd and Bmax of DNA binding to NPAS2:BMAL1 in a redox level-dependent manner [31]. In contrast, later report did not find differential DNA binding of the bHLH domain of CLOCK in isothermal calorimetry experiments with or without dinucleotides regardless of oxidation or phosphorylation levels [32]. Importantly, the concentration of free NAD+ is estimated to be in the micromolar range, which calls into question the biological conditions under which NAD(P)+/NAD(P)H might be sensed. Overall, the mode and manner by which the dinucleotides modulate DNA binding remain elusive [31].
NmrA like redox sensor 1 (NMRAL1)
A similar redox sensor protein has also been described for the NADP+/NADPH redox pair. NMRAL1 (also known as HSCARG 1) is a protein resembling short-chain dehydrogenases but is devoid of enzyme activity. NMRAL1 contains an allosteric binding site for NADPH, showing an approximately 30-fold higher affinity for NADPH compared to NADP+ (Kd 0.486 and 13.5 mM for NADPH and NADP+, respectively) [33, 34]. Under normal NADPH concentrations, NMRAL1 exists as an asymmetric dimer, with one monomer binding a single NADPH molecule. Dimerization requires NADPH binding, and thus decreased NADPH levels (and increased NADP+/NADPH ratio) lead to a restructuring of the dinucleotide binding Rossmann fold. Under such conditions, the dimer dissociates, exposing a binding site for argininosuccinate synthetase (ASS1), the rate-limiting enzyme in nitric oxide synthesis. Association with ASS1 inhibits synthetase activity, leading to decreased nitric oxide production and thereby protecting cells from undergoing apoptosis [35].
Redox regulator (Rex)
Rex is a dimeric gene regulator of several respiratory pathways identified in Streptomyces coelicolor, but homologs are also found in other Gram-positive bacteria such as Staphylococcus aureus, Streptococcus pneumonia, Bacillus subtilis (B-Rex), and Thermus aquaticus (T-Rex) [36, 37]. As described above, Rex proteins were used to generate genetically encoded fluorescent NAD(H) biosensors. The protein contains an N-terminal winged helix DNA-binding domain and a C-terminal Rossmann fold, that can bind both NAD+ and NADH. Thus, Rex proteins serve as direct sensors of NAD+/NADH ratio. Allosteric binding of two NADH molecules to the T-Rex dimer results in a conformational change of the DNA binding domain, releasing the dimer from DNA [36]. The dinucleotide binding sites of a DNA-bound Rex dimer are placed within ~7Å of each other, and as a result of both charge repulsion of the pyridinium rings and rearrangement of nearby residues, only one NAD+ can complex a T-Rex dimer. However, DNA binding capability is retained, leading to the differential gene expression as a function of NAD+/NADH ratio [37].
Sirtuins and use of NAD+ as a co-substrate
Yeast Sir2 silences transcription [38, 39] and was originally shown to possess ADP-ribosyltransferase activity using NAD+ [40, 41]. Because overexpression of Sir2 also resulted in histone hypoacetylation [42, 43], NAD+ was hypothesized to act as co-factor for the proposed deacetylation activity of Sir2 [44]. Using purified, recombinant Sir2 enzyme and histone-based peptide substrates containing various N-acetylated lysine patterns, NAD+-dependent deacetylase activity was found in vitro. Furthermore, neither NADH, NADP+, nor NADPH possessed this property as a “co-factor” or inhibited the deacetylase activity. The stoichiometry of the deacetylation experiments suggested that Sir2 was acting catalytically and failure to inhibit this activity using potent HDAC inhibitor trichostatin A (TSA) further supported that Sir2 had a different mechanism of action than the Zn2+-dependent histone deacetylases. A mouse SIR2 homolog, mSir2, was also characterized and exhibited potent deacetylase activity as well as ADP-ribosyltransferase activity, showing that mammalian Sir2 proteins possess the same enzymatic activities [44].
In the time since the initial discovery, the human sirtuins (SIRT1–7) have been shown be NAD+-dependent deacylase enzymes [45]. NAD+ is a co-substrate in all of the different deacylation reactions catalyzed by sirtuins (Figure 3). The importance of NAD+ as a participating co-substrate may be demonstrated by applying a thin layer chromatography(TLC)-based assay that monitors the consumption of 32P-radio-labeled NAD+ and shows the appearance of radioactive spots corresponding to the O-acyl-ADP-ribose byproducts formed in the respective deacylation reactions [46]. This assay was applied for the discoveries of desuccinylation and deglutarylation by SIRT5 [47, 48], hydrolysis of hydrocarbon-based acyl groups of varying length by the action of SIRT1–3 and 6 [49, 50], and most recently for the discovery of SIRT4 deacylase activity [51].
Figure 3.
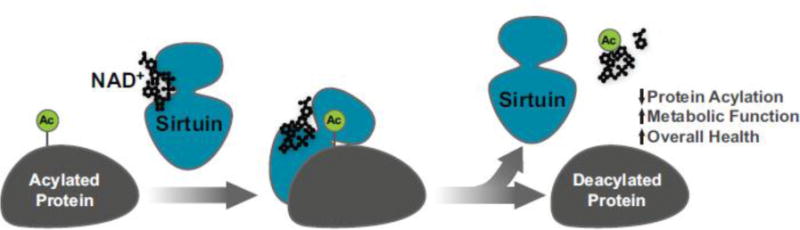
Schematic of sirtuin enzymatic activity. Mammalian sirtuins SIRT1-SIRT7 are NAD+-dependent protein deacylases on a wide range of cellular substrates. Using NAD+ as a co-substrate (black) the sirtuins (blue) catalyze a deacylation reaction (green) to form nicotinamide, O-acyl-ADP-ribose, and a deacylated protein.
Furthermore, Michaelis-Menten constants (Km) measured in vitro against peptide substrates for SIRT1 deacetylation (~750 μM) [50], SIRT2 demyristoylation (~40 μM) [52], as well as, SIRT5 desuccinylation (~150 μM) [53] and deglutarylation (~200 μM) [50] are in the range of measured free NAD+ concentrations, which supports the idea that sirtuin deacylase activity is regulated by NAD+ [19].
Sirtuins and their interactions with NADH
Further investigations into the initial observation that NADH did not significantly inhibit sirtuin deacetylation activity [44] further supported this notion. Using an enzymatically acetylated histone 4 peptide as a substrate, NADH (0–300 μM) inhibited deacetylase activity of yeast Sir2 and human SIRT1 via an apparent NAD+-competitive mechanism [5]. Further studies investigated a wider range of NAD+ analogs and derivatives, including NADH, nicotinamide, NMN+, and NAAD+ and found that yeast Sir2, HST2, and human SIRT2 mediated deacetylation of a H3K14 acetylated peptide were inhibited by NADH at high concentrations (IC50 values = 15 mM, 28 mM, and 11 mM, respectively). Importantly, these studies show these sirtuins have an approximate 1000-fold lower affinity for NADH compared to NAD+ [54].
With the newly discovered long chain deacylase activities of human SIRT1–3 and SIRT6, as well as the dicarboxyl-derived deacylase activities of SIRT5, we recently elaborated on these reports [50]. We investigated deacetylation by SIRT1, 2, and 3; demyristoylation by SIRT1, 2, 3, and 6; and desuccinylation and deglutarylation by SIRT5 using H3K9 acylated peptide substrates. Consistent with deacetylation, were found millimolar concentrations of NADH are required to inhibit the enzymatic deacylation. Similarly, using short pseudopeptide substrates, millimolar concentrations were needed to inhibit deoctanoylation by SIRT1, 2, 3, and 6. We also found that NADH inhibits both SIRT1-mediated deacetylase and SIRT5-mediated desuccinylase activities via a non-competitive mechanism for both peptide substrate and NAD+ [50]. Based on these findings, we could conclude that millimolar concentrations of NADH are required to bind to and/or inhibit deacetylase and deacylase activities of the tested mammalian sirtuins.
Molecular dynamics simulations revealed that binding in the so-called A and C pockets on the sirtuins, which accommodate the dinucleotides and are required for catalysis, favored NAD+. In the simulations with NADH, the nicotinamide moiety lacked the hydrogen bonding observed for NAD+ to residues in the C pocket and hence did not stay in this pocket during simulations [50]. Interestingly, the simulations applying SIRT1 agreed particularly with differences in preferred conformations of the two dinucleotides determined by density functional theory (DFT) calculations, showing that the pyramidalization of the pyridyl nitrogen (N1) and C4 of the 1,4-dihydropyridine affects rotation of both the ribose moiety and the exocyclic primary amide functionality of the nicotinamide [55, 56]. Thus, the C-O bond of the ribose moiety adopts a perpendicular arrangement with respect to the dihydropyridine ring in NADH and an in-plane conformation with respect to nicotinamide in NAD+ (Figure 4). Furthermore, the energetic barrier of rotation of the exocyclic primary amide is two-fold higher for NADH [55], and it stays in the favored coplanar conformation in both reported simulations [50], whereas this functionality is rotated slightly in both NAD+ simulations and engages in the hydrogen bonding in the C pocket mentioned above (Figure 4). These differences may also explain the lower binding affinities indicated by the Ki values for NADH compared to Km for NAD+ in sirtuins [50], rendering it unlikely for the sirtuins tested to bind NADH to a significant extent under physiological conditions.
Figure 4.
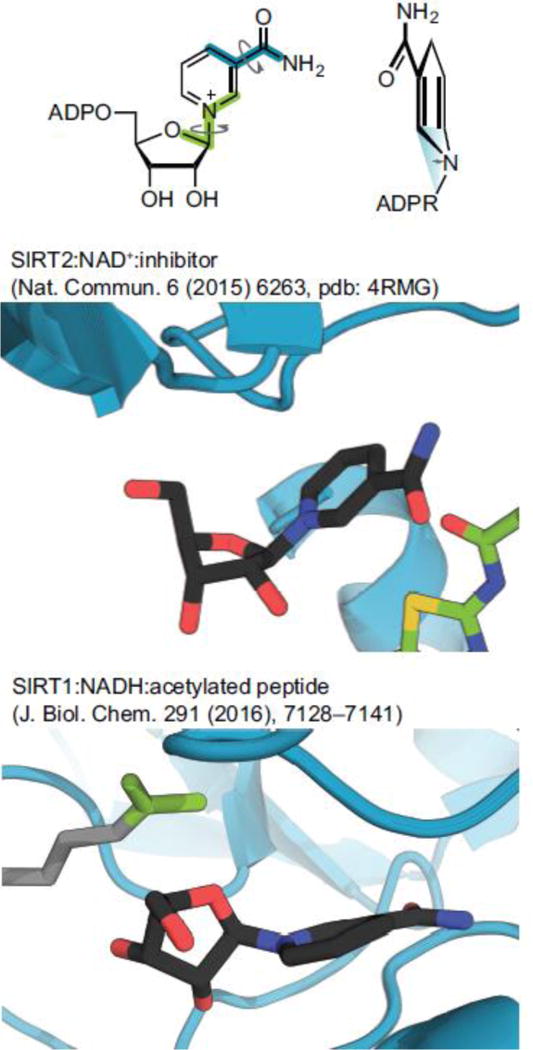
Conformations of NAD+ vs. NADH. Top panel, dihedral angles: Glycosidic bond (green) and exocyclic amide (blue), pyramidalization of nitrogen (blue triangle); Middle panel, crystal structure of SIRT2 in complex with NAD+ and SirReal2–a SIRT2 selective inhibitor (pdb: 4RMG) showing almost parallel orientation of the planar pyrimidine moiety and the C–O bond in the ribose ring; Bottom panel, structure from molecular modeling of SIRT1 in complex with NADH and an acetylated substrate showing almost perpendicular orientation of the dihydropyridine ring, with significant pyramidalization of the nitrogen atom.
Sirtuins as sensors of NAD+
Because sirtuins use NAD+ as a co-substrate, numerous studies have suggested that the sirtuins ability to sense changes in NAD+ is the primary regulatory link between energy state and sirtuin function. As described above, the basal free NAD+ concentrations in the nuclear–cytosolic and mitochondrial compartments are 10–100 μM and ~230 μM, respectively. Since these levels fall within the range of measured sirtuin Km values for NAD+, sirtuins could function as sensors of NAD+. The question remains: do changes in intracellular NAD+ concentration occur that are large enough to be sensed? Numerous studies have reported intracellular NAD+ concentration changes under various physiological conditions with magnitudes in the range of 2–5-fold. For example, NAD+ levels fluctuate daily via activation of the NAD+ salvage pathway as part of a mechanism to synchronize energy metabolism with circadian rhythms and the light-dark cycle [57]. As another example, NAD+ concentrations change in response to nutritional and environmental conditions. Specifically, levels increase in response to conditions associated with low nutrient availability such as calorie restriction, fasting, glucose or serum deprivation, and exercise [23, 58–62]. In contrast, NAD+ levels decrease from over-nutrition such as occurs from high-fat diet feeding [63]. NAD+ levels also decline during aging and senescence of human cells and in rodents [64–66].
Studies into how changes in NAD+ concentration occur are ongoing. The control of basal levels of NAD+ and of concentration changes result from a dynamic balance between NAD+ synthesis and NAD+ consumption. Enzyme classes including PARPs, ARTs, and SIRTs consume NAD+, enough in some situations to deplete NAD+ pools [1]. The consuming enzymes generate NAM as a byproduct of their enzymatic activities that feedbacks to regulate their activity by directly binding the NAD+ binding pocket to inhibit activity and as a biosynthetic precursor to NAD+ production via NAMPT. The repression or induction of Nampt expression often parallels the observed reduction or increase in NAD+ concentration and is thought to be the mechanism behind altered NAD+ levels in many contexts.
In addition, studies have linked changes in NAD+ to altered activity of sirtuins under various physiological conditions. NAMPT-driven NAD+ synthesis and SIRT1 were shown to regulate part of the core circadian machinery, CLOCK/BMAL, in an interlocked transcriptional-enzymatic feedback loop [67]. In peripheral tissue, NAMPT and NAD+ levels cycle with a 24-hr rhythm controlled by the clock. In this cycle, NAD+ functions as a metabolic oscillator that periodically activates SIRT1 deacetylase activity that in turn represses CLOCK/BMAL-mediated transcription of CLOCK target genes, including Nampt itself [67]. Oscillations in the level of SIRT1 deacetylase activity are in phase with the NAD+ oscillations, consistent with SIRT1 functioning as an NAD+ sensor.
CLOCK-dependent NAD+ cycling also generates rhythms in mitochondrial SIRT3 deacetylase activity [57]. SIRT3-dependent modulation of mitochondrial protein acetylation, in turn, contributes to the synchronization of oxidative metabolism across the 24-hr fasting and feeding cycle. Knockout of the circadian clock component Bmal in mice abolishes the rhythmic oscillation of NAD+ levels, reduces the total endogenous NAD+ concentration in liver, and correspondingly diminishes SIRT3 activity [57]. These findings are consistent with the idea that SIRT3 activity is impaired in Bmal−/− liver due to reduced NAD+ and that SIRT3 thus functions as an NAD+ sensor in response to circadian cycling of NAD+ levels.
NAD+ levels have been shown to decrease with aging in rats and mice and in primary and cultured cells, with effects on SIRT1 activity observed in some cases. For example, a 2–4 fold decrease in NAD+ levels in ex vivo samples of hippocampus, cortex, cerebellum, and brain stem from aging rats was reported along with a proportionate decrease in SIRT1 deacetylase activity [65]. In another study, a 2–3-fold decrease in nuclear NAD+ in aging mouse skeletal muscle was shown to coincide with decreased SIRT1 activity [64]. These events were linked to increased HIF-1, blocked TFAM activity, and reduced expression of OXPHOS components in the mitochondria, creating reduced OXPHOS and a pseudohypoxic, Warburg-like state. In a third aging study, a magnetic resonance-based in vivo NAD+ assay was used to noninvasively measure NAD+, NADH, and NAD+/NADH content in intact human brain of healthy volunteers [68]. An age-dependent increase in intracellular NADH and age-dependent decreases in total NAD, NAD+, and the NAD+/NADH ratio were reported. Although the changes, observed across individuals 20–65 years old, were subtle and gradual the finding follows the trend of ex vivo studies showing much higher NAD+ changes in rodent brain from different age groups. Effects on sirtuin activity were not mentioned in the study.
On the contrary, sirtuin activity changes are not always clearly linked to changes in NAD+. For example, a rapid cAMP/PKA-mediated phosphorylation of SIRT1 on serine434 was shown to increase SIRT1 deacetylase activity by decreasing affinity of the enzyme for NAD+[69]. Phospho-SIRT1 modulates gene expression to promote fatty acid oxidation independently of changes in NAD+ levels. Interestingly, during calorie restriction (CR) or fasting, SIRT1 mRNA and protein levels are upregulated in various metabolic tissues by cAMP/PKA/CREB signaling. Conversely, in the absence of metabolic stress, ChREBP represses SIRT1 expression [70]. CR and fasting are two well-studied metabolic contexts leading to increased NAD+ levels that parallel increased SIRT1 activity. Thus, increased SIRT1 activity during CR and fasting, purportedly due to increased NAD+, could be influenced by increases in SIRT1 expression.
In another study, SIRT1 was shown to bind to and recruit NMNAT1 to specific gene promoters in the breast cancer cell line MCF-7 [71]. The SIRT1 at these target promoters required NMNAT1 for deacetylase activity. Though the precise mechanism of SIRT1 activation by NMNAT1 was not defined, NMNAT1 was hypothesized to promote more efficient use of NAD+ by SIRT1 via substrate channeling or by creating a high local concentration of NAD+ specifically for SIRT1. In this scenario, SIRT1 responds to NAD+ produced at the site of SIRT1 function, as opposed to sensing an NAD+ concentration change in the nuclear pool. An alternative mechanism suggested was the allosteric activation of SIRT1 activity by NMNAT1, as NMNAT1 has been shown to bind to and allosterically activate PARP1 in other contexts [72]. This later type of mechanism might operate independently of NAD+ altogether.
Thus, while the physiological contexts in which changing NAD+ has been described (e.g. CR, fasting, HFD-feeding, aging), how these complex metabolic events change free NAD+ is not yet known. Despite the notion that sirtuin regulation can occur independently of changes in NAD+, we conclude that strong evidence exists for sirtuins to be sensors of NAD+.
Sirtuins as sensors of NAD+/NADH
In contrast to NAD+, little evidence exists for sirtuins to sense the NAD+/NADH ratio. As described above, the free NAD+ concentration in the nuclear–cytoplasmic pool is several orders of magnitude higher than that of NADH (Figure 2), so that the free NAD+/NADH ratio is ~700:1 [9]. Because the concentration of NAD+ is so much higher compared to NADH, the conversion of a small amount of NAD+ to NADH would be expected to result in a relatively large change in the NADH, but a small change in NAD+. It follows that NADH can provide a more sensitive reflection of a change in redox state compared to NAD+ and thus the best sensors of NAD+/NADH sense NADH at a lower Km than NAD+. The free NADH concentration in the nuclear-cytoplasmic pool is ~100 nM. Since the sirtuin Km for NADH is about 1000-fold above this level, the sirtuins are unlikely to serve as very good sensors of the NAD+/NADH ratio within the nuclear–cytoplasmic compartment. In contrast, the nuclear NADH sensor is the transcription corepressor CtBP described above binds NADH with an affinity close to the physiological free NADH concentration, and binding to NADH increases its ability to interact with E1A [28]. The same logic could be applied to mitochondria, where the NAD+ concentration is much higher compared to NADH, although with an NAD+/NADH ratio of 8, the difference is not great compared to that of the nuclear–cytosolic pool. Thus, the current knowledge about the physiological levels of free NAD+ and NADH in different organelles, combined with the observed IC50 values, strongly suggest that sirtuins are not sensors of the NAD+/NADH ratio.
Outstanding questions
While much is known about how the sirtuins bind NAD+ and NADH, several questions remain regarding their role as sensors. For example: does NAD+ fluctuate in compartments or local sirtuin-resident environments enough to influence sirtuin activity? Several physiological conditions have been described that influence the intracellular NAD+ or NADH levels. However, in many of these studies, the changes in NAD(H) were determined by quantification of total NAD+ and NADH levels. If, and to what degree, these changes in total levels influenced the free NAD+ concentration in specific subcellular compartments were not determined. A wide range of pathophysiological conditions including oxidative phosphorylation deficiencies and cardiac and renal diseases have also been associated with alterations in NAD+ and/or NADH levels [recently reviewed in [1, 2]], but again, effects on the free NAD+ concentration were often not determined. Now that direct quantification of free NAD+ in subcellular compartments of live cells is possible with genetically encoded biosensors [19], a challenge exists in extending this technology to other relevant physiological/pathophysiological contexts that might influence fluctuations in NAD(H). Studies along this line should lead to a clearer picture of sirtuin—NAD+-sensing opportunities. Finally, boosting NAD+ using precursor supplementation (NMN, NR) or small molecular inhibitors of NAD+-consuming enzymes increases NAD+ levels [1]. The role of sirtuin activation in these NAD+-centric strategies has not yet been studied and represents another important area of future investigation.
Conclusions
Sirtuins are often described as “NAD+ sensors” that link the cellular energy state to a response since they require NAD+ as co-substrate for protein deacylase activity. Free NAD+ concentrations in nuclear-cytosolic and mitochondrial compartments under basal conditions fall within the range of measured sirtuin Km values for NAD+, indicating the possibility of sirtuins to function as sensors. Numerous studies report changes in NAD+ concentrations in response to various physiological conditions with a magnitude of 2–5-fold. In many of these studies, for example, in response to circadian oscillations of NAD+, as well as in response to nutritional or environmental challenges, changes in NAD+ concentrations have been linked to corresponding changes in sirtuin activity, consistent with the behavior of an NAD+ sensor. Collectively these findings support the notion that sirtuins sense NAD+. In contrast, little evidence supports the sirtuins as sensors of the NAD+/NADH ratio. Since the physiological free concentration of NAD+ is so much higher compared to NADH, the best sensors of NAD+/NADH sense NADH. The sirtuin Km for NADH is 1000-fold above the physiological free concentration of NADH and therefore sirtuins are unlikely to serve as good sensors of NADH or of the NAD+NADH ratio. Interestingly, although sirtuins likely sense NAD+, mechanisms beyond changes in NAD+ concentrations could regulate sirtuin activity, including posttranslational modification or changes in expression levels. Future studies integrating NAD+ sensing with other mechanisms of activation to alter sirtuin activity will undoubtedly lead to exciting new discoveries.
Highlights.
NAD+ and NADH are ubiquitous cellular redox cofactors
Enzymes outside those participating directly in redox sense these dinucleotides
The sirtuin family of protein deacylases use NAD+ to remove protein modifications
Sirtuins sense NAD+, but neither NADH nor the ratio
Acknowledgments
We would like to thank the entire Hirschey lab for feedback. Work in the Hirschey lab is supported by the American Heart Association grants 12SDG8840004 and 12IRG9010008, The Ellison Medical Foundation, the National Institutes Aging (NIA) grant R01AG045351, the NIAAA grant R01AA022146, the Duke Pepper Older Americans Independence Center (OAIC) Program in Aging Research supported by the National Institute of Aging (P30AG028716-01). Work in the Olsen lab is supported by the Danish Independent Research Council–Technology and Production Sciences (Sapere Aude Grant 12-132328; ASM), The Carlsberg Foundation (CF15-0115; CAO), The Novo Nordisk Foundation (NNF15OC0017334; CAO), and the European Research Council (ERC-CoG-725172, SIRFUNCT; CAO)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hershberger KA, Martin AS, Hirschey MD. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nat Rev Nephrol. 2017;13:213–225. doi: 10.1038/nrneph.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cantó C, Menzies KJ, Auwerx J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conforti L, Janeckova L, Wagner D, Mazzola F, Cialabrini L, Di Stefano M, Orsomando G, Magni G, Bendotti C, Smyth N, Coleman M. Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J. 2011;278:2666–2679. doi: 10.1111/j.1742-4658.2011.08193.x. [DOI] [PubMed] [Google Scholar]
- 4.Frederick DW, Loro E, Liu L, Davila A, Jr, Chellappa K, Silverman IM, Quinn WJ, 3rd, Gosai SJ, Tichy ED, Davis JG, Mourkioti F, Gregory BD, Dellinger RW, Redpath P, Migaud ME, Nakamaru-Ogiso E, Rabinowitz JD, Khurana TS, Baur JA. Loss of NAD Homeostasis Leads to Progressive and Reversible Degeneration of Skeletal Muscle. Cell Metab. 2016;24:269–282. doi: 10.1016/j.cmet.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 2004;18:12–16. doi: 10.1101/gad.1164804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15:241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 7.Easlon E, Tsang F, Dilova I, Wang C, Lu SP, Skinner C, Lin SJ. The dihydrolipoamide acetyltransferase is a novel metabolic longevity factor and is required for calorie restriction-mediated life span extension. J Biol Chem. 2007;282:6161–6171. doi: 10.1074/jbc.M607661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilhelm F, Hirrlinger J. The NAD+/NADH redox state in astrocytes: independent control of the NAD+ and NADH content. J Neurosci Res. 2011;89:1956–1964. doi: 10.1002/jnr.22638. [DOI] [PubMed] [Google Scholar]
- 9.Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in cytoplasm and mitochondria of rat liver. Biochemical J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen CE, Karlsson M, Winther JR, Jensen PR, Lerche MH. Noninvasive in-cell determination of free cytosolic [NAD+]/[NADH] ratios using hyperpolarized glucose show large variations in metabolic phenotypes. J Biol Chem. 2014;289:2344–2352. doi: 10.1074/jbc.M113.498626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson GH, Knobel SM, Arkhammar P, Thastrup O, Piston DW. Separation of the glucose-stimulated cytoplasmic mitochondrial NAD(P)H responses in pancreatic islet β cells. Proc Natl Acad Sci U S A. 2000;97:5203–5207. doi: 10.1073/pnas.090098797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang QH, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science. 2002;295:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 13.Vishwasrao HD, Heikal AA, Kasischke KA, Webb WW. Conformational dependence of intracellular NADH on metabolic state revealed by associated fluorescence anisotropy. J Biol Chem. 2005;280:25119–25126. doi: 10.1074/jbc.M502475200. [DOI] [PubMed] [Google Scholar]
- 14.Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, Duchen MR. Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat Commun. 2014;5:3936. doi: 10.1038/ncomms4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao Y, Yang Y. Profiling metabolic states with genetically encoded fluorescent biosensors for NADH. Curr Opin Biotechnol. 2015;31:86–92. doi: 10.1016/j.copbio.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Hung YP, Albeck JG, Tantama M, Yellen G. Imaging cytosolic NADH-NAD+ redox state with a genetically encoded fluorescent biosensor. Cell Metab. 2011;14:545–554. doi: 10.1016/j.cmet.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y, Jin J, Hu Q, Zhou HM, Yi J, Yu Z, Xu L, Wang X, Yang Y, Loscalzo J. Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab. 2011;14:555–566. doi: 10.1016/j.cmet.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bilan DS, Matlashov ME, Gorokhovatsky AY, Schultz C, Enikolopov G, Belousov VV. Genetically encoded fluorescent indicator for imaging NAD+/NADH ratio changes in different cellular compartments. Biochim Biophys Acta. 2014;1840:951–957. doi: 10.1016/j.bbagen.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cambronne XA, Stewart ML, Kim D, Jones-Brunette AM, Morgan RK, Farrens DL, Cohen MS, Goodman RH. Biosensor reveals multiple sources for mitochondrial NAD+ Science. 2016;352:1474–1477. doi: 10.1126/science.aad5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen WW, Freinkman E, Wang T, Birsoy K, Sabatini DM. Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell. 2016;166:1324–1337. doi: 10.1016/j.cell.2016.07.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blinova K, Carroll S, Bose S, Smirnov AV, Harvey JJ, Knutson JR, Balaban RS. Distribution of mitochondrial NADH fluorescence lifetimes: Steady-state kinetics of matrix NADH interactions. Biochemistry. 2005;44:2585–2594. doi: 10.1021/bi0485124. [DOI] [PubMed] [Google Scholar]
- 22.Yu Q, Heikal AA. Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J Photochem Photobiol, B. 2009;95:46–57. doi: 10.1016/j.jphotobiol.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, Lamming DW, Souza-Pinto NC, Bohr VA, Rosenzweig A, de Cabo R, Sauve AA, Sinclair DA. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell. 2007;130:1095–1107. doi: 10.1016/j.cell.2007.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pittelli M, Formentini L, Faraco G, Lapucci A, Rapizzi E, Cialdai F, Romano G, Moneti G, Moroni F, Chiarugi A. Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J Biol Chem. 2010;285:34106–34114. doi: 10.1074/jbc.M110.136739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Magrane M. UniProt Consortium, UniProt Knowledgebase: a hub of integrated protein data. Database. 2011;2011 doi: 10.1093/database/bar009. bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hua YH, Wu CY, Sargsyan K, Lim C. Sequence-motif detection of NAD(P)-binding proteins: discovery of a unique antibacterial drug target. Sci Rep. 2014;4:6471. doi: 10.1038/srep06471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar V, Carlson JE, Oghi KA, Edwards TA, Rose DW, Escalante CR, Rosenfeld MG, Aggarwal AK. Transcription Corepressor CtBP is an NAD+-regulated Dehydrogenase. Mol Cell. 2002;10:857–869. doi: 10.1016/s1097-2765(02)00650-0. [DOI] [PubMed] [Google Scholar]
- 28.Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci U S A. 2003;100:9202–9207. doi: 10.1073/pnas.1633591100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nardini M, Spanò S, Cericola C, Pesce A, Massaro A, Millo E, Luini A, Corda D, Bolognesi M. CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J. 2003;22:3122–3130. doi: 10.1093/emboj/cdg283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science. 2001;293:506–509. doi: 10.1126/science.1060699. [DOI] [PubMed] [Google Scholar]
- 31.Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–514. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Wu Y, Li L, Su XD. Intermolecular recognition revealed by the complex structure of human CLOCK-BMAL1 basic helix-loop-helix domains with E-box DNA. Cell Res. 2013;23:213–224. doi: 10.1038/cr.2012.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dai X, Li Y, Meng G, Yao S, Zhao Y, Yu Q, Zhang J, Luo M, Zheng X. NADPH is an allosteric regulator of HSCARG. J Mol Biol. 2009;387:1277–1285. doi: 10.1016/j.jmb.2009.02.049. [DOI] [PubMed] [Google Scholar]
- 34.Zheng X, Dai X, Zhao Y, Chen Q, Lu F, Yao D, Yu Q, Liu X, Zhang C, Gu X, Luo M. Restructuring of the dinucleotide-binding fold in an NADP(H) sensor protein. Proc Natl Acad Sci U S A. 2007;104:8809–8814. doi: 10.1073/pnas.0700480104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao Y, Zhang J, Li H, Li Y, Ren J, Luo M, Zheng X. An NADPH sensor protein (HSCARG) down-regulates nitric oxide synthesis by association with argininosuccinate synthetase and is essential for epithelial cell viability. J Biol Chem. 2008;283:11004–11013. doi: 10.1074/jbc.M708697200. [DOI] [PubMed] [Google Scholar]
- 36.McLaughlin KJ, Strain-Damerell CM, Xie K, Brekasis D, Soares AS, Paget MSB, Kielkopf CL. Structural basis for NADH/NAD+ redox sensing by a Rex family repressor. Mol Cell. 2010;38:563–575. doi: 10.1016/j.molcel.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Brekasis D, Paget MS. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2) EMBO J. 2003;22:4856–4865. doi: 10.1093/emboj/cdg453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rine J, Herskowitz I. Four genes responsible for a position effect on expression from HML and HMR in Saccharomyces cerevisiae. Genetics. 1987;116:9–22. doi: 10.1093/genetics/116.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gottlieb S, Esposito RE. A new role for a yeast transcriptional silencer gene, SIR2, in regulation of recombination in ribosomal DNA. Cell. 1989;56:771–776. doi: 10.1016/0092-8674(89)90681-8. [DOI] [PubMed] [Google Scholar]
- 40.Frye RA. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem Biophys Res Commun. 1999;260:273–279. doi: 10.1006/bbrc.1999.0897. [DOI] [PubMed] [Google Scholar]
- 41.Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- 42.Braunstein M, Sobel RE, Allis CD, Turner BM, Broach JR. Efficient transcriptional silencing in Saccharomyces cerevisiae requires a heterochromatin histone acetylation pattern. Mol Cell Biol. 1996;16:4349–4356. doi: 10.1128/mcb.16.8.4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braunstein M, Rose AB, Holmes SG, Allis CD, Broach JR. Transcriptional silencing in yeast is associated with reduced nucleosome acetylation. Genes Dev. 1993;7:592–604. doi: 10.1101/gad.7.4.592. [DOI] [PubMed] [Google Scholar]
- 44.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 45.Anderson KA, Green MF, Huynh FK, Wagner GR, Hirschey MD. SnapShot: Mammalian Sirtuins. Cell. 2014;159:956. doi: 10.1016/j.cell.2014.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu A, Su X, Lin H. Detecting sirtuin-catalyzed deacylation reactions using 32P-labeled NAD and thin-layer chromatography. Methods Mol Biol. 2013;1077:179–189. doi: 10.1007/978-1-62703-637-5_12. [DOI] [PubMed] [Google Scholar]
- 47.Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH, He B, Chen W, Zhang S, Cerione RA, Auwerx J, Hao Q, Lin H. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science. 2011;334:806–809. doi: 10.1126/science.1207861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Mühlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feldman JL, Baeza J, Denu JM. Activation of the Protein Deacetylase SIRT6 by Long-chain Fatty Acids and Widespread Deacylation by Mammalian Sirtuins. J Biol Chem. 2013;288:31350–31356. doi: 10.1074/jbc.C113.511261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Madsen AS, Andersen C, Daoud M, Anderson KA, Laursen JS, Chakladar S, Huynh FK, Colaço AR, Backos DS, Fristrup P, Hirschey MD, Olsen CA. Investigating the Sensitivity of NAD+-Dependent Sirtuin Deacylation Activities to NADH. J Biol Chem. 2016;291:7128–7141. doi: 10.1074/jbc.M115.668699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anderson KA, Huynh FK, Fisher-Wellman K, Stuart JD, Peterson BS, Douros JD, Wagner GR, Thompson JW, Madsen AS, Green MF, Sivley RM, Ilkayeva OR, Stevens RD, Backos DS, Capra JA, Olsen CA, Campbell JE, Muoio DM, Grimsrud PA, Hirschey MD. SIRT4 Is a Lysine Deacylase that Controls Leucine Metabolism and Insulin Secretion. Cell Metab. 2017;25:838–855. doi: 10.1016/j.cmet.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galleano I, Schiedel M, Jung M, Madsen AS, Olsen CA, Continuous A. Fluorogenic Sirtuin 2 Deacylase Assay: Substrate Screening and Inhibitor Evaluation. J Med Chem. 2016;59:1021–1031. doi: 10.1021/acs.jmedchem.5b01532. [DOI] [PubMed] [Google Scholar]
- 53.Madsen AS, Olsen CA. Substrates for Efficient Fluorometric Screening Employing the NAD-Dependent Sirtuin 5 Lysine Deacylase (KDAC) Enzyme. J Med Chem. 2012;55:5582–5590. doi: 10.1021/jm300526r. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt MT, Smith BC, Jackson MD, Denu JM. Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J Biol Chem. 2004;279:40122–40129. doi: 10.1074/jbc.M407484200. [DOI] [PubMed] [Google Scholar]
- 55.Wu YD, Houk KN. Theoretical study of conformational features of NAD+ and NADH analogs: Protonated nicotinamide and 1,4-dihydronicotinamide. J Org Chem. 1993;58:2043–2045. [Google Scholar]
- 56.Wu YD, Houk KN. Theoretical evaluation of conformational preferences of NAD+ and NADH: An approach to understanding the stereospecificity of NAD+/NADH-dependent dehydrogenases. J Am Chem Soc. 1991;113:2353–2358. [Google Scholar]
- 57.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen D, Bruno J, Easlon E, Lin SJ, Cheng HL, Alt FW, Guarente L. Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 2008;22:1753–1757. doi: 10.1101/gad.1650608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayashida S, Arimoto A, Kuramoto Y, Kozako T, Honda S, Shimeno H, Soeda S. Fasting promotes the expression of SIRT1, an NAD+ -dependent protein deacetylase, via activation of PPARα in mice. Mol Cell Biochem. 2010;339:285–292. doi: 10.1007/s11010-010-0391-z. [DOI] [PubMed] [Google Scholar]
- 60.Cantó C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, Nyakas C, Radak Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech Ageing Dev. 2010;131:21–28. doi: 10.1016/j.mad.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshino J, Mills KF, Yoon MJ, Imai SI. Nicotinamide Mononucleotide, a Key NAD+ Intermediate, Treats the Pathophysiology of Diet- and Age-Induced Diabetes in Mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, Mercken EM, Palmeira CM, de Cabo R, Rolo AP, Turner N, Bell EL, Sinclair DA. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braidy N, Poljak A, Grant R, Jayasena T, Mansour H, Chan-Ling T, Guillemin GJ, Smythe G, Sachdev P. Mapping NAD+ metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology. 2014;15:177–198. doi: 10.1007/s10522-013-9489-5. [DOI] [PubMed] [Google Scholar]
- 66.Borradaile NM, Pickering JG. NAD+, sirtuins, and cardiovascular disease. Curr Pharm Des. 2009;15:110–117. doi: 10.2174/138161209787185742. [DOI] [PubMed] [Google Scholar]
- 67.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu XH, Lu M, Lee BY, Ugurbil K, Chen W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc Natl Acad Sci U S A. 2015;112:2876–2881. doi: 10.1073/pnas.1417921112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gerhart-Hines Z, Dominy JE, Jr, Blättler SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP, Puigserver P. The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD+ Mol Cell. 2011;44:851–863. doi: 10.1016/j.molcel.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Noriega LG, Feige JN, Canto C, Yamamoto H, Yu J, Herman MA, Mataki C, Kahn BB, Auwerx J. CREB and ChREBP oppositely regulate SIRT1 expression in response to energy availability. EMBO Rep. 2011;12:1069–1076. doi: 10.1038/embor.2011.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics. 2009;8:215–225. doi: 10.1074/mcp.M800187-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Berger F, Lau C, Ziegler M. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci U S A. 2007;104:3765–3770. doi: 10.1073/pnas.0609211104. [DOI] [PMC free article] [PubMed] [Google Scholar]